









-
Kiwifruit is an economically valuable crop with global significance due to its exceptionally high and balanced nutritional composition, including vitamins, chlorophyll, polyphenols, amino acids, carotenoids, anthocyanins, flavonoids, dietary fibre, calcium, iron, potassium, and other beneficial metabolites[1]. However, kiwifruit is a representative climacteric fruit. During the initial storage period, the main life characteristics are the consumption of dry matter and the hydrolysis of starch, but the appearance and quality of fruit, especially the firmness change a little. In the late stage, the peak of respiration and ethylene production appear almost simultaneously, cell walls expand and dissolve, and the fruit firmness decreases rapidly, losing storability[2,3]. The maturity rate largely depends on storage conditions and post-harvest processing. So improving the quality of post-harvest fruits is considered one of the most indispensable links in the kiwifruit production procedures.
Phytohormones play fundamental roles in plant growth and development, which coordinates these processes with internal and external stimuli[4,5]. The gibberellin (GA) is essential for a number of important developmental processes ranging from seed sprouting to the inducement of flowering[6,7]. Exogenous GA has been studied in plants since the 1970s. Preharvest application of exogenous GA increased the thickness of the wax layer and reduced the rate of fruit water loss, leading to a significant increase in fruit storage capacity[8]. Post-harvest GA treatment effectively retained the firmness of okra fruits and delayed fruit hardening during storage of okra[9]. Post-harvest storage of persimmon fruit impregnated with GA3 induces physiological changes that delay persimmon fruit softening and reduce ABS development thereby enhancing storability[10]. Vacuum-impregnating post-harvest lemon fruit with GA delays senescence and colour change[11]. The application of GA, either as a pre-harvest spray or as a post-harvest dipping treatment, effectively preserved the colour of the 'Oroblanco' green fruit[12]. GA reduced ascorbic acid content in kiwifruit leaves, and delayed post-harvest ripening of kiwifruit[13,14]. DELLA proteins act as an extremely important negative regulatory role in GA signalling transduction[15,16]. It is classified as the plant-specific GRAS family and is named after its N-terminal DELLA structural domain[17]. The DELLA family has been discovered in many plants, such as Arabidopsis, maize, rice, wheat, corn, and tomato[18−20]. DELLA proteins are located in the nucleus and function as transcriptional repressors[21]. It is universally recognized that GA signalling occurs when the GA receptor GID1 first senses and binds GA, and then binds to DELLA proteins to form GA-GID1-DELLA polymers[22]. However, the DELLA protein lacks a DNA-binding domain and is unable to bind directly to DNA. Therefore, DELLA often interacts with regulatory proteins in different signalling pathways to regulate plant growth and development[23].
DELLA proteins serve as a vital function in various aspects of plant growth and development, they play a key integrative role in the phytohormone signal response network[24]. In Arabidopsis, DELLA proteins chelated MYBL2 and JAZ inhibitors to promote anthocyanin biosynthesis[25]. In lettuce, knockdown of LsRGL1 promoted tillering, while tillering was inhibited as well as GA and growth hormone biosynthesis in the overexpression lines[26]. In tomato, the DELLA protein SlDELLA interacted with the growth hormone signalling component SlARF7/SlIAA9 to induce fruit germination[27], and silencing of DELLA resulted in solitary fruits which produced a smaller and distinctive elongated shape[28]. As for ethylene, it can regulate Arabidopsis development in part through alteration of the properties of DELLA protein[29]. DELLA protein can recognise and bind to the ethylene signaling transduction component EIN3/EIL1, affecting the formation of apical curved hooks in Arabidopsis[30]. However, few researchers have focused on the function of DELLA on ripening regulation, and the potential interaction between DELLA and ethylene deserves more attention.
In this study, eight DELLAs were classified from kiwifruit, and the relationship between DELLAs expression and kiwifruit postharvest ripening was investigated. The DELLA domain was used to identify members of the DELLA gene family in the Actinidia chinensis cultivar Red5 genome. The physicochemical properties, chromosomal localization, gene structure, protein structural domains, distribution of conserved motifs, and phylogenetic tree were carried out to explore the characteristics of AcDELLA proteins. Real-time PCR presented that five of eight AcDELLA expression were induced by GA3 treatment, and four of them showed significantly high negative correlations with ethylene production. The above study enriches our understanding of DELLAs function in kiwifruit postharvest ripening and lays the foundation for subsequent functional study of AcDELLA genes.
-
In September 2022, 'Cuixiang' (Actinidia chinensis) mature kiwifruit was harvested from a commercial orchard in Yantai, Shandong province, China. The initial total soluble solid (TSS) of its fruit was 5.5%. Four hundred evenly sized healthy fruits without apparent imperfections were randomly distributed into two groups. One group was the control group (evenly sprayed with distilled water three times every 8 h at 24 °C) and another one was gibberellin-treated group (evenly sprayed with 0.3 g/L GA3 solution three times every 8 h at 24 °C). After the treatment, the fruits were kept at room temperature (24 °C). Each sampling point had nine fruits, one replicate for every three fruits. The pulp of kiwifruit samples were collected excluding the skin, seeded portion, and centre column. They were chopped into tiny pieces, which were snap-frozen in liquid nitrogen, and then preserved at −80 °C for further experiments. Leaf, stem, and flower tissue parts of 'Cuixiang' (Actinidia chinensis) kiwifruit were collected separately from the same commercial orchard, in May 2022.
Measurements of fruit firmness, total soluble solids (TSS), and total acid
-
Firmness was assessed with an AMETEK Brookfield CT3 10K texture analyzer (Middleboro, USA) equipped with a 6.0 mm diameter probe suppression distance and speed of 4.0 and 2.0 mm/s, respectively. The firmness of kiwifruit was measured at two opposite positions at the fruit equator and the maximum force was recorded to represent the fruit firmness, expressed as the peak force (N). Digital handheld refractometers were used to measure total suspended solids and total acids.
Determination of starch content
-
A bead mill (Qiagen TissuLyzer II) was used to grind 0.03 g of the frozen pulp sample into a fine powder, and starch was determined using the Solarbio Starch Content Assay Kit instructions with three biological replicates.
Determination of ethylene production
-
The amount of ethylene released from kiwifruit was measured according to previous studies[14]. In brief, nine fruits were classified into three replicates, and the fruits were placed in a 1 L sealed container for 1 h before a basin of water and a 5 mL syringe were prepared for ethylene collection. The Shimadzu GC-2014 gas chromatography (GC) system was used to inject 1,000 microliters of gas into the gas chromatograph. Ethylene yield was normalized by fruit weight, with ethylene standard gas used as a control. Three individual biological replicates were performed at each sampling point.
Determination of cell wall components
-
Cell wall components were derived and segregated as discussed in a previous method[31]. Three g kiwifruit flesh was frozen in 20 mL 80% ethanol solution (v/v), vortexed well, heated in a boiling water bath for 20 min, cooled, centrifuged at 3,900 rpm for 10 min at room temperature, and the supernatant was discarded. The precipitates were successively rinsed with 6 mL of 80% ethanol (v/v), trichloromethane/methanol (1:1, v/v), and acetone, then centrifuged to place the white precipitate in a 40 °C oven for 24 h, and cell wall material (CWM) was collected and weighed. Water soluble pectin (WSP), ionic soluble pectin (ISP), and covalently bound pectin (CBP) were obtained respectively.
Detection of gibberellins
-
Extraction process: by grinding the kiwifruit flesh and adding a specific extraction reagent (70% ethanol containing 0.1% formic acid), combined with ultrasonic and vortex extraction techniques to achieve effective extraction of gibberellins. Purification process: MAX and C18 (CNWLC-C18200mg) were used as purification materials according to the mass ratio of 1:2 to 1:4 to ensure the purity of the samples. Detection and analysis: ultra performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS) was used to determine the stability of the gibberellins by calculating the content of the gibberellins to be measured. Gibberellins contents were determined by MetWare (
www.metware.cn ) based on the AB Sciex QTRAP 6500 LC-MS/MS platform.Identification of members of the kiwifruit DELLA protein family
-
DELLA genes were identified from Actinidia chinensis Red5 kiwifruit genome database (
http://kiwifruitgenome.org/ ), and the genome sequence (DNA FASTA), protein sequence file (Protein sequence FASTA) and gene structure annotation file (GTF) were downloaded. The putative DELLA-related gene domain was checked, and the candidate genes were further confirmed. All the identified protein sequences of the DELLA family contained GRAS and DELLA domains.Bioinformatic analysis of the kiwifruit DELLA protein gene family
-
The intron, exon, and genomic localization information of the DELLA family genes in kiwifruit were all sourced from the Actinidia chinensis Red5 kiwifruit genome database (
https://kiwifruitgenome.org/organism/3 ). By downloading the kiwifruit gene structure annotation file from the kiwifruit gene database, specific positional information of eight DELLA gene family members on chromosomes was obtained, and chromosome localization maps were drawn by TBtools. TBtools was also used to draw a schematic diagram of gene structure. Prot Param was used to analyze the primary structure and physicochemical properties of DELLA protein (https://web.expasy.org/protparam ). PRABI (www.prabi.fr ) was carried out to predict the secondary structure of proteins. According to the online software SWISS-MODEL (https://swissmodel.expasy.org/interactive ), protein tertiary structure analysis was performed. MEGA-X and GENEDOC were used for protein comparison analysis.Multi-species sequence alignment and conserved domain database analysis
-
DELLA protein sequences were downloaded from some species genomes through keyword search functions on NCBI, and then the protein sequence data was imported into TBtools software to construct a phylogenetic tree and conserved domain analysis was performed on DELLA proteins in species, including Arabidopsis thaliana (AtGAI, AtRGA, AtRGL1, AtRGL2, AtRGL3), Brassica napus (BnRGA1, BnRGA2), Malus domestica (MdRGL1a, MdRGL1b), Triticum aestivum (Rht-D1), Hordeum vulgare (SLN1), Cucumis sativus (CsGAIP-B), Lactuca sativa Linn (LsDELLA1), Triticum aestivum (Rht-D1), Solanum lycopersicum (PROCERA), Gossypium hirsutum (GhGAI), Solanum tuberosum (StGAI), Pyrus × bretschneideri (PbGAI-like), Glycine max (GmGAI), Juglans regia (JrGAI), Vitis vinifera (VvGAI), Prunus avium (PaGAI), Cucumis melo (CmGAI-P), Fragaria vesca (FveRGA1), Sugarcane (ScGAI), Oryza sativa (OsSLR1), Zea mays (Zm-D8), Prunus mume (PmGAIP-B), and Prunus persica (PpGAIP-B) (Supplementary Table S1).
Extraction of RNA from kiwifruit
-
The RNA was extracted using an RNA extraction kit (TIANGEN), following the instructions. The extracted RNA was subjected to 1% agarose gel electrophoresis, and the RNA concentration was detected with a micro spectrophotometer. Then, using the RNA as a template, the RNA was reverse transcribed into cDNA using a reverse transcription kit (Vazyme), and the cDNA was stored in a −20 °C freezer for later use.
Analysis of DELLA gene expression characteristics in kiwifruit
-
Real-time PCR was conducted using reversed cDNA at fruit different sampling periods and stems, leaves, and flowers as templates, with AcActin (Genbank No. EF063572) as the internal reference gene. The amplification system and program were developed according to Vazyme SYBR following the instructions. Three biological replicates were performed. The primers are shown in Supplementary Table S2, using 2−ΔCᴛ to calculate the relative expression levels of each gene. The results were statistically analyzed by using DPS 7.05 software.
Statistical analysis
-
One-way ANOVA followed by Student's t-test (** p < 0.05, ** p < 0.01, *** p < 0.001) or LSD (least significant difference) analyses were used. Heatmaps were drawn using TBtools software. Figures for Pearson's correlation tests and correlation analyses were generated using Origin 2022 (Microcal Software Inc., Northampton, MA, USA).
-
In this study, 300 mg/L GA3 was used to treat 'Cuixiang' kiwifruit. All samples were stored at room temperature (24 °C). During storage, the fruit status of the 'Cuixiang' kiwifruit in the gibberellin treated group and the control group are shown in Fig. 1a. Throughout the storage process, the firmness of both GA-treated and control group fruits showed a continuous decreasing trend (Fig. 1b). The firmness of control fruits decreased rapidly during post-harvest storage, while the firmness of GA treated fruits decreased relatively slowly. The initial firmness of kiwifruit is 67.01%. After 7 d, the firmness of the control group decreased to 42.86 N, while the GA treated group kept on 64.05 N. Even after 15 d of treatment with GA, it still maintained firmness with 49.27 N. GA treatment effectively maintained the firmness of kiwifruit in the later storage period. The TSS content of kiwifruit during post-harvest storage showed an increasing trend. In the control group, it increased rapidly during the 7−10 d period and reached a peak on the 15th day. However, in gibberellin treated kiwifruit, TSS increased relatively slowly and reached its peak on the 25th day (Fig. 1c). Throughout the storage process, the titratable acid content of both the control and the gibberellin-treated fruits showed a continuous decreasing trend. The decrease rate in kiwifruit treated with gibberellin is relative slow (Fig. 1d). The ethylene production of the fruit at day 0 is 0.38 μL/g/h. The control group fruits reached a respiratory peak at day 15, with the maximum at 48.44 μL/g/h. While, the GA-treated group reached its peak until day 25, with a lower ethylene production of 36.45 μL/g/h (Fig. 1e). In terms of starch and cell wall material (CWM) content, they both exhibited a similar trend with firmness (Fig. 1f & g). Among cell wall components, the CBP content changes were consistent with the CWM trend, after 15 d of storage, the CBP content decreased to 37% and 54% of the level measured at day 0, respectively (Fig. 1i). In contrast, ISP and WSP contents increased during storage and were lower in GA3-treated fruit than in control fruit (Fig. 1h & j). Moreover, the variation tendency of ISP and WSP contents were also similar.
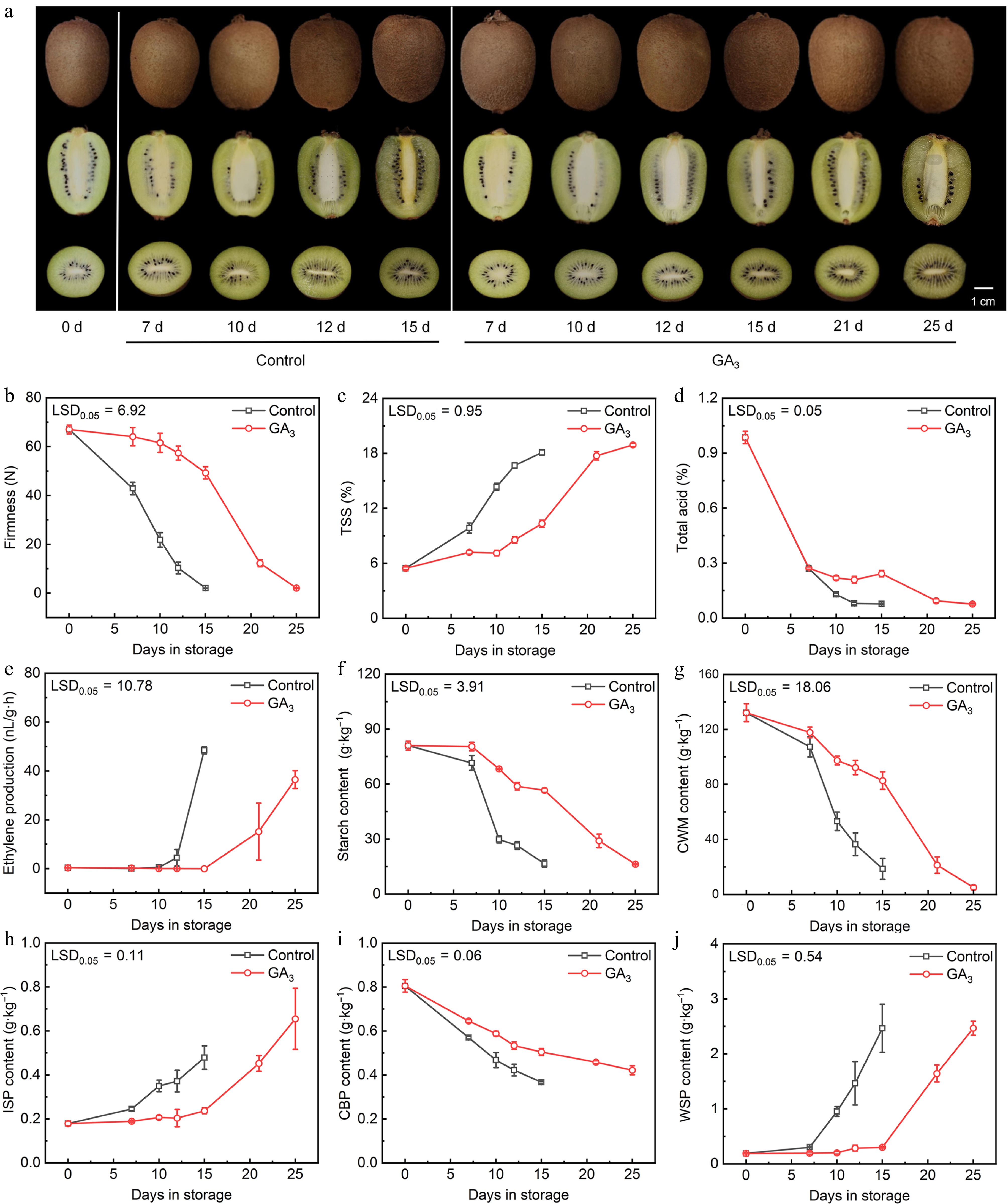
Figure 1.
The alterations in performance and physiological attributes of kiwifruit following treatment with gibberellin has been conducted. (a) Photographs of kiwifruit in different groups. (b) Firmness of kiwifruit in different groups. (c) Total soluble solids (TSS) of kiwifruit in different groups. (d) Total acid of kiwifruit in different groups. (e) Ethylene production of kiwifruit in different groups. (f) Starch content of kiwifruit in different groups. (g) Cell-wall material (CWM) content of kiwifruit in different groups. (h) Ionic soluble pectin (ISP) content of kiwifruit in different groups. (i) Covalent binding pectin (CBP) content of kiwifruit in different groups. (j) Water soluble pectin (WSP) content of kiwifruit in different groups. Error bars: ± SE from nine replicates (for firmness, TSS, and total acid) or three replicates (for ethylene production, starch content, CWM, ISP, CBP, and WSP). LSD: least significant difference at p = 0.05.
Determination of gibberellin levels
-
The endogenesis gibberellin content in kiwifruit at 0, 7, and 12 d after harvest was measured by UPLC-MS/MS. The results suggested that GA1, GA3, GA4, GA5, GA6, GA7, GA8, GA9, GA12, GA15, GA19, GA20, GA24, GA29, GA34, GA44, and GA53 could be detected in kiwifruit samples, and among them, the content of GA3 was the highest (Fig. 2). GA3 content was 178.7 ng/g at day 0, and reduced to 45.3 and 28.9 ng/g in the control group on the 7th day and the 12th day, respectively. By contrast, GA3 content increased to 258.7 and 217.9 ng/g in the GA3 treated group at day 7 and day 12, respectively. In other words, the endogenesis GA3 content in the GA3-treated group was five times more than in the control group on the 7th day and seven times on the 12th day. The content of GA1 showed a similar tendency as GA3. Overall, the results showed that the gibberellins content in the GA3-treated group was significantly higher than that in the control group (Fig. 2).

Figure 2.
Determination of endogenous gibberellin contents in different group kiwifruit at 0, 7, and 12 d of post-harvest storage. Error bars: ± SE from three replicates.
Identification and cloning verification of members of the DELLA protein gene family in the whole genome of kiwifruit
-
Eight AcDELLA gene family members were identified from the kiwifruit genome database, which were named AcDELLA1 to AcDELLA8 by the order of chromosome localization (Fig. 3a). The AcDELLA genes were located on six chromosomes, with AcDELLA1 and AcDELLA2 on LG1, AcDELLA3 and AcDELLA4 on LG9, AcDELLA5 on LG12, AcDELLA6 on LG20, AcDELLA7 on LG25, and AcDELLA8 on LG27 (Fig. 3a). The gene structure map of the DELLA gene family in kiwifruit showed that all the eight AcDELLA genes contained only one exon, no intron, and no significant difference in CDS sequence location (Fig. 3b). The analyses of amino acid length, molecular weight, and isoelectric point of eight AcDELLA proteins were exhibited in Supplementary Table S3. The longest sequence in the coding region was AcDELLA2 (1,746 bp) and the shortest was AcDELLA6 (1,560 bp). The amino acid number of AcDELLA proteins ranged from 519 to 581 aa. Besides, the molecular weights ranged from 57,203.13 to 62,891.92 Da, and the theoretical isoelectric points ranged from 4.95 to 5.40.
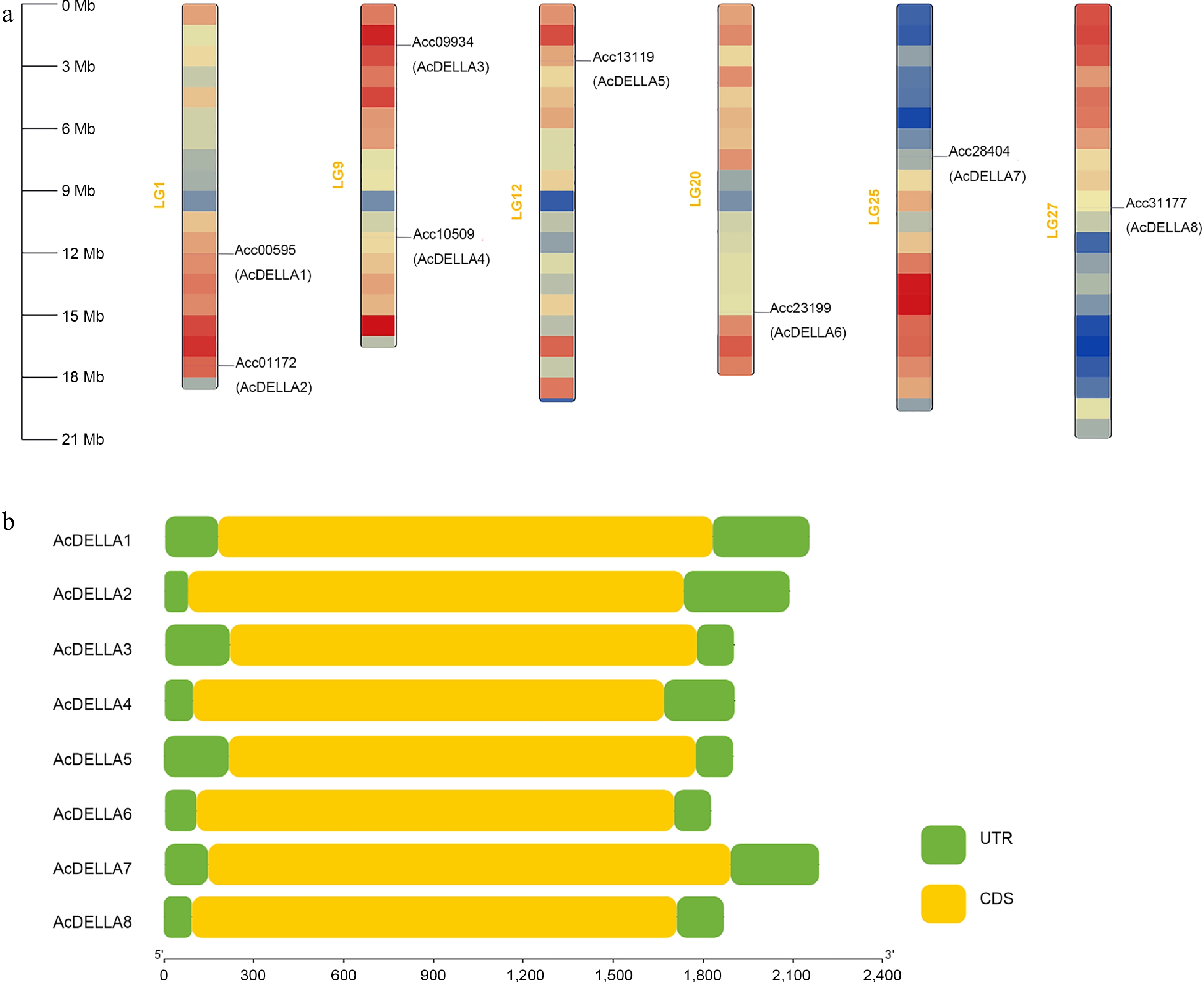
Figure 3.
(a) Chromosome localization of kiwifruit AcDELLA gene family, and (b) schematic diagram of gene structure of kiwifruit AcDELLA gene family.
Phylogenetic tree and amino acid sequence analysis of AcDELLA gene family in kiwifruit
-
To further understand the evolutionary relationship between kiwifruit AcDELLA and DELLA proteins from other species, DELLA proteins of kiwifruit, Arabidopsis thaliana, wheat, rice, tomato, grape, and potato were subjected to phylogenetic tree construction (Fig. 4a). The results showed that AcDELLA2 and AcDELLA4 were the closest to Solanum tuberosum (StGAI) and Solanum lycopersicum (PROCERA), and AcDELLA7 and AcDELLA8 were the closest relatives to Vitis vinifera (VvGAI) (Fig. 4a). It was found that these eight DELLA genes all contained the typical characteristic structural domains of DELLA, with only slight differences in their positions (Fig. 4b). Eight AcDELLA proteins were found to contain the same conserved motifs: DELLA and VHYNP at N-terminal, LHRI (leucine enriched region I), VHIID, LHRII (leucine enriched region II), PFYRE, and SAW by amino acid sequence analysis (Fig. 4c).
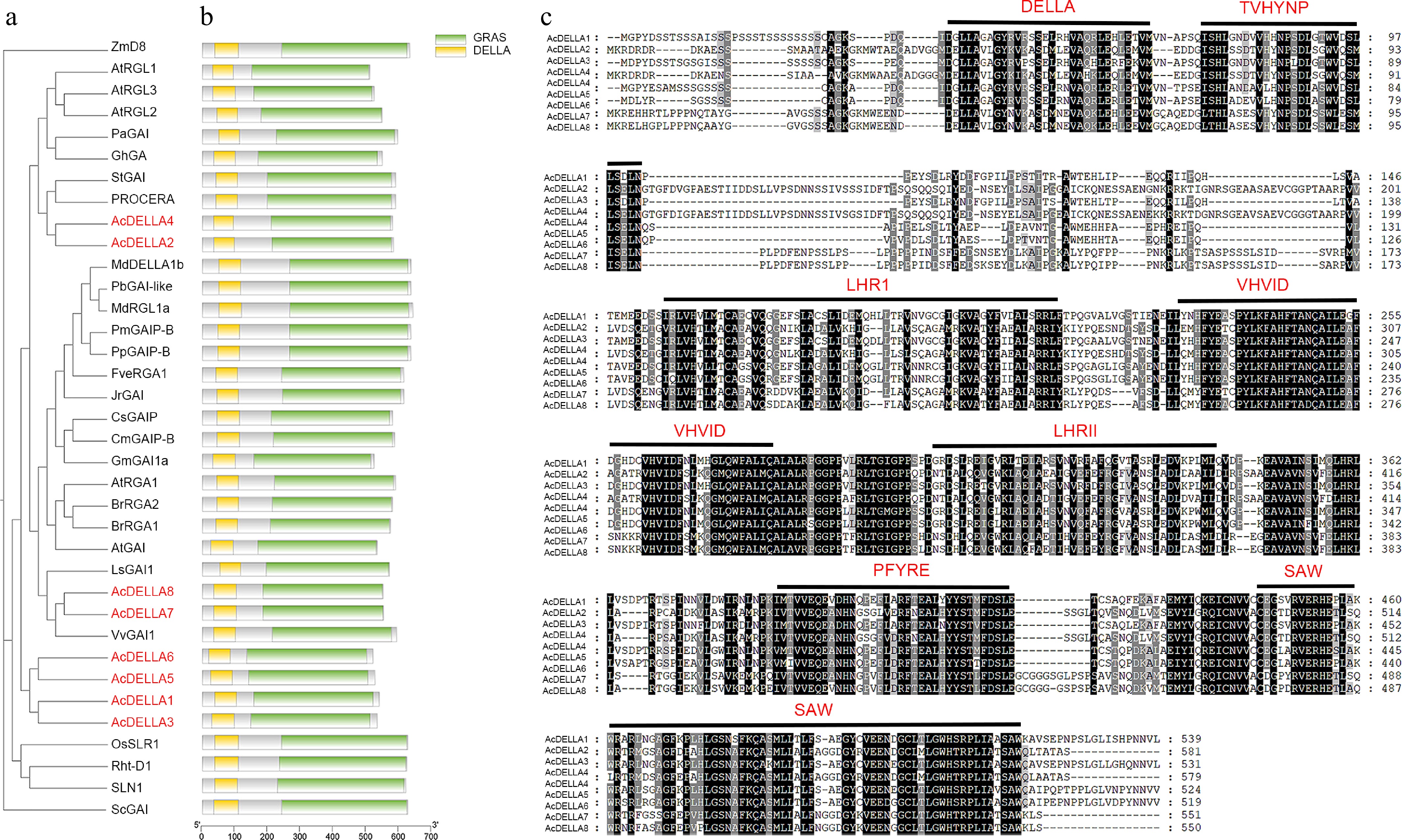
Figure 4.
Phylogenetic analysis, protein domain, conserved motif analysis of AcDELLA proteins. (a) Phylogenetic analysis of kiwifruit DELLA proteins with other species DELLA proteins. (b) Multiple comparison of amino acid sequences of kiwifruit and other species DELLA proteins. (c) Conserved motif analysis of AcDELLA proteins. Conserved residues are indicated by dark and gray boxes and variant residues are shown by light gray boxes. Black lines represent conserved motifs.
Predicted secondary and tertiary structure of kiwifruit AcDELLA proteins
-
Analysis of the secondary structure of kiwifruit DELLA proteins revealed that the secondary structures of all eight kiwifruit DELLA proteins were dominated by α-helixes, with a proportion of 47.32% to 49.82%, and β-turns were fewer, with a proportion of 4.15% to 6.11% (Fig. 5a). Three-dimensional structure modelling showed that the protein molecular conformations formed by the three-dimensional spatial arrangement of its proteins were diverse, and the protein conformations of AcDELLA5 and AcDELLA6, AcDELLA7 and AcDELLA8 were more similar (Fig. 5b).
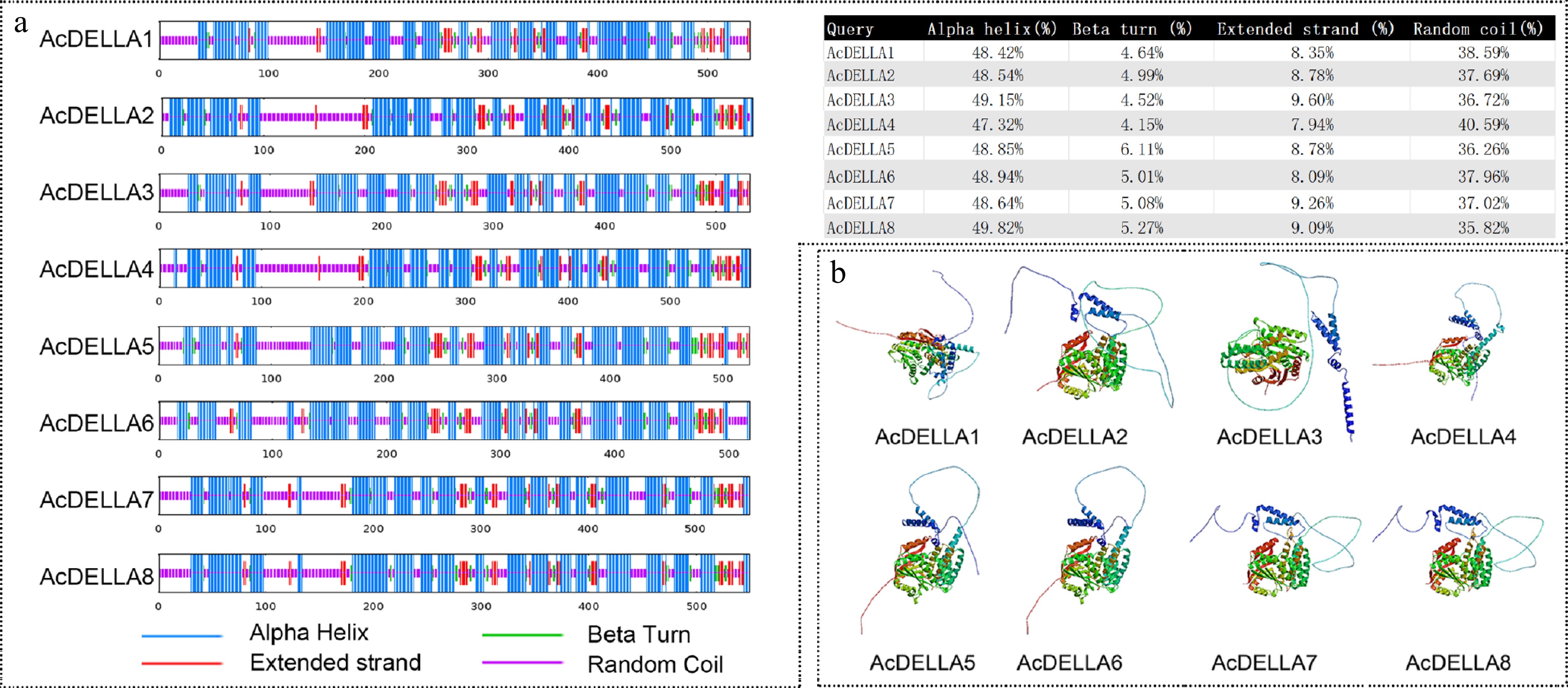
Figure 5.
Structure prediction of eight AcDELLA proteins in kiwifruit. (a) Secondary structure of AcDELLA proteins. (b) Tertiary structure of AcDELLA proteins.
The expression of AcDELLA genes in different treatments and ripening stages
-
To understand the regulation role of AcDELLA genes in different treatments and ripening stages of kiwifruit and various tissues, the expression of eight AcDELLA genes was analysed by qRT-PCR (Fig. 6). Except for AcDELLA1 and AcDELLA2, a total of six AcDELLA genes showed significant difference and different expression patterns between the GA3 treatment group and the control group. The expression levels of AcDELLA1 and AcDELLA2 were very low, and there were no significant differences between the two groups (Fig. 6a, b). The expression of AcDELLA3, AcDELLA4, AcDELLA5, and AcDELLA6 were induced by GA3 to varying degrees (Fig. 6c−f), while, GA3 delayed the expression of AcDELLA7, whose expression is closely related to kiwifruit ripening (Fig. 6g). AcDELLA8 maintained higher level in GA3 treated group than in control group during the whole storage period (Fig. 6h). Except for AcDELLA1, all of the DELLAs expressed in stem, leaf, and flower. AcDELLA2, AcDELLA4, AcDELLA7 showed high expression levels in leaves. AcDELLA7 had the highest expression level in flowers and AcDELLA8 in stems (Fig 6i).
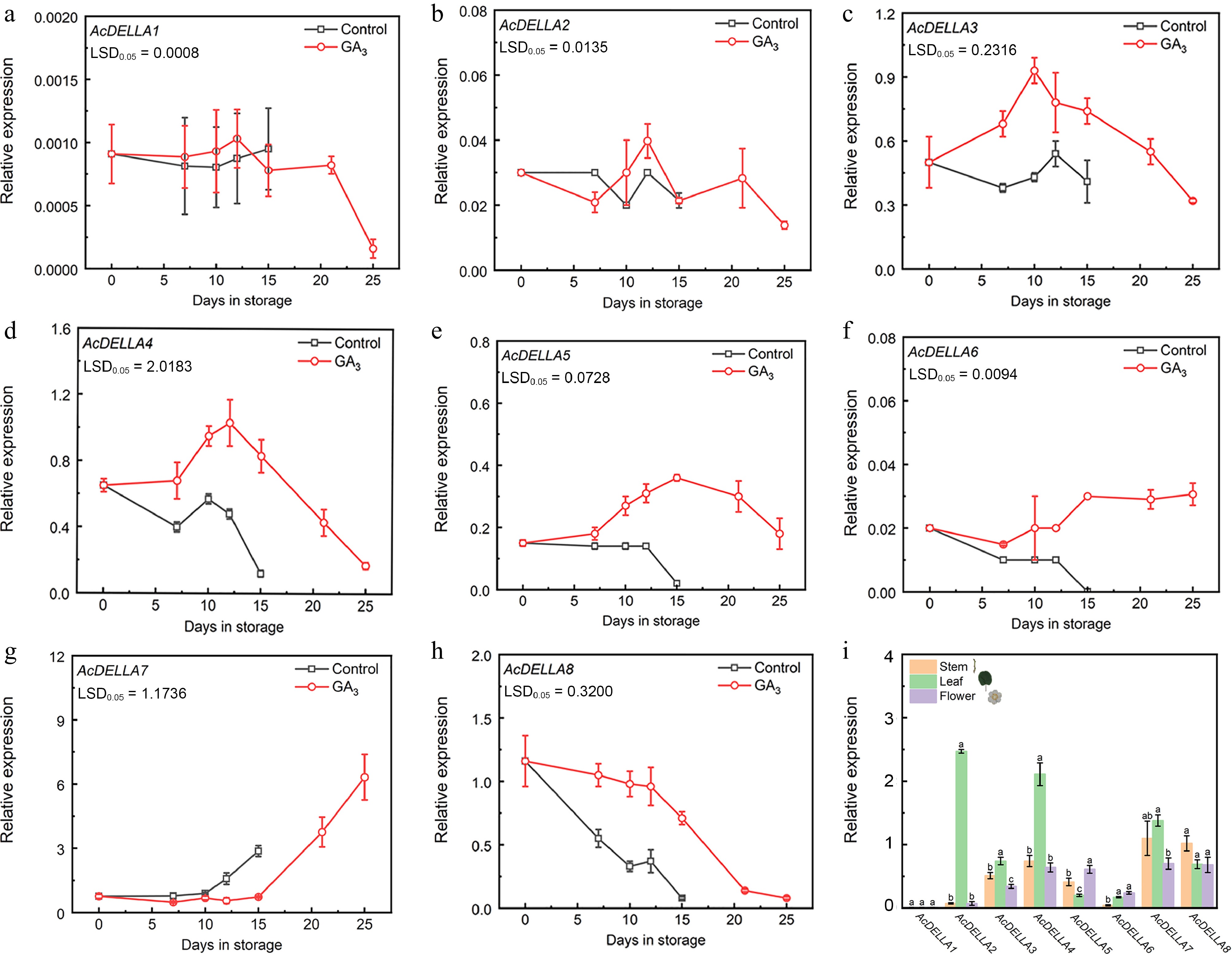
Figure 6.
qRT-PCR analysis of eight DELLAs expression patterns in 'Cuixiang' kiwifruit. (a) AcDELLA1. (b) AcDELLA2. (c) AcDELLA3. (d) AcDELLA4. (e) AcDELLA5. (f) AcDELLA6. (g) AcDELLA7. (h) AcDELLA8. (i) Expression of eight DELLAs at different tissues. Error bars indicate SE from three replicates. LSD values indicate LSD at p = 0.05. Analysis of variance (ANOVA) conducted using SPSS indicated that the observed differences between letters (a−c) at p < 0.05 were statistically significant.
Relationships between physiological properties and AcDELLA genes in kiwifruit
-
To ascertain a comprehensive comprehension of the interrelation between physiological indices (firmness, TSS, total acid, ethylene, starch, cell wall components, WSP, ISP, CBP) and eight AcDELLA genes, an in-depth correlation analysis was undertaken (Fig. 7). Consequently, the physiological attributes and genes demonstrated distinct correlation patterns, with notable high correlations observed across a multitude of physiological attributes and genes. (|r| > 0.90 and p < 0.001). There was a significant negative correlation between firmness and TSS, ethylene, ISP, and WSP. On the contrary, there was a significant positive correlation between firmness and total acid, starch, CWM, and CBP. AcDELLA3 and AcDELLA4 were positively correlated with firmness, starch, CWM, and CBP, and negatively correlated with TSS, ethylene, WSP, and ISP. At the gene level, AcDELLA3 and AcDELLA4 were positively correlated with AcDELLA5 and AcDELLA8, and negatively correlated with AcDELLA7. AcDELLA7 was negatively correlated with firmness, total acid, starch, CWM and CBP, and positively correlated with TSS, ethylene, WSP, and ISP. The correlation between AcDELLA8 and physiological traits was exactly opposite to that of AcDELLA7. AcDELLA8 was positively correlated with firmness, total acid, starch, CWM, and CBP, and negatively correlated with TSS, ethylene, WSP, and ISP (Fig. 7).
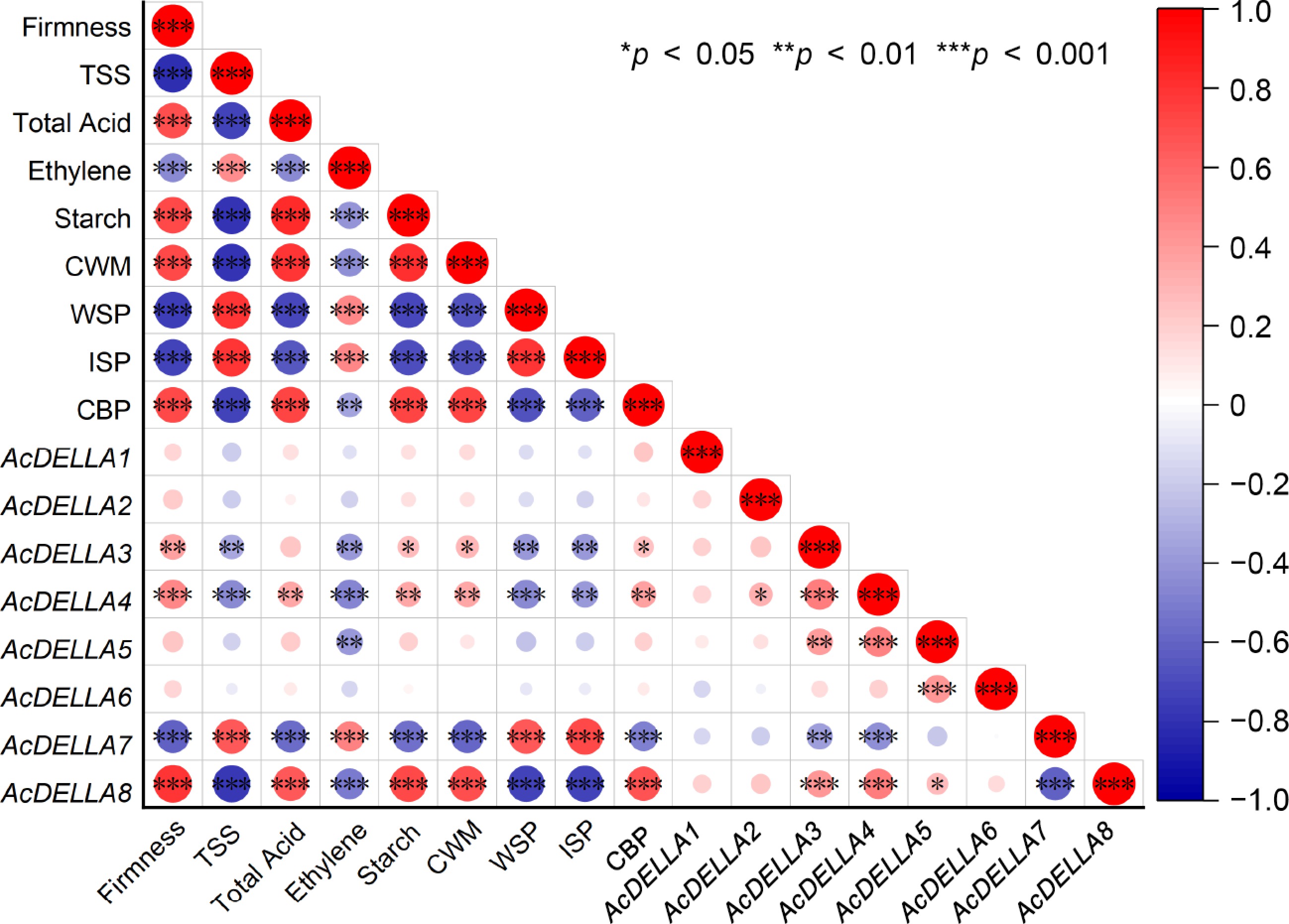
Figure 7.
Correlation analysis of AcDELLA genes expression with physiological characteristics and cell wall composition during storage in kiwifruit. The presence of positive and negative correlations is denoted through the use of color coding, with red signifying a positive correlation and blue indicating a negative one (*, p < 0.05, **, p < 0.01 and ***, p < 0.001).
-
Fruit ripening is a complex procedural process. As a respiratory climate fruit, kiwifruit undergoes a rapid post-ripening and softening process[32]. Gibberellin affects various physiological processes of plant life, including the elongation and division of plant cells, the stimulation of seed germination, and fruit growth[33,34]. In recent years, Gibberellin has been known to play an important role in the fruit ripening process. GA delayed ripening and senescence in various fruits, such as cherry, plum, peach, dates, and banana[35−39]. As for kiwifruit, gibberellin treatment delayed the post-ripening process of post-harvest 'Hongyang'[14]. Here, exogenous GA3 was used to treat 'Cuixiang' kiwifruit at the post-ripening stage, and it was found that GA3 treatment maintained fruit firmness, inhibited ethylene production, and delayed ripening (Fig. 1). Previous studies concluded that exogenous gibberellin treatment increased endogenous gibberellin content and delayed fruit ripening and senescence in tomato[40]. When grape berries treated with exogenous GA3, the endogenous GA3, IAA, and CTK content increased, while ABA content decreased[41]. In our study, it is interesting that exogenous GA3 treatment can induce an increase in endogenous GA1, GA3, and GA19 content, especially GA3 (Fig. 2). In summary, GA treatments were effective in delaying postharvest ripening of fruit and presented similar effects in different varieties of kiwifruit.
DELLA proteins play a key role in the signal transduction pathway of gibberellins. DELLA is characterised by the N-terminal domain of DELLA and the C-terminal domain of GRAS[42]. The N-terminal domain includes two highly conserved acidic domains, the DELLA domain, and the TVHYNP domain, but the amino acid sequence is less similar. In the middle position, it is a nuclear localization signal (NLS), followed by a highly conserved amino acid domain VHVID, two leucine repeats of the LHR sequence, and the ploy S/T/V. In the C-terminal position, it is a highly conserved GRAS domain, which is the functional domain of DELLA[43,44]. These conserved domains not only constitute the complex three-dimensional structure of DELLA protein, but also play important roles in the regulation of plant growth and development. It was found that DELLA and TVHYNP play key roles in the perception of GA signals in plants and belong to the regulatory domains; Poly S/T/V is a modification site for phosphorylation and glycosylation of DELLA protein and is a regulatory domain for inhibitory activity; LHRs act in protein-protein interactions and belongs to the dimerization domain; VHVID, PFYRE, and SAW distributed at the C-terminus of DELLA protein belong to the deterrence domain[45,46]. However, the DELLA gene family has not been found in kiwifruit. In this study, eight AcDELLAs were identified in the kiwifruit genome, and they are distributed on six different chromosomes (Fig. 3a). Moreover, AcDELLAs have complete motifs of DELLA, TVHYNP, LHR, VHVID, PFYRE, and SAW (Fig. 4c), so they may possess powerful functions.
In plants, DELLA proteins are not only involved in GA signalling, but also in the signalling and biosynthesis of other hormones, such as abscisic acid (ABA), ethylene, and jasmonate (JA)[47]. Besides, DELLA proteins are capable of integrating a wide range of hormonal and environmental signals to regulate plant growth and development[48]. In tomato, the DELLA protein PROCERA (PRO) not only promotes ABA-induced stomatal closure and gene transcription by up-regulating the ABA transporter protein AIT1.1[46], but also mediates crosstalk between the GA and growth hormone pathways with the SlARF7/SlIAA9 complex to regulate fruit development[27]. In pea, DELLA proteins can activate the expression of KNOX and BELL transcription factors, which are involved in cytokinin metabolism[49]. In Arabidopsis, DELLA protein can recognise and bind to the ethylene signaling component EIN3/EIL1 protein, thereby affecting the formation of apical curved hooks[29]. However, there is few reports on the association of DELLA with fruit post-ripening development.
In this study, we found that the gene expression of AcDELLA3, AcDELLA4, AcDELLA5, AcDELLA6, and AcDELLA8 were induced by GA3 (Fig 6c−f), and AcDELLA3, AcDELLA4, AcDELLA5, AcDELLA8 showed significantly high negative correlations with ethylene production (Fig. 7). Therefore, we hypothesised that DELLA might be involved in the regulation of kiwifruit ripening by interacting with ethylene phytohormones. Compared to these GA3-induced DELLAs, AcDELLA3/4/5/6/8, the expression of AcDELLA7 remained at a low level in the earlier-storage period, but increased with the prolongation of the storage time in both control and GA3 treated samples (Fig. 6f). Moreover, the expression trend of AcDELLA7 extremely significant negatively correlated with firmness, whereas showed a high positive correlation with the trend of TSS, ethylene, WSP, and ISP (Fig. 7). In other words, the expression of AcDELLA7 was positively correlated with kiwifruit softening and ripening. It seemed that AcDELLA7 did not respond to GA3 treatment, but to ripeness. The phenomenon of AcDELLA7 indicated that the expression of some DELLAs or the stability of DELLA proteins could be regulated by other factors, not only by gibberellins. Therefore, we deduced that part of AcDELLAs participate in kiwifruit postharvest ripening and prolong storage time by responding to GA signals, ethylene signals, or other ripening signals. The identification and expression of AcDELLA provided new targets and might be particularly important for kiwifruit post-ripening regulation.
-
In this study, GA3 treatment effectively preserved kiwifruit fruit firmness and delayed fruit ripening. Eight AcDELLA genes were identified from the kiwifruit genome database. Kiwifruit AcDELLA genes are distributed on six different chromosomes, and they contain only one exon. Conserved domains and secondary and tertiary structural analyses showed the similarity of DELLA on the same evolutionary meristem. Phylogenetic analyses demonstrated the homology of the kiwifruit AcDELLA proteins within and among species, to better understanding the evolutionary mechanisms. In addition, gene expression analyses revealed that several AcDELLAs may be involved in the GA signaling pathway to inhibit kiwifruit ripening and softening, especially AcDELLA3, AcDELLA4, and AcDELLA5. The present study comprehensively investigated the genetic characterization of the kiwifruit AcDELLA gene family, which lays the foundation for understanding the kiwifruit AcDELLA genes, and provided a reference for the postharvest preservation of kiwifruit.
This work was supported by the Natural Science Foundation of Shandong Province (ZR2023MC007), and the Key Research and Development Program of Shandong Province (2024TZXD004). The funders had no role in the material creation, designing the study, analysis data and in writing the manuscript.
-
The authors confirm contribution to the paper as follows: data curation: Zhang X, Zhao K, Wang W, Li J, Alabd A; formal analysis: Zhang X, Zhao K, Wang W, Li J, Alabd A, Zhang A; software, visualization, writing - original draft: Zhang X; methodology, writing - review & editing: Zhao K, Wang W, Li J, Alabd A, Zhang A; conceptualization, resources, funding acquisition: Zhang A. All authors reviewed the results and approved the final version of the manuscript.
-
The datasets generated during the current study are available from the corresponding author on reasonable request.
-
The authors declare that they have no conflict of interest.
- Supplementary Table S1 DELLA proteins from different plant species.
- Supplementary Table S2 Primers for real-time PCR.
- Supplementary Table S3 Basic information of the kiwifruit DELLA gene family.
- Copyright: © 2025 by the author(s). Published by Maximum Academic Press on behalf of Chongqing University. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Zhang X, Zhao K, Wang W, Alabd A, Li J, et al. 2025. Gibberellin delays kiwifruit postharvest ripening via the AcDELLA gene family. Plant Hormones 1: e005 doi: 10.48130/ph-0025-0005 

Gibberellin delays kiwifruit postharvest ripening via the AcDELLA gene family
- Received: 02 December 2024
- Revised: 23 January 2025
- Accepted: 11 February 2025
- Published online: 04 March 2025
Abstract: Gibberellin (GA) has a significant regulatory role in the growth and development of higher plants. DELLA proteins are the key negative regulators of the gibberellin signalling pathway. However, the genomic overview of the kiwifruit DELLA gene family and their function on kiwifruit ripening has not yet been studied. Exogenous GA3 can effectively maintain postharvest kiwifruit firmness and delay the ripening process. Eight AcDELLAs genes were identified from the kiwifruit genome and named as AcDELLA1 to AcDELLA8 by the order of chromosomal localization. Sequence analysis confirmed that the N terminus of all eight AcDELLA proteins contained high-conservation DELLA and TVHYNP motifs. Phylogenetic analysis showed that AcDELLA1, and AcDELLA3 were highly homologous with AcDELLA5 and AcDELLA6. The gene expression of AcDELLA3/4/5/6 were significantly induced by GA3 treatment, while, GA3 delayed the expression of AcDELLA7, and AcDELLA8 maintained higher level in GA3 group than in control. Correlation analysis between physiological parameters and AcDELLAs gene expression demonstrated that AcDELLA3/4/5/7/8 had a marked correlation with firmness, TSS, and ethylene production. All these results showed that GA3 treatment could inhibit kiwifruit postharvest ripening and prolong storage time, and these GA-responsive DELLAs may play important roles in this process.
-
Key words:
- Kiwifruit /
- Gibberellin /
- DELLA family /
- Fruit ripening