









-
Corydalis DC. is the largest and most diverse genus of Papaveraceae, comprising more than 500 species, which are mainly divided into four subgenera (subg. Bipapillatae, subg. Cremnocapnos, subg. Sophorocapnos, and subg. Corydalis) and 39 sections[1]. Corydalis is widely distributed in the Northern Hemisphere with a few species extending into East Africa[2]. Moreover, this genus demonstrates remarkable diversity in China, particularly in the Himalaya–Hengduan Mountains and the adjacent regions[2,3]. As a genus with spectacular radiation, Corydalis exhibits an extremely high level of diversity in its leaves, subterranean organs, fruits, seeds, flower color, and the length of spurs, and can adapt to diverse habitats, such as riversides, forests, shrubs, grasslands, screes, or even cliffs. Previous studies speculated that Corydalis had an ancient origin, and underwent rapid radiation after the middle Miocene, which was most likely promoted by the continuous orogenesis and climate change associated with the uplift of the Qinghai-Tibetan Plateau (QTP)[4−7].
The sequencing of plant genomes has greatly advanced our understanding of the underlying diversification mechanisms[8−10]. Although considerable progress has been made recently in the classification and phylogeny of Corydalis, the underlying diversification mechanisms remain poorly understood. Particularly, polyploidy is common in this genus, suggesting that polyploidization possibly contributed to the diversification of Corydalis. Polyploidy could drive dramatic changes in the genome landscapes, such as genome size and structural variation, providing a genetic basis for the adaptation and diversification of species-rich groups. While more and more studies link polyploidization events with speciation, fewer studies have documented chromosomal variation in plant groups that underwent rapid radiations, thus the extent to which it may have contributed to radiation is still elusive, especially in biodiversity hotspots. In Corydalis, only the genomes of C. tomentella and C. yanhusuo have been released to date[11,12], which has hindered our understanding of its diversification and adaptation.
Moreover, a large number of Corydalis species have been frequently used in folk medicine due to their antibacterial, antiviral, and anticancer activities. For instance, C. yanhusuo, C. bungeana, and C. decumbens are the most famous medicinal plants recorded in the Pharmacopoeia of China (
http://db.ouryao.com/yd2020/ ). High medicinal value of C. sheareri, C. hendersonii, C. incisa, C. repens, C. edulis, C. racemosa, and C. pallida, also have been reported[2]. Previous phytochemical investigations have isolated various components from Corydalis, including alkaloids, coumarins, flavonoids, anthraquinones, triterpenes, steroids, and organic acids[13]. Of particular interest are alkaloids, especially benzylisoquinoline alkaloids (BIAs), which have been reported to play a crucial role in sedation, releasing pain, promoting blood circulation, and inhibiting cancer cells[13]. Compared with other genera in Papaveraceae, Corydalis can produce multiple types of BIAs, such as cavidines, apocavidine, tetrahydropalmatine, corydalis, protopine, dehydroapocavidine, and dehydrocavidine. Notably, it was also reported that species-specific BIAs were also isolated in different species of Corydalis[13−16], which indicates the remarkable diversity of BIAs in these plant groups. However, the limited genomic resources hampered an in-depth understanding of the diversity of BIAs in Corydalis. Fortunately, biosynthesis pathways of BIAs have been proposed in Corydalis[12,17], as well as in other Papaveraceae species, such as Macleaya cordata[18], Eschscholzia californica[19], and Papaver somniferum[20−22]. The biosynthesis of BIAs in Corydalis involves a series of enzymes, namely berberine bridge enzyme (BBE), berberine bridge enzyme-like (BBEL), C-methyltransferase (CMT), cytochrome P450 (CYP), norcoclaurine synthase (NCS), N-methylcoclaurine 30-hydroxylase (NMCH), coclaurine N-methyltransferase (CNMT), 4-hydroxyphenylpyruvate decarboxylase (HPPDC), O-methyltransferase (OMT), tyrosine aminotransferase (TAT), tetrahydroprotoberberine N-methyltransferase (TNMT), tyrosine decarboxylase (TYDC), and tyramine 3-hydroxylase (TYR).In this study, PacBio long read sequencing, chromosome conformation capture (3C)-based Hi-C sequencing, and Illumina short-read sequencing were used to assemble a high-quality genome of Corydalis sheareri, one species from subg. Corydalis, the largest and most diverse lineages of Corydalis. Genomic comparison was then carried out between C. sheareri and C. tomentella, one previously reported diploid genome from subg. Sophorocapnos. Additionally, we identified the candidate BIAs biosynthesis genes in representative species in Papaveraceae and traced their evolution history by combing phylogenetic reconstruction, chromosomal location, and gene duplication analyses. Our study will provide more insights into the genome evolution as well as the BIAs diversity in Corydalis.
-
Fresh leaves, stems, rhizomes, and flowers at different developmental stages of C. sheareri were collected from Zhongshan Botanical Garden (Nanjing, China) (Fig. 1a). After collection, these samples were immediately frozen in liquid nitrogen or dried in silica gel followed by preservation at −80 °C in the laboratory. The silica gel-dried leaves were used for the flow cytometry measurement, and the material stored in liquid nitrogen was used for genome and transcriptome sequencing. Genomic DNA was extracted using a modified CTAB method. Total RNA was extracted from leaves, stems, rhizomes, and flowers at different developmental stages using RNAprep Pure Plant Kit (Tiangen, Beijing).
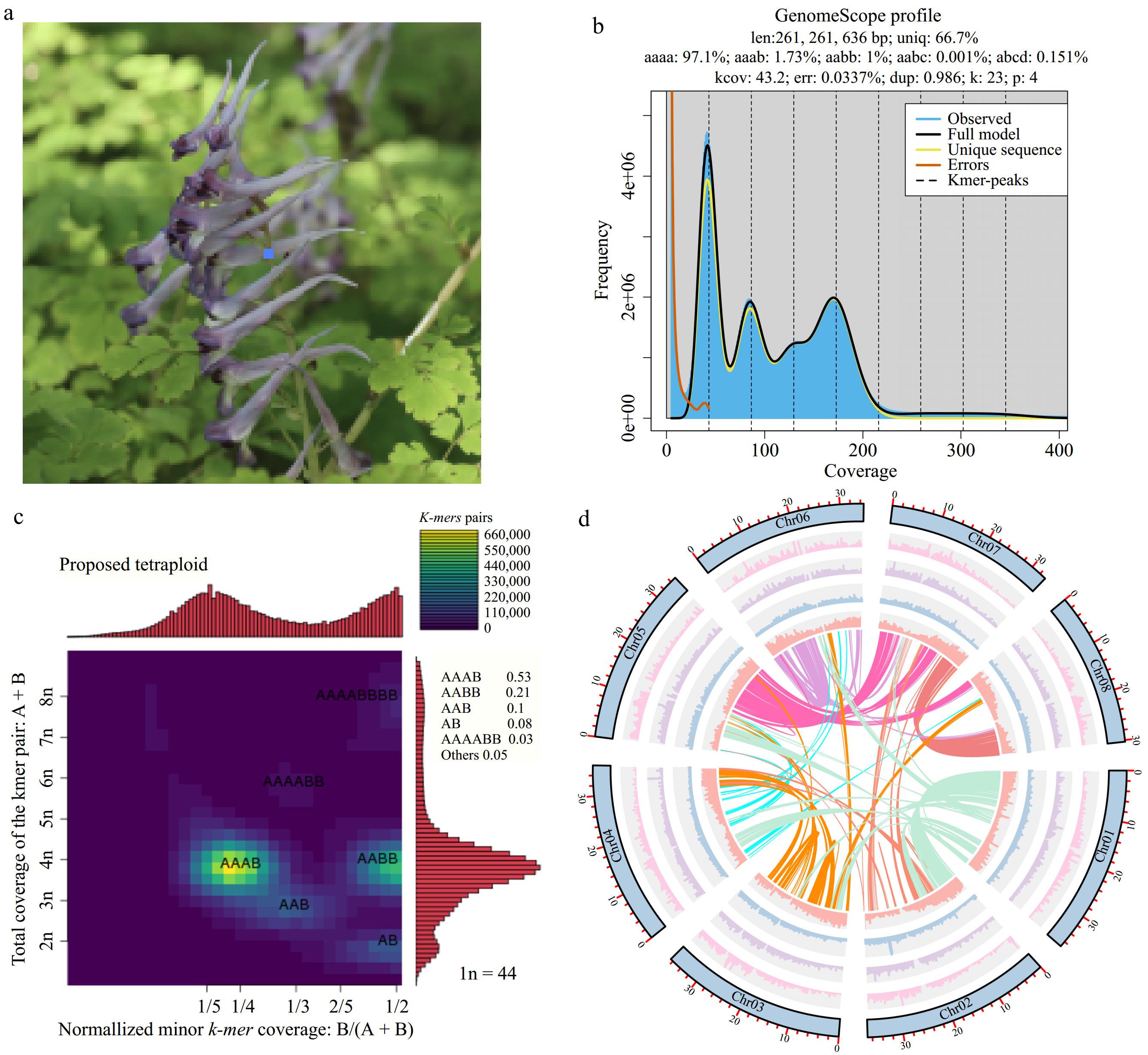
Figure 1.
Overview of Corydalis sheareri genome. (a) Photo of C. sheareri. (b) Genome survey of C. sheareri. (c) Heatmap for coverage pattern of heterozygous k-mer pairs, in which X axis indicates the normalized minor k-mer coverage, while Y axis indicates the total k-mer pairs coverage. (d) Genomic features of eight pseudochromosome. The outermost circle (blue) represents each chromosome of the genome. The bar charts of the second to fifth circles suggest gene density, LTR density, Copia, and Gypsy density, respectively. The inner circular shows inter-chromosomal synteny.
For the genome survey, the 350 bp paired-end library was constructed according to the Illumina protocols and sequenced on the Illumina Novaseq 6000 platform. For PacBio HiFi sequencing, the DNA SMRT bell library with an insert size of approximately 15 kb was prepared using SMRTbell® Express Template Prep Kit 2.0 (Pacific Biosciences, PN 101-853-100), and subsequently sequenced on the PacBio Sequel II platform (Pacific Biosciences, USA). For high-throughput 3C-based Hi-C sequencing, the library was generated using the standard procedures and sequenced on the Illumina Novaseq 6000 platform. For transcriptome sequencing, RNA from different tissues was pooled equally for library construction to obtain more expressed genes. Thereafter, the cDNA library with an insert size of 300−500 bp was prepared using VAHTS mRNA-seq v2 Library Prep Kit for Illumina (Vazyme) and sequenced on the Illumina Novaseq 6000 platform to generate paired-ends reads. All sequencing was carried out in Berry Genomics Company (
www.berrygenomics.com ), Beijing, China. The raw Illumina data for genome survey, Hi-C, and RNA-seq reads, was trimmed using Fastp v0.23.2[23] to remove the adaptors and low-quality paired reads. The HiFi reads were generated through CCS software (https://github.com/PacificBiosciences/ccs ) with the following parameters: --min-passes = 3 --min-rq = 0.99.Genome survey
-
To guide genome sequencing and assembly, flow cytometry[24], and genome survey were used to estimate the genome size. For flow cytometry, the dried material was chopped with a razor blade and then the DNA content was measured following the protocol of CyStain®PI Absolute P (Sysmex-Partec, Germany) with an Elite flow cytometer (BD FACSCalibur, USA) at the Institute of Botany, Chinese Academy of Sciences (China). Finally, the genome size was inferred based on the external reference standards (Glycine max = 1,100 Mb). For genome survey, the obtained clean reads were used to estimate the genome size based on the k-mer method with KMC v3[25], and Genomescope v2.0[26]. Given that multiple peaks were detected in the k-mer spectrum, Smudgeplot[26] was further employed to visualize and evaluate the ploidy and genome structure through the analysis of heterozygous k-mer pairs.
Genome assembly and annotation
-
Hifiasm v0.14.2[27] was used for de novo assembly of the draft contig genome of C. sheareri, which contained unphased contigs from two homolog genomes (csh v1.0). Purge_dups v1.2.3[28] was used to improve the assembly by removing duplications. The filtered Hi-C reads were aligned to the draft genome (csh v1.0) using Juicer v1.6.2[29], and scaffolding was performed on the contigs with 3D-DNA v180922[30]. Juicebox v1.9.8[31] was used to visualize the Hi-C heatmap, and the scaffolds were manually adjusted to get the chromosomal-level assembly of C. sheareri (csh v2.0). BWA v0.7.15[32] and minimap2[33] were used to assess the quality of the genome by aligning the Illumina short reads and the long HiFi reads to the genome, respectively. BUSCO v4.14[34] was performed to evaluate the integrity of the assembled genome by searching against the 1,614 conserved single-copy genes obtained from the embryophyta_odb10 database.
LTR_Finder v1.07[35], MITE-Hunter v1.0[36], and Repeat-Masker v4.1.0 (
www.repeatmasker.org ) were used to predict the repeat sequences. Protein-coding genes were predicted by combining the results of ab initio-based, homology-based, and RNAseq-based predictions. For ab initio prediction, Augustus v3.2.2[37], Snap v6.0[38], Glimmer hmm v3.0.4[39], and GeneMark-ET v4.57[40] were utilized to predict the gene structure in the repeat-masked genome. GeMoMa v1.7.1[41] was used to perform homology prediction with Arabidopsis thaliana, Oryza sativa, Macleaya cordata, and Papaver rhoeas as references. For RNAseq-based prediction, transcriptomic data from different tissues were assembled de novo using trinity v2.2.0[42]. PASA r20140417[43] was used to predict the gene structure based on the obtained transcript. All predicted protein-coding genes were annotated by blast against five databases, including KEGG (www.genome.jp/kegg/brite.html ), Gene Ontology (GO) terms, NR (https://ftp.ncbi.nlm.nih.gov/blast/db/FASTA/nr.gz ), SwissProt (https://ftp.uniprot.org/pub/databases/uniprot/current_release/knowledgebase/complete/uniprot_sprot.fasta.gz ), and eggNOG (http://eggnog5.embl.de/#/app/home ). The tRNA (transfer RNA) was predicted by tRNAscan-SE v2.0[44]. Other types of noncoding RNAs (ncRNAs), including rRNA (ribosomal RNA), miRNA (microRNA), and snRNA (small nuclear RNA), were annotated by BLAST against the Rfam database (https://ftp.ebi.ac.uk/pub/databases/Rfam/14.1/ ).Phylogenomic analysis and whole-genome duplication (WGD) identification
-
Eight species from Papaveraceae were selected for phylogenomic analysis, including C. sheareri, C. tomentella, Eschscholzia californica, Macleaya cordata, Papaver somniferum, Capnoides sempervirens, Ceratocapnos vesicaria, and Hypecoum procumbens, with Aquilegia coerulea (Ranunculaceae) as outgroup. The genomes or transcriptomes were directly retrieved from Genome Warehouse in National Genomics Data Center (
https://ngdc.cncb.ac.cn/gwh ), National Center for Biotechnology Information (www.ncbi.nlm.nih.gov/datasets/genome ), and ONEKP database (https://db.cngb.org/onekp ). Gene families were clustered using protein sequences by OrthoFinder v2.2.7[45] with the parameter '-S diamond' . Each gene family was aligned with MAFFT[46]. Single or low-copy gene families (one or two copies in the polyploid species Papaver somniferum, while one copy in the other eight species) with more than 50 amino acids were retained to reconstruct the phylogenetic tree by RAxML v8.1.17[47] with 1,000 bootstrap replicates under PROTCATWAG model using protein sequences. Divergence time was estimated under a relaxed molecular clock model by the MCMCTree implement in the PAML v4.8[48]. Calibration points retrieved from TimeTree (http://timetree.org ) were used as priors in divergence time estimation, including the split of Papaveraceae and Ranunculaceae (103~117 Ma), the crown age of Papaveraceae (65~111 Ma), the split age of Hypecoideae and Fumarioideae (63~96 Ma) and the crown age of Fumarioideae (38~44 Ma). Gene family expansion and contraction were inferred by CAFE v4.1[49], with an input species tree constructed from the single or low-copy orthologs. Meanwhile, we performed functional enrichment analysis for the expanded gene families by BLAST against the KEGG and GO databases.The whole-genome duplication (WGD) and whole-genome triplication (WGT) events were identified by Ks method, syntenic analyses, and phylogenomic methods. For Ks method, the homologous gene pairs were firstly identified by the all-against-all BLASTP search (e-value cutoff < 1e-5). Then, YN00 in PAML v4.8[48] was called by WGDI[50] to calculate the synonymous substitution rate (Ks) of each gene pair between two species or within a single species. For syntenic analyses, collinear blocks for intra- and interspecies comparisons were detected using MCScanX v0.8[51] with '-s 15', meaning that each block contained at least 15 collinear gene pairs. JCVI v0.8.12[52] was used to draw dotplots of C. sheareri, C. tomentella, V. vinifera, and A. trichopoda with the default parameters. TBtools[53] was used to visualize the synteny between C. sheareri and C. tomentella. Additionally, structural variants between C. sheareri and C. tomentella, i.e., inversion, translocation, and duplication, were identified with SyRI v1.6.3[54]. For phylogenomic methods, gene families were firstly identified by OrthoFinder v2.2.7[45] and multiple sequence alignments were performed by MAFFT[46]. Then, Maximum-Likelihood (ML) trees were constructed using RAxML[47], with bootstrap values estimated from 100 replicates using the PROTCATWAG model. The WGD/WGT event was identified by tree2GD (
https://sourceforge.net/projects/tree2gd/ ) with the default parameter and WGD/WGT events were considered to have occurred according to any of the following conditions: (1) gene duplication (GD) > 500, of which the number of (AB)(AB) type is over 250; (2) GD > 1,500, of which (AB)(AB) type is over 100, and at the same time, the sum of (AB)(AB) type and (AB)A or (AB)B type are over 1,000[55].Evolution of BIA biosynthesis-related gene family
-
To gain more insights into the diversity of BIAs in Corydalis, 12 gene families involved in BIAs biosynthesis, i.e., BBE, BBEL, CMT, CYP719, CYP80B, CYP82N, NCS, NMT, OMT, TAT, TYDC, and TYR, were identified. All BIA biosynthesis genes reported in C. tomentella genome were firstly retrieved by the gene ID reported previously[12]. Then, TBLASTN was conducted to identify the candidate BIA-related genes in C. sheareri and seven other species of Papaveraceae, including Chelidonium majus, Capnoides sempervirens, Ceratocapnos vesicaria, Hypecoum procumbens, Eschscholzia californica, Macleaya cordata, Papaver somniferum, with Aquilegia coerulea as the outgroup. A sequence is regarded as a candidate gene if it encompasses the entire domain region and the pairwise amino acid identity between the queries and the targets exceed 40%. The annotation for each candidate gene was manually checked according to the blast result. All candidate sequences were confirmed by BLAST against the InterPro (
www.ebi.ac.uk/interpro/ ) and the NCBI Conserved Domain Database (CDD,www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi ) with e < 1e-10.To infer the expansion mechanism of the BIAs biosynthesis genes, phylogenetic reconstruction, chromosomal location, and gene duplication analyses were performed. ML analyses were conducted based on the protein sequences by IQ-TREE v1.6.8[56] using the JTT model and 100 bootstrap replications. Gff files, gene files, and targeted BIA biosynthesis gene IDs of C. sheareri and C. tomentella were downloaded or extracted. The chromosome location information was obtained from the genome annotation file, and visualized by TBtools[53]. Gene duplication events were analyzed using MCScanX[51] with a BLASTp search (e < 1e-10). The synonymous (Ks) and nonsynonymous (Ka) values of the identified species-specific tandem duplicated gene pairs were calculated by the M0 model in PAML v4.8[48].
-
Flow cytometry indicated that C. sheareri had a genome size of approximately 580 Mb (Supplementary Fig. S1a). Based on the obtained 66.29 Gb short paired-end reads (Supplementary Table S1), the 23-mer distribution showed that the estimated genome size of C. sheareri was 261 Mb (Fig. 1b), and the k-mer spectrum exhibited four distinct peaks at ~40, 80, 120, 160, which was highly similar to the results for autotetraploids (M. sativa and S. spontaneum)[26]. Moreover, nucleotide heterozygosity analysis showed 1.73% aaab and 1% aabb, which was consistent with the expectation that the heterozygous rate of autotetraploid AAAB would be greater than that of AABB[26]. The Smudgeplot analysis also revealed that more heterozygous k-mer pairs concentrated at 1/4 for the normalized coverage of minor k-mer and 4n for the total coverage of k-mer pairs, and the prevalence of AAAB (53%) was considerably greater than AABB (21%) (Fig. 1c). All these results suggested that the genome of C. sheareri exhibited complex genome structure and possibly an autotetraploid.
PacBio Sequel II sequencing yielded approximately 33.02 Gb of high quality Hifi reads, and the average length and N50 of filtered subreads (1,990,989) were 16,584 bp and 16,614 bp, respectively (Supplementary Table S1). De novo assembly generated the draft genome of C. sheareri (csh v1.0, ~550 Mb) with contig N50 of 9.18 Mb, and the longest contig was approximately 25.09 Mb (Table 1). The assembly was further scaffolded with 67.96 Gb Hi-C data (Supplementary Table S1). Finally, the high-quality genome of C. sheareri (csh v2.0, ~282 Mb) comprised eight pseudochromosomes and 36 contigs (Supplementary Fig. S1b, Supplementary Table S2), with contig N50 of 11.39 Mb (Table 1). The minimum length of the chromosome was greater than 30 Mb (Supplementary Table S2). Approximately 97.62% of DNA reads and 88.32% of RNA-seq reads could be mapped to the assembly genome. BUSCO analysis revealed that 97.1% (1,567/1,614) of the core eukaryotic genes were completely present in the C. sheareri genome, of which 91.4% were single copy (1,475) and 5.7% were duplicated (92), while 0.9% (15), and 2.0% (32) were partially present or missing, respectively.
Table 1. Genome assembly and annotation of Corydalis sheareri.
Analytical process Characteristic C. sheareri Genome survey Genome size (flow cytometry) (Mb) 580 Genome size (k-mer spectrum) (Mb) 261 Assembly_csh v1.0 Number of contigs 2,713 Assembly size (Mb) 550 Contig N50 (Mb) 9.18 Shortest contig (bp) 16,037 Largest contig (bp) 25,090,490 Assembly_csh v2.0 Total number of contigs 179 Assembly size (Mb) 282 Contig N50 (Mb) 11.39 Number of pseudochromosomes 8 GC content 37.11% Annotation Number of protein-coding genes 26,287 Mean gene length (bp) 5,092 Mean CDS length (bp) 1,440 Complete BUSCOs (C) 1,567 Percentage of repeat sequences (%) 44.47 Approximately 44.47% (125,404,725 bp) of the C. sheareri genome was annotated as transposable elements (TEs), of which 26.47% were retrotransposons and 7.37% were transposons (Supplementary Table S3). For retrotransposons, a total of 84,322 LTR elements were identified, of which 53,097 (7.27%) belonged to the Copia superfamily and 29,747 (5.05%) belonged to the Gypsy superfamily (Supplementary Table S3). Based on a combination of homology search, de novo prediction, and RNA-seq based prediction, a total of 26,287 protein-coding genes were confidently annotated for C. sheareri, and the mean lengths of the predicted gene and coding sequence were 5,092 and 1,440 bp, respectively (Table 1). Approximately, 92.39% (24,286/26,287) of the genes were functionally annotated, of which, 24,257, 19,162, 8,566, 5,531, and 23,330 genes showed high similarity to known proteins in the NR, SwissProt, KEGG, GO, and eggNOG databases, respectively (Supplementary Fig. S2). In addition, a total of 2,746 noncoding RNA genes were identified in the genome of C. sheareri, including 1,014 tRNA genes, 919 rRNA genes, 609 snRNA genes, and 86 miRNA genes (Supplementary Table S4).
Genome evolution
-
A total of 28,545 orthologous groups (OGs) were identified in nine selected species (Fig. 2b, Supplementary Table S5). Of them, 7,769 gene families were shared by all species, while 1,714 gene families were specific to Corydalis. Additionally, 1,025 single or low-copy nuclear gene families were identified in these species. After removing the OGs less than 50 aa, 1,003 single or low-copy nuclear gene families were retained to infer a high-confidence species tree. As expected, three subfamilies were recovered in the phylogenomic analysis, and Hypecoideae was strongly supported as a sister to Fumarioideae. Within Fumarioideae, C. sheareri and C. tomentella formed a highly supported clade, while the relationship of Ceratocapnos vesicaria, Capnoides sempervirens, and Corydalis was not fully resolved. Within Papaveroideae, Eschscholzia californica diverged firstly, and Papaver somniferum was sister to Macleaya cordata (Fig. 2a). Molecular dating indicated that the divergence of C. sheareri and C. tomentella was dated to 24.92 Ma, with a 95% confidence interval (95% CI) of 16.35~33.22 Ma (Fig. 2a). Gene family expansion and contraction analyses revealed that 674 and 566 gene families expanded and contracted in Corydalis, respectively (Fig. 2a). GO and KEGG enrichment analyses showed that the significantly expanded gene families were mainly related to response to stimulus, membrane, cell periphery, response to chemical, and cellular response to stimulus, which primarily enriched in secondary metabolites pathways such as phenylpropanoid biosynthesis, pentose, and glucuronate interconversions, photosynthesis, flavonoid biosynthesis, and monoterpenoid biosynthesis (Supplementary Fig. S3). Specifically, cytochrome P450 (CYP) and photosynthesis proteins were also enriched in the KEGG analysis (Supplementary Fig. S3).
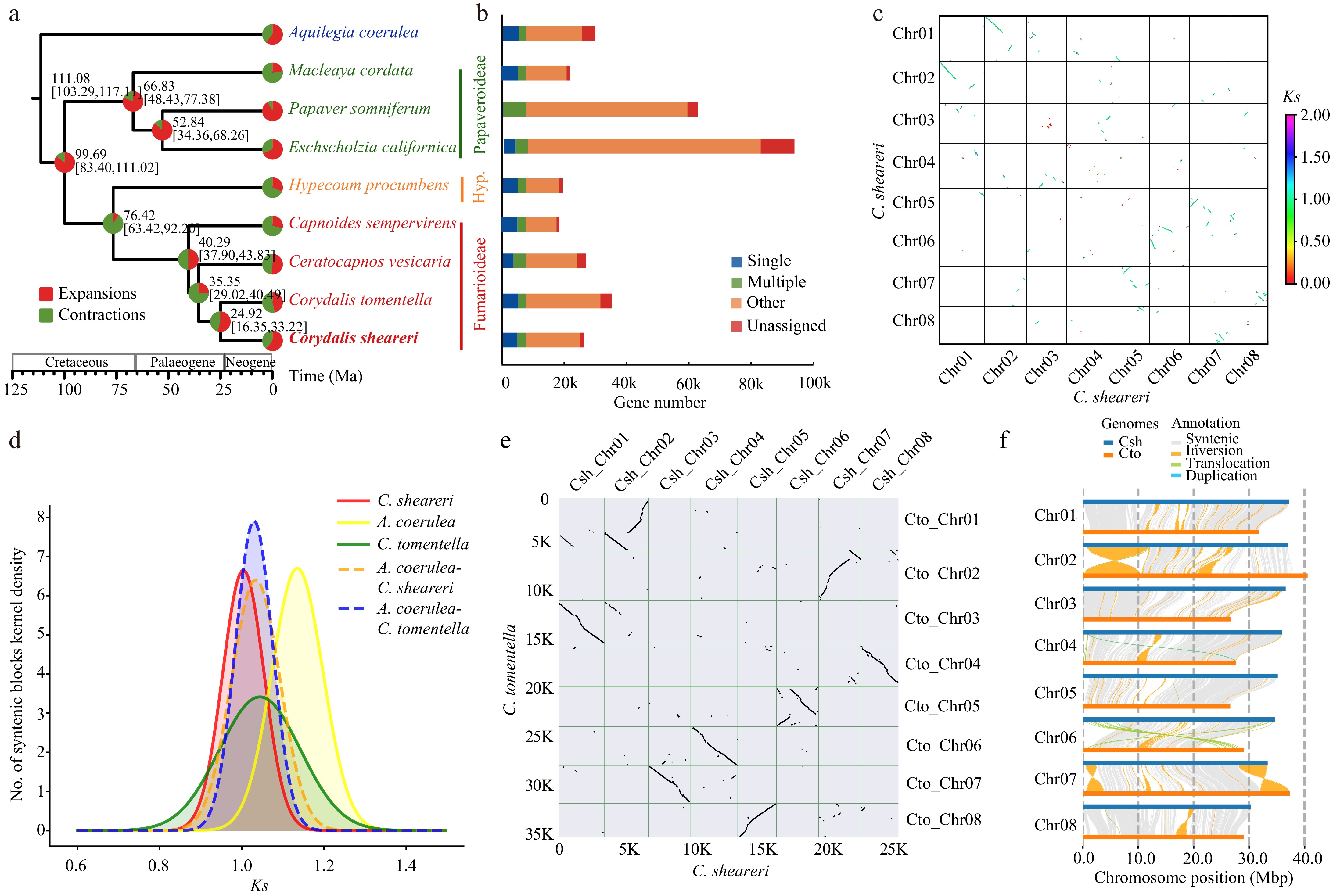
Figure 2.
Genome evolution of Corydalis sheareri. (a) Phylogenetic tree and divergence time estimation. Pal., Palaeogene; Neo., Neogene; Hyp., Hypecooideae. (b) Gene families identified in each species. (c) Collinearity within the genome of C. sheareri. (d) Ks distributions of anchor pairs for the paralogous genes of C. sheareri, C. tomentella and Aquilegia coerulea, and for orthologous genes between C. sheareri and C. tomentella, A. coerulea, respectively. (e) Collinearity analysis between C. sheareri and C. tomentella. (f) Structural variation detection between C. sheareri and C. tomentella performed by SyRI.
WGD events play a crucial role in the duplication and retention of genes. Intra-genomic colinearity analyses uncovered remnants of one WGD event in C. sheareri (Fig. 2c). Obviously, one signature peak of synonymous substitutions per synonymous site (Ks) distribution was detected for the C. sheareri genome at approximately 1.0 (Fig. 2d), indicating an ancient WGD event. Similarly, previously sequenced genomes of Ranunculales species, including C. tomentella, and A. coerulea, also showed a signature peak at 1.0~1.2 in their genomes (Fig. 2d), suggesting this WGD event was probably shared by all Ranunculales species. Comparison of the C. sheareri paralogue Ks distribution between Corydalis and Aquilegia coerulea also indicated a WGD occurred in Ranunculales (Fig. 2d). Previous studies reported that the Vitis vinifera genome had an ancestral hexaploidization[57] and the Amborella trichopoda genome shows no evidence of more recent lineage-specific genome duplications[58]. The inter-genomic comparison and syntenic depths analyses among Corydalis sheareri, Vitis vinifera, and Amborella trichopoda further confirmed that this ancient WGD event, in which two paralogous segments in the Corydalis sheareri genome correspond to one and three orthologous regions in the Amborella trichopoda (with no WGD), and Vitis vinifera (with one WGT) genomes, respectively (Supplementary Fig. S4). The tree2GD analyses revealed 1,281 GDs with 934 (73%) (AB)(AB) retention type, which also strongly suggested the identified WGD event was shared by the ancestor of Ranunculales (node 5) (Supplementary Fig. S5). The genome of Corydalis sheareri and C. tomentella exhibited good collinearity (Fig. 2e, f, Supplementary Fig. S6). A number of large structural variations were detected, including multiple invasions across the genome, and a small fraction of translocations on chromosomes 6 (Fig. 2f). To further verify the authenticity of these structural variations, several specific inversion regions were randomly checked by mapping the HiFi reads to the assembled genome with Integrative Genomics Viewer (IGV)[59]. Our results confirmed that these inversions are genuine and not assembly artifacts (Supplementary Fig. S7).
Chromosomal location and gene duplication of BIAs biosynthesis genes in Corydalis
-
A total of 172 genes involved in the BIAs biosynthesis have been identified in Corydalis sheareri, which is notably larger than that found in C. tomentella (125), Macleaya cordata (128), and Aquilegia coerulea (138), while smaller than that observed in Papaver somniferum (364) and Eschscholzia californica (197) (Fig. 3b; Supplementary Table S6), which has undergone two WGD events[12]. For each gene family, the upstream BIA biosynthetic genes, such as TAT, TYDC, TYR, BBE, and CYP80B (NMCH), generally possess small copy numbers in Corydalis. Conversely, the downstream BIA biosynthetic genes, including OMT, CMT, NMT, and BBEL, typically have a higher copy number (Fig. 3b; Supplementary Table S6). The only exception is NCS, which encodes enzymes to catalyze the condensation of dopamine and 4-hydroxyphenylacetaldehyde (4-HPAA) to produce norcoclaurine, in the upstream of BIA biosynthesis, which also has a greater number of copies (Fig. 3a, b).
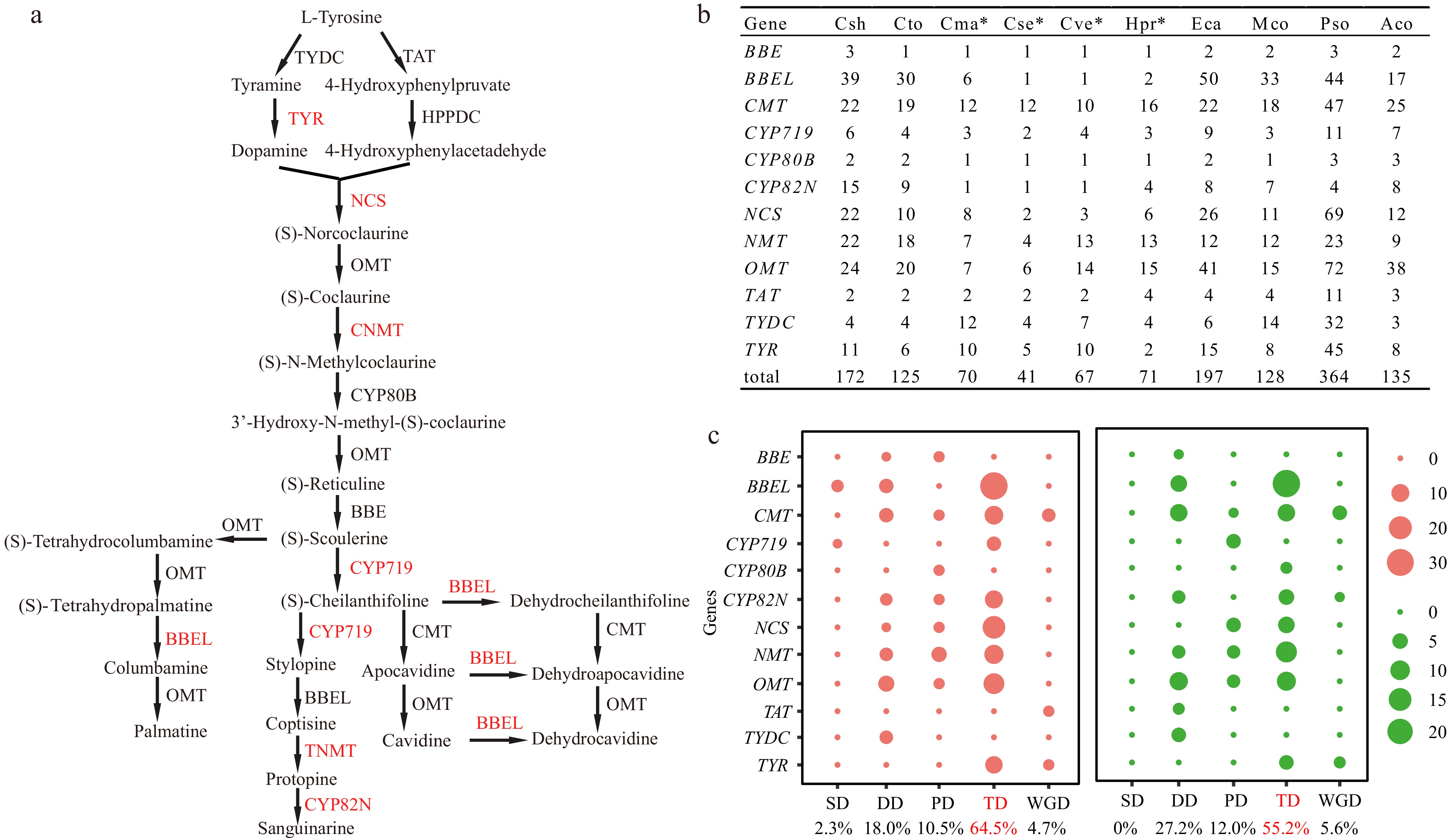
Figure 3.
BIAs biosynthesis related gene families identified in this study. (a) The biosynthetic pathway of benzylisoquinoline alkaloids (BIAs) in Corydalis. The species-specific tandem duplicated gene pairs or clusters in Corydalis identified in Fig. 5 and Table 2 were highlighted with red color. (b) The copy number of BIAs biosynthesis related gene families. Four species with transcriptome sequence are represented by asterisks. (c) Gene duplication type identified in C. sheareri (left) and C. tomentella (right). Abbreviations: BBE, berberine bridge enzyme; BBEL, berberine bridge enzyme-like; CMT, C-methyltransferase; CNMT, coclaurine N-methyltransferase; CYP, Cytochrome P450; HPPDC, 4-hydroxyphenylpyruvate decarboxylase; NCS, norcoclaurine synthase; NMT, N-methyltransferase; OMT, O-methyltransferase; TAT, tyrosine aminotransferase; TNMT, tetrahydroprotoberberine N-methyltransferase; TYDC, tyrosine decarboxylase; TYR, tyramine 3-hydroxylase. Aco, Aquilegia coerulea; Cma, Chelidonium majus; Cse, Capnoides sempervirens; Csh, Corydalis sheareri; Cto, C. tomentella; Cve, Ceratocapnos vesicaria; Eca, Eschscholzia californica; Hpr, Hypecoum procumbens; Mco, Macleaya cordata; Pso, Papaver somniferum. SD, Singleton; DD, dispersed duplications; PD, proximal duplications; TD, Tandem duplications; WGD, segmental/WGD duplications.
Chromosomal location analysis indicated that some BIA biosynthetic genes are unevenly distributed among chromosomes. For instance, all 22 NCS genes of C. sheareri are located on chromosome 6, whereas, in C. tomentella, all members are mapped on the chromosome 5. Additionally, in C. sheareri, 34 out of the 39 BBEL genes are located on chromosome 4, 17 out of the 22 NMT genes are mapped on the chromosome 6, and nine out of 15 CYP82N genes are found on chromosome 3 (Fig. 4a). Similarly, in C. tomentella, 25 out of 31 BBEL genes are found on chromosome 6, and 15 out of the 20 NMT genes are located on chromosome 5 (Fig. 4b). On the contrary, some other genes are widely distributed throughout genomes but are uneven among chromosomes in both two species. For instance, seven out of eight chromosomes (except for chr02) harbor the 22 CMT genes in C. sheareri, and 11 of them are located on chromosome 4. Furthermore, five chromosomes (1, 2, 4, 6, and 8) contain the OMT genes, and both chromosome 2 and 4 harbor nine members (Fig. 4a). In C. tomentella, 20 OMT genes are distributed across five chromosomes (1, 2, 4, 5, and 6). and chromosome 6 harbors eight members, chromosome 1 harbors four (Fig. 4b). Nineteen CMT genes are located on seven chromosomes (except for chr01), and 9 of them are located on chromosome 8 (Fig. 4b). Interestingly, a gene cluster including seven NCS genes and ten NMT genes, was identified in chromosome 6 within a 270-kb region of C. sheareri genome (Fig. 4a).
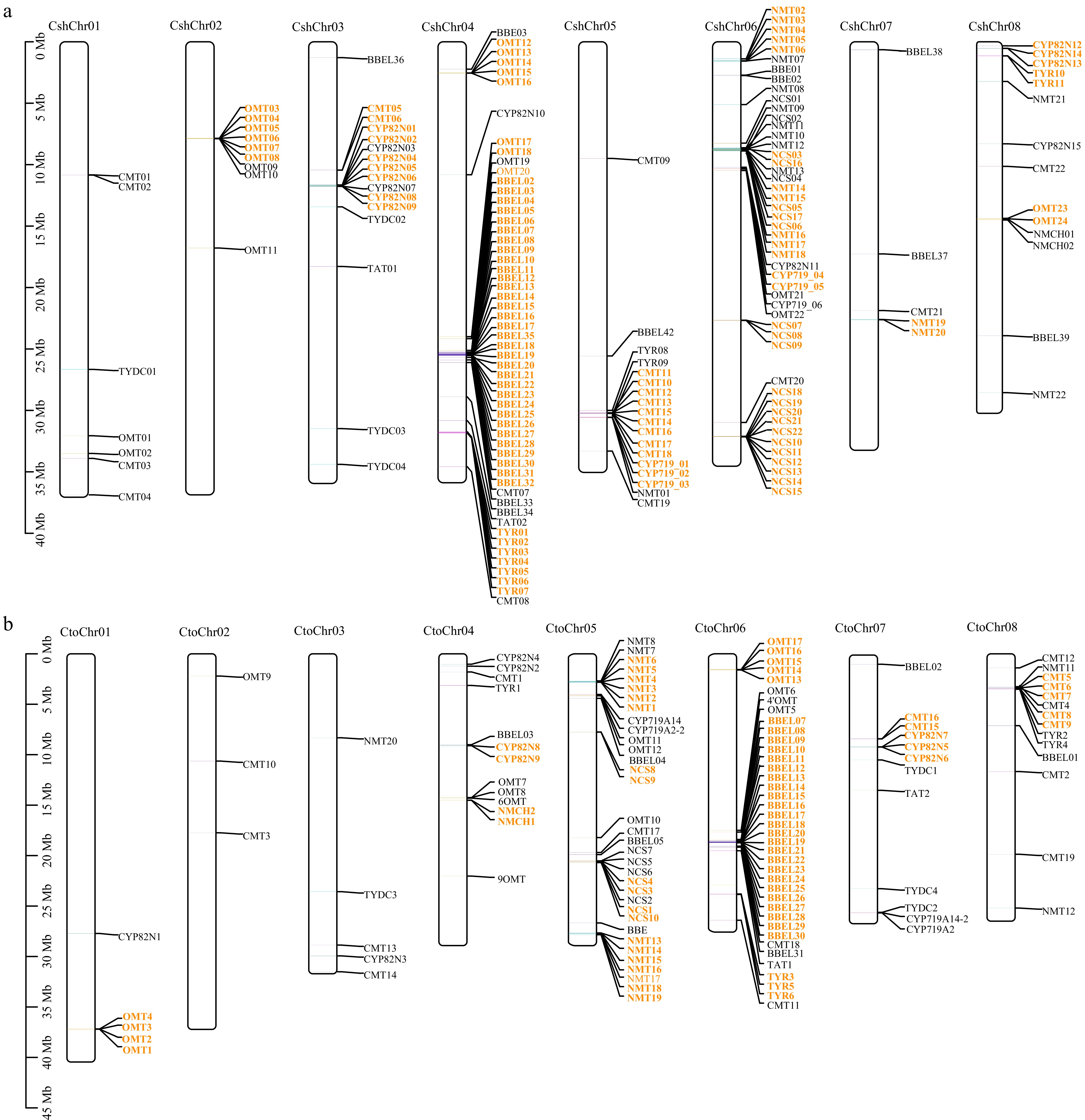
Figure 4.
Chromosome location and duplication event analyses of BIA biosynthesis genes in (a) C. sheareri and (b) C. tomentella. The chromosome number is indicated at the top of each chromosome. Tandem duplicated genes are indicated with orange color. The scale bar on the left indicates the length (mb) of chromosomes. The gene and species abbreviations were the same as Fig. 3. Chr, chromosome.
Gene duplication analysis revealed that the majority of BIAs biosynthesis genes were generated through gene duplication events in Corydalis. Over half of these genes were identified as tandem duplications, with 55.2% in C. tomentella and 64.5% in C. sheareri, respectively (Fig. 3c; Supplementary Table S6). Additionally, 18% to 27.2% of genes were identified as dispersed duplications, and one-tenth of genes, corresponding to 10.5% to 12.0%, were recognized as proximal duplications (Fig. 3c; Supplementary Table S6). Meanwhile, a small fraction, ranging from 4.7% to 5.6%, were identified as segmental/WGD (Fig. 3c; Supplementary Table S6). Specifically, BBEL genes exhibit an extremely high probability of tandem duplication in both C. sheareri and C. tomentella. For instance, 31 BBEL genes (79.4%) of C. sheareri were found to be tandemly duplicated on chromosome 4, while 24 members (80%) of C. tomentella tandemly duplicated on chromosome 6 (Figs 3c, 4; Supplementary Table S6). Moreover, NCS, CYP719, and TYR genes also demonstrated an extremely high rate of tandem duplication in C. sheareri, with 19 out of 22 NCS genes (86%), five out of six CYP719 genes (83%) and nine out of 11 TYR genes (82%) were observed to be tandemly duplicated (Figs 3c, 4a; Supplementary Table S6).
Phylogeny of genes involved in BIAs biosynthesis
-
Among the genes associated with the BIAs biosynthesis, the largest is the BBEL family, with 39 members in Corydalis sheareri and 30 members in C. tomentella, respectively (Fig. 3b; Supplementary Table S6). Phylogenetic analysis of BBE and BBEL revealed three monophyletic clades (BBE, I, and II). Clade II has undergone a significant expansion in Corydalis, encompassing 34 members from C. sheareri and 23 from C. tomentella. Seven C. sheareri or C. tomentella-specific monophyletic groups were identified in clade II, and one of them comprised a notably high number of 14 members from C. sheareri (Fig. 5a).
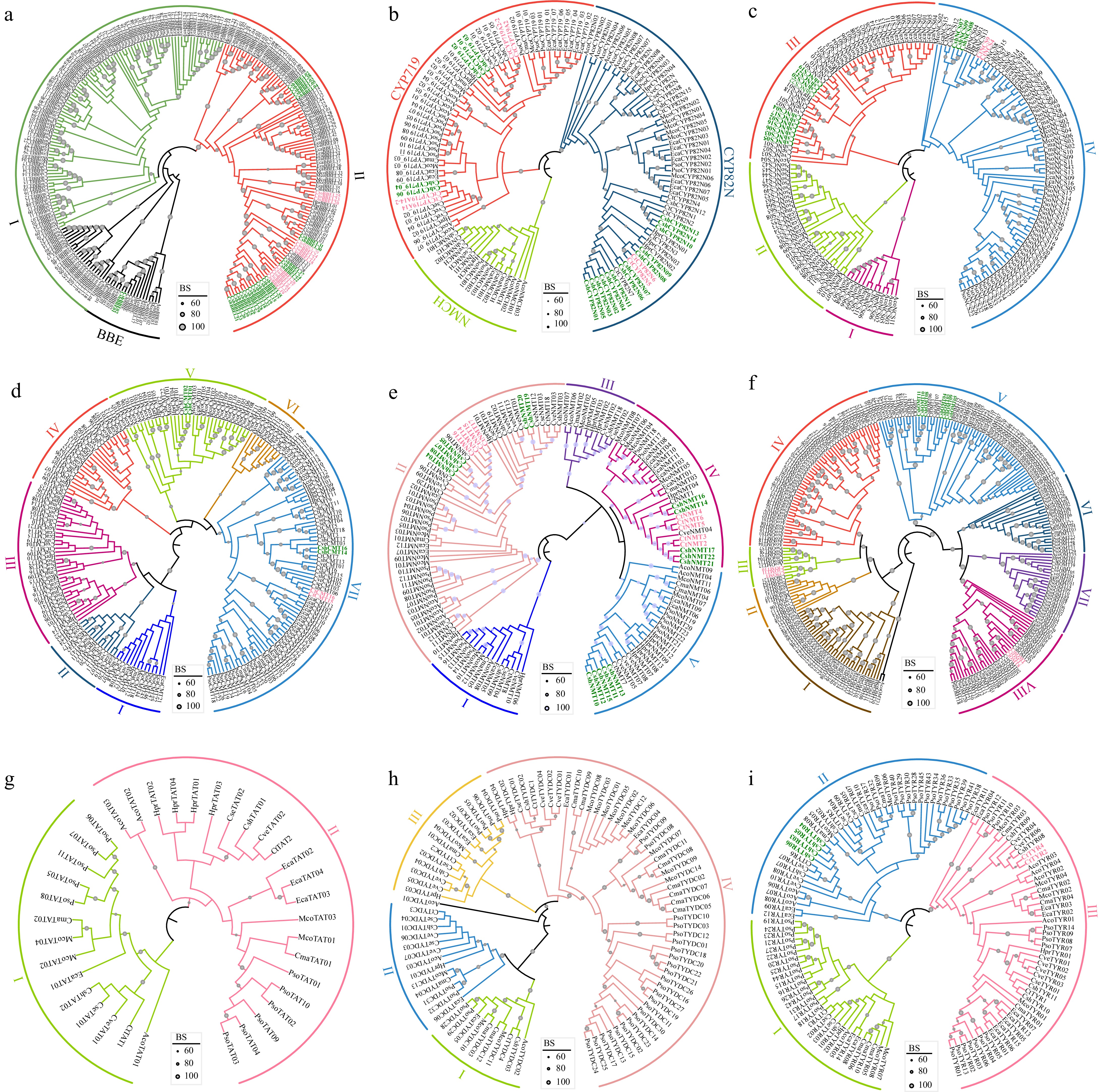
Figure 5.
Phylogenetic trees of BIAs biosynthesis genes. (a) BBE and BBEL, (b) CYP719, CYP80B (NMCH), and CYP82N, (c) NCS, (d) CMT, (e) NMT, (f) OMT, (g) TAT, (h) TYDC, and (i) TYR. Corydalis sheareri and C. tomentella – specific monophyletic groups are highlighted with green and pink color, respectively. The gene and species abbreviations were the same as Fig. 3.
CYPs are important for determining chemical diversity in metabolism, in which, CYP719, CYP80B (NMCH), and CYP82N, have been identified as key components of BIA biosynthesis[12,60,61]. Phylogenetic analysis of these three CYP subfamily suggested that 13 CYP82N genes of C. sheareri (a total of 15 members) were clustered into four monophyletic groups, and two members of C. tomentella (CtoCYP82N5 and CtoCYP82N6) were clustered into one highly supported monophyletic group (Fig. 5b). For CYP719 genes, two C. sheareri specific monophyletic groups were identified, consisting of three (CshCYP719_01, CshCYP719_02, and CshCYP719_03) and two members (CshCYP719_04 and CshCYP719_06), respectively. Similarly, four members of C. tomentella clustered into two monophyletic groups in the phylogenetic tree (Fig. 5b).
Phylogenetic analysis of the NCS genes revealed four highly supported monophyletic clades (I−IV). In clade III, two C. sheareri specific monophyletic groups were identified, comprising seven (CshNCS02, CshNCS03, CshNCS04, CshNCS05, CshNCS06, CshNCS16 and CshNCS17), and three (CshNCS19, CshNCS20 and CshNCS21) members, respectively (Fig. 5c). In clade IV, one C. sheareri specific monophyletic group, and one C. tomentella specific monophyletic group were identified, containing three (CshNCS07, CshNCS08 and CshNCS09) and two members (CtoNCS4 and CtoNCS9), respectively (Fig. 5c).
Phylogeny the CMT genes revealed seven monophyletic clades (I−VII), and the majority of Corydalis members are scattered throughout the phylogenetic tree (Fig. 5d). Conversely, phylogenetic tree of the NMT genes revealed five highly supported clades (I−V) and eight species-specific monophyletic groups were recognized in Corydalis (Fig. 5e). In clade II, three species-specific monophyletic groups were identified, comprising four C. sheareri members (CshNMT04, CshNMT05, CshNMT07, and CshNMT08), four C. tomentella members (CtoNMT14, CtoNMT15, CtoNMT16, and CtoNMT17), and two C. sheareri members (CshNMT19 and CshNMT20), respectively. In clade IV, four species-specific monophyletic groups, with three C. sheareri members (CshNMT17, CshNMT21, and CshNMT22), three C. tomentella members (CtoNMT4, CtoNMT5, and CtoNMT6), two C. sheareri members (CshNMT14 and CshNMT16) and two C. tomentella members (CtoNMT2 and CtoNMT3), respectively. In clade V, one monophyletic group, containing five C. sheareri members (CshNMT10, CshNMT11, CshNMT12, CshNMT13, and CshNMT15), was identified. Similarly, phylogenetic analysis of the NMT genes revealed eight well-supported clades (I−VIII). In clade III, three C. tomentella members (Cto9OMT, CtoOMT10, CtoOMT11) formed one well-supported monophyletic group. In clade VIII, another three C. tomentella members (Cto6OMT, CtoOMT7, and CtoOMT8) also formed one well-supported monophyletic group. While in clade V, two C. sheareri - specific monophyletic groups were identified, containing four (CshOMT03, CshOMT04, CshOMT05, and CshOMT09), and three (CshOMT08, CshOMT10, and CshOMT11) members, respectively.
Notably, the close relationship of the species-specific tandem duplicated gene pairs or clusters with high sequence similarity was confirmed in the phylogenetic analyses, such as CshBBEL03 and its paralogs (CshBBEL04 and CshBBEL05), CshBBEL30 and its paralog CshBBEL31, CshCYP719_01 and its paralogs (CshCYP719_02 and CshCYP719_03), CshNCS07 and its paralogs (CshNCS08 and CshNCS09), CshNCS19 and its paralogs (CshNCS20 and CshNCS21), CshNMT19 and its paralog CshNMT20, CshTYR03 and its paralogs (CshTYR04, CshTYR05, and CshTYR06), CtoCYP82N5 and its paralog CtoCYP82N6, and CtoCYP719A2 and its paralog CtoCYP719A2-2 (Fig. 5). The Ka/Ks ratio of these gene pairs ranged from 0.15543 to 0.43823 with an average of 0.292909 (Table 2), suggesting that purifying selection was the primary evolutionary force on these species-specific tandem duplicated gene pairs or clusters.
Table 2. Selective pressure and sequence similarity of the species-specific tandem duplicated gene pairs or clusters in Corydalis.
Gene pairs or clusters Ka Ks Ka/Ks Similarity CshBBEL03, CshBBEL04, CshBBEL05 0.1656 0.4321 0.38315 97.54% CshBBEL30, CshBBEL31 0.1090 0.3349 0.32543 98.73% CshCYP719_01, CshCYP719_02,
CshCYP719_030.0841 0.3688 0.22803 98.81% CshNCS07, CshNCS08, CshNCS09 0.0546 0.2236 0.24430 99.11% CshNCS19, CshNCS20, CshNCS21 0.1325 0.8527 0.15543 94.61% CshNMT19, CshNMT20 0.1435 0.4799 0.29900 98.68% CshTYR03, CshTYR04, CshTYR05, CshTYR06 0.1635 0.373 0.43823 97.79% CtCYP719A2, CtCYP719A2-2 0.0841 0.3808 0.22096 97.73% CtCYP82N5, CtCYP82N6 0.0913 0.2672 0.34165 97.79% -
In this study, by combining data from PacBio long-read sequencing, 3C-based Hi-C sequencing, and Illumina short-read sequencing, we assembled the genome of Corydalis sheareri, one species from subg. Corydalis, the largest and most diverse lineages of Corydalis. Genome survey showed that its genome is extremely complex (Fig. 1b). Both the GenomeScope and Smudgeplot analyses implied that the genome structure of C. sheareri might be an autotetraploid (a special tetraploid with three homologous chromosomes and one non-homologous chromosome) with a special AAAB karyotype (Fig. 1b, c). Intriguingly, the karyotype of C. sheareri is remarkably similar to that of C. yanhusuo[11], which also belongs to subg. Corydalis.
As previously reported, the diploid C. tomentella has a genome size of 258 Mb[12], and the estimated tetraploid C. sheareri genome size was 580 Mb (Supplementary Fig. S1a), which is nearly twice that of C. tomentella. This indicates that polyploidization might have played a significant role in the genome evolution within this genus. It is noteworthy that both C. sheareri and C. yanhusuo are tetraploid, yet their genome sizes differ by more than threefold. In Corydalis, both the genome size (
https://cvalues.science.kew.org/search/angiosperm ) and the chromosome number (https://ccdb.tau.ac.il/ ;http://legacy.tropicos.org/Project/IPCN ) varied considerably. We deduce that diploidization following polyploidization, chromosomal rearrangements, including inversions, translocations, and changes in chromosome number via fusion and fission, as well as gene loss might be common and could thus trigger the genome size diversity in Corydalis. In our study, the detection of large-scale chromosomal structural variants, especially multiple inversions, between the genomes of C. sheareri and C. tomentella (Fig. 2e, f), seems to provide strong evidence in support of this hypothesis.Tandem duplications drive the diversity of BIA biosynthetic genes in Corydalis
-
Polyploidization and structural variations might not only lead to changes in genome size but also substantially affect the gene content. In this study, we identified 172 candidate genes involved in the BIAs biosynthesis within the C. sheareri genome and traced their evolutionary history in Papaveraceae. As previously reported, BBELs, CMTs, NMTs, OMTs, and CYPs (CYP719, CYP82N) are the key enzymes downstream of the BIA biosynthetic pathway (Fig. 3a). In the Corydalis genome, the genes that encode these downstream enzymes, such as BBEL, CMT, NMT, and OMT, tend to possess a larger number of copies than the upstream genes of the BIA biosynthetic pathway (Fig. 3a, b), which enables plants to synthesize various BIAs. Interestingly, NCS, one upstream gene family, also has a relatively high copy number (Fig. 3b). NCS catalyzes the condensation of dopamine and 4-HPAA to produce norcoclaurine, which was identified as one of rate - limiting enzymes in BIA biosynthesis in opium poppy[62].
Furthermore, the chromosomal location and gene duplication analyses demonstrated that these genes frequently appeared as tandem duplications scattered throughout the genome (Figs 3c, 4). All these findings corroborated that tandem duplications might play a key role in the diversity of BIA biosynthetic genes in Corydalis. What's more, our comprehensive phylogenetic analyses, which cover representative species throughout Papaveraceae, revealed that clade I and II of NCS, clades I, II, and VI of CMT, clades I and III of NMT, clades II, VI, and VII of OMT, two members of TAT, four members of TYDC, and clade I of TYR, each retain a single copy in Corydalis (Fig. 5). In addition, the fact that their homologs are shared by the majority of species suggests that they likely originate before the divergence of Papaveraceae or even Ranunculales. By contrast, the identification of C. sheareri or C. tomentella - specific monophyletic groups in BBEL, CYP719, CYP82N, clades III and IV of NCS, clades V and VI of CMT, clades II, IV and V of NMT, clades V and VIII of OMT, clade III of TYR (Fig. 5) indicate that recent gene duplications might occur frequently for these members in Corydalis. Particularly, the close relationship of some tandem duplicated gene pairs or clusters was confirmed in our phylogenetic analyses (Fig. 5), strongly suggesting that these genes may be generated from the recent duplication events that occurred within C. sheareri, or C. tomentella. Additionally, more C. sheareri-specific tandem duplication events are identified compared to those in C. tomentella, which is likely associated with the significant expansion of BIAs biosynthetic genes in C. sheareri.
Until recently, with the burst of plant genome sequencing projects and the advancement of bioinformatic tools, biosynthetic pathways for many natural products have been elucidated[63−65]. Numerous studies have stated that tandem duplication is a major factor contributing to the diversity of secondary metabolites biosynthesis, by recruiting novel genes and potentially introducing new metabolic pathways. For instance, Papaver somniferum has undergone significant tandem duplication events, which result in the emergence of morphinan and noscapine biosynthesis pathways[22,66]. The divergence and expansion of CYP genes strongly contribute to the alkaloid diversity in Coptis[67]. Tandem duplications are also common for the triterpene biosynthetic genes in Aralia elata, especially for CYP72A, CSLM, and UGT73, which may drive the diversity of triterpenoids[68]. In the case of Scutellaria, tandem duplications of the CYP82D subfamily shape the flavonoid diversification[69]. In Corydalis, each species was found to contain a particular set of BIAs, some of which are common to other species but not in the same combinations. BBEL genes have been reported to be expanded and considered to be related to the appearance of cavidines and coptisine in C. tomentella[12]. In this study, tandem duplications are also found to be evident in other genes involved in the biosynthesis of BIAs, particularly NCS, CMT, NMT, OMT, and CYP. Despite not conducting the functional experiment, we still have reason to believe that tandem duplications may drive the expansion of BIA biosynthesis genes, which is conducive to the complexity of biosynthesis pathway and further contributes the BIAs diversity in Corydalis. The incorporation of sequencing data from more Corydalis species, in conjunction with the conduction of multi-omics landscape investigations and the performance of functional experiment studies related to these recently duplicated genes in the future, may lead to an updated canvas for illustrating the genetic mechanisms underlying the diversity of BIAs.
-
In this study, we proposed that C. sheareri might be a complex autotetraploid with a special AAAB karyotype. Genomic comparison detected large syntenic blocks between C. sheareri and its relatives C. tomentella, and also uncovered large-scale chromosomal structural variations (particularly inversions) between these two genomes, which might have profound effects on the divergence of Corydalis. Furthermore, we identified 172 candidate genes involved in BIAs biosynthesis in C. sheareri and traced their evolution history in Papaveraceae. We deduce that tandem duplication has played a prominent role in the expansion of the BIAs biosynthesis genes, especially for BBEL, CMT, NMT, OMT, and NCS. Moreover, the identification of the species-specific tandem duplication events implies that the growth in the number of gene members is likely to complicate the metabolic pathway, and consequently, has further contributed to the diversification of BIAs biosynthesis, ultimately leading to the diversity of BIAs in Corydalis. Our study provides more insights into the genome evolution for species-rich taxa with radiation, as well as the mechanisms underlying the diversity of the BIA biosynthetic pathway. It is also of great value for future genetic studies and medicinal applications of Corydalis.
We thank Dr Yuan-Yuan Feng (Institute of Botany, Chinese Academy of Sciences) for her help in the flow cytometry measurement. We also thank Mr Hai-Kuan Zhang (Berry Genomics Company) for his help in the genome assembly and annotation. This research was funded by National Natural Science Foundation of China (32000170) and Xinyang Academy of Ecological Research Open Foundation (2023XYMS05).
-
The authors confirm contribution to the paper as follows: study conception and design: Liu YY, Peng D, Li JM; data collection: Liu YY, Peng D, Yu CL, Liu YJ, Chen M, Kan SL, Cao YN; analysis and interpretation of results: Liu YY, Peng D, Yu CL, Liu YJ, Kan SL, Cao YN; draft manuscript preparation: Liu YY, Peng D, Wang HW, Li JM, Peng D. All authors reviewed the results and approved the final version of the manuscript.
-
The whole-genome sequence data, including Illumina short reads, PacBio HiFi reads, Hi-C interaction reads, transcriptome data, and genome annotation files, have been deposited in The National Genomics Data Center (NGDC), under the project number: PRJCA035358.
-
The authors declare that they have no conflict of interest.
- Supplementary Table S1 Statistics of genome sequencing of C. sheareri.
- Supplementary Table S2 Statistics of chromosomes of the C. sheareri genome.
- Supplementary Table S3 Transposable elements (TE) in the C. sheareri genome.
- Supplementary Table S4 Statistics of non-coding RNA (ncRNA) in the C. sheareri genome.
- Supplementary Table S5 Gene families used in the phylogenomic analysis.
- Supplementary Table S6 The BIA biosynthetic genes identified in this study.
- Supplementary Fig. S1 Genome survey of Corydalis sheareri.
- Supplementary Fig. S2 Detail information of functionally annotated genes based on various databases.
- Supplementary Fig. S3 GO and KEGG enrichment analyses of the expanded gene family in Corydalis.
- Supplementary Fig. S4 The inter-genomic comparison and syntenic depths analyses among C. sheareri, Vitis vinifera and Amborella trichopoda.
- Supplementary Fig. S5 The detection of whole-genome duplication based on tree2GD analysis.
- Supplementary Fig. S6 Inter-genomic comparison between C. sheareri and C. tomentella based on the protein-coding genes.
- Supplementary Fig. S7 Structural variation detection in C. sheareri performed by IGV.
- Copyright: © 2025 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Liu YY, Yu CL, Liu YJ, Kan SL, Chen M, et al. 2025. An autotetraploid genome of Corydalis sheareri provides insight into the evolution and benzylisoquinoline alkaloids diversity of Corydalis. Genomics Communications 2: e002 doi: 10.48130/gcomm-0025-0002 

An autotetraploid genome of Corydalis sheareri provides insight into the evolution and benzylisoquinoline alkaloids diversity of Corydalis
- Received: 26 November 2024
- Revised: 20 January 2025
- Accepted: 22 January 2025
- Published online: 25 February 2025
Abstract: Corydalis DC. is the largest and most diverse genus of Papaveraceae, which has undergone rapid diversification. Corydalis is notable for its diverse benzylisoquinoline alkaloids (BIAs). However, limited genomic resources hinder the understanding of its evolution and BIA diversity. Here, we report a high-quality genome of Corydalis sheareri and provide a comparative analysis of Corydalis genomes. Genome survey shows that the genome of C. sheareri might be an autotetraploid. De novo assembly yields a single homolog genome of 282 Mb, with a contig N50 of 11.39 Mb. Repetitive DNA accounts for 44.47% of the genome, and a total of 26,287 protein-coding genes are predicted. When comparing this genome with the previously reported diploid genome of C. tomentella, large-scale structural variations are detected, which might have dramatic effects on the evolution of Corydalis. Furthermore, a total of 172 candidate genes involved in benzylisoquinoline alkaloids (BIAs) biosynthesis are identified in the genome of C. sheareri. Duplication and phylogenetic analyses indicate that tandem duplications play a prominent role in the evolution of the BIA biosynthesis genes, and might be associated with the BIA diversity in Corydalis.Our study provides more insights into the genome evolution and secondary metabolites diversity of Corydalis, and will accelerate genomic and evolutionary radiation study of species-rich plant genera.
-
Key words:
- Corydalis /
- Comparative genomics /
- Polyploid /
- Tandem duplication