










-
One of the great challenges facing agriculture is to ensure food security by increasing productivity for the world's growing population. An important cause of food insecurity is the current instability of food production. Therefore, increasing crop yield and stability is crucial. The yield of a crop depends on three key factors: the panicle/spike number per unit area; the grain number per panicle/spike; and the thousand-grain weight[1]. The number of grains per spike of all grains is influenced by the structure of inflorescence. The uncertainty of inflorescence, the branching pattern, the growth pattern, and the number of spikelets and florets all have important effects on the final panicle number[2]. Therefore, inflorescence development is a key determinant of crop yield potential.
Members of the MADS-box family are widespread transcription factors in the plant kingdom, and have many different functions in floral organ differentiation and spikelet development. These genes are summarized according to function into the classic ABCDE model[3−5]. They interact with each other and play a crucial role in inflorescence development. Class A genes, including rice OsMADS14/15/18 and maize ZAP1, play crucial roles in regulating inflorescence meristem (IM) and spikelet meristem (SM) identity[3,6]. Classes B, C, and D genes, such as rice OsMADS32/4/16/3/13/2/58, and maize SI1, ZAG1, and ZAG2 genes, are involved in reproductive stamen, carpel, and lodicule development[3,7,8]. The class E genes, OsMADS8/7/5/1/34, can determine SM and floral meristem (FM) formation, and floral organ differentiation[3,9]. Previous studies have shown that a total of 70 MADS genes were identified in foxtail millet. SiMADS51 and SiMADS64 may be involved in the ABA-dependent pathway of drought response[10].
CLAVATA3 (CLV3) is an important regulator of meristem homeostasis and floral organ number[11]. The CLAVATA-WUSCHEL (CLV–WUS) pathway is a very important negative-feedback loop[12]. WUS positively regulates stem cell maintenance by repressing CLV[13−15]. In Arabidopsis, CLV3 signaling can bind to several different receptors, including CLV1, CLV2/CORYNE (CRN), and BARELY ANY MERISTEM (BAM)/RECEPTOR-LIKE PROTEIN KINASE2 (RPK2), to negatively regulate WUS to maintain stem apical meristem (SAM) homeostasis[16]. In rice, FLORAL ORGAN NUMBER (FON4) and OsMADS1 synergistically maintain FM identity[17]. In maize, CLE peptide FON2-LIKE CLE PROTEIN1 (FCP1) is signaled by the LRR receptor-like protein FASCIATED EAR3 (FEA3) to inhibit WUS1 expression, thereby regulating inflorescence meristem maintenance[18].
Increasing numbers of studies show that inflorescence development is influenced by many factors. KNOX (KNOTTED1-like HOMEOBOX) protein regulates the maintenance and formation of SAM. Loss of KNOX gene function, such as SHOOT MERISTEMLESS (STM) in Arabidopsis thaliana, KNOTTED1 (KN1) in maize, and homeobox 1 (OSH1) in rice, can cause defects in inflorescence morphology[19,20]. Plant hormones such as cytokinin (CK), auxin, and gibberellic acid (GA) play an important role in inflorescence development by regulating cell differentiation and proliferation. Local synthesis of cytokinin plays an essential role in the maintenance of a functional apical meristem. For example, the reduction of OsCKX2 can produce bigger panicles, thus increasing grain yield in rice[21]. Spikelet is the basic unit of the gramineous inflorescence, and the transition process from spikelet meristem to floret meristem is also crucial. AP2 transcription factor, FZP/BRANCHED FLORETLESS 1 (BFL1) in maize, and ETHYLENE RESPONSE FACTOR (ERF) in rice, are essential to this process[22−24].
However, the regulatory networks and molecular mechanisms of foxtail millet inflorescence development are less reported compared with major cereal crops. To date, only a few genes involved in the development of foxtail millet inflorescences have been identified. SiMADS34 regulates inflorescence development through a variety of pathways and affects foxtail millet yield[25]. SiBOR1 plays an essential role in grain yield maintenance through regulating the development of panicle primary branches[26]. The SiNOG1 gene can lead to complete sterilization of foxtail millet at the heading stage[27]. In Setaria viridis, BSL1 can regulate spikelet meristem determinacy[28]. AP1/FUL-like genes can regulate meristem determinacy and flowering time. SvFUL2 can significantly delay flowering time and disrupt multiple levels of meristem determinacy in panicles of the C4 model panicoid grass, Setaria viridis[29]. SGD1, an important RING-type E3 ligase, is involved in regulating grain weight, grain size, and panicle size[30]. The CLV3 homolog can regulate the size of the inflorescence meristem[31]. SvAUX1 was found to affect the branching of the inflorescence and spikelet number[32].
In this study, the transcriptome landscape was analayzed at three stages of foxtail millet inflorescence development to investigate the possible regulatory networks and transcriptional processes of foxtail millet inflorescence development. The cell cycle genes, plant hormones, and key genetic regulatory factors may play key roles during the development of foxtail millet inflorescence. The present study found that 270 genes were specifically expressed in the double ridge meristem (DR) period, 172 genes specifically expressed in the bristle primordium meristem (BP) period, and 1,088 genes were specifically expressed in the green anther (GrA) period. To further evaluate the function of SiMADS1, knockout lines of SiMADS1 were generated by CRISPR/Cas9-mediated gene editing. Five different phenotypes of the simads1 inflorescence were discovered. Subsequently, the downstream regulatory genes of SiMADS1 were identified by RNA-seq and DAP-seq analysis. The present study constructs the transcriptome of the developing inflorescence based on RNA-seq, providing valuable genetic resources for future understanding of the process of foxtail millet inflorescence development.
-
The foxtail millet variety 'Jingu 21' was planted in the Shenfeng experimental field in Jinzhong, Shanxi, China, from May to October, 2023 for RNA-seq. Samples from four groups of tissues (the double ridge meristem stage, the bristle primordium meristem stage, the green anther stage, and two-week-old seedling leaves) were collected. All samples were collected at the same time on July 10, 2023. There were three repetitions in each group. Samples were quickly frozen in liquid nitrogen before processing and stored at −80 °C. Different developmental stages of inflorescence tissues were photographed with a Stereo microscope (SZX16, Olympus, Japan).
Ci846 and simads1 were planted in the Shenfeng experimental field in Jinzhong, Shanxi, China, from May to October 2024 for RNA-seq and DAP-seq. RNA-seq samples from two stages of tissues (the green anther stage and panicle stage) were collected. All samples were collected at the same time on July 15, 2024.
RNA-seq analysis
-
OE biotech Co., Ltd performed the analysis of the RNA-seq. Sequence alignment of Clean Reads to a specified reference genome (
https://phytozome-next.jgi.doe.gov ), by means of hisat2[33]. The number of reads aligned to protein-coding genes in each sample was determined using htseq-count software[34]. The criteria used to screen for differentially expressed genes (DEGs) were p adjustment < 0.05, and Fold change > 2. According to gene expression levels, the genes were divided into six groups. Very high expression FPKM > 300, high expression FPKM ranging from 100 to 300, medium expression FPKM ranging from 30 to 100, low expression FPKM ranging from 10 to 30, extremely low expression FPKM < 10, and no expression FPKM < 1.Analysis of DEGs, GO enrichment analysis, and KEGG pathways
-
Gene expression levels were estimated using DESeq2[35] software based on FPKM values. The significance of differential expression was evaluated by employing a negative binomial distribution test, subsequent to the calculation of fold changes. Finally, differential protein coding genes were screened according to the multiple difference and significance difference test results. Following the identification of differentially expressed genes (DEGs), Gene Ontology (GO) enrichment analysis was performed to characterize their functions. Kyoto Encyclopedia of Genes and Genomes (KEGG)[36] is the main public database on Pathway. Hierarchical cluster, GO, and KEGG pathway enrichment analysis were performed using the OE Biotech cloud analysis tools (
https://cloud.oebiotech.cn ).Quantitative RT-qPCR
-
Sequencing companies provided the RNA of samples. The Union Script First-strand cDNA Synthesis Kit (Genesand Biotech, Beijing, China) was used to reverse transcribe the RNA into cDNA. Gene expression analysis was performed using gene-specific primers and SYBR green reagents on the Bio-Rad CFX connect real-time PCR assay system. The actin gene was used as an internal reference for normalizing the numbers of cDNA among the samples. The primers used in this experiment are shown in Supplementary Table S1.
Statistical analysis
-
Statistical analysis was performed using Prism 7.0 (GraphPad Software, INC., USA). Duncan's multiple comparison range test and t-test were used to evaluate the statistical significance, which was considered significant at the p-value < 0.05 level.
DAP-seq
-
DNA affinity purification (DAP)[37] was performed and analyzed by Boyuan Biology, China. Trimmomatic software[38] was used for data filtering to cut off the end adapter sequence and retain the minimum length of 20 bp. The filtered data were processed by FastQC (version: 0.11.5). Clean reads data were compared to the reference genome using bwa software[39], using default parameters. The resulting reads were uniquely mapped and preprocessed for subsequent analysis. The genome region was divided into exon, intron, intergenic, promoter (transcript start site upstream 2k), and other regions, and the peaks shared by the two biological repeats were used for subsequent analysis.
-
To explore the molecular mechanism of inflorescence development, genes associated with foxtail millet inflorescence development by transcriptome analysis were searched on the basis of the stages of inflorescence development[40]. RNA-seq samples were collected at three developmental stages: the double ridge meristem (DR); the bristle primordium meristem (BP); and the green anther (GrA). These were subsequently compared with the transcriptome of two-week-old seedling leaves (SE) (Fig. 1a–c). A total of 12 samples, consisting of three biological replicates per group, were collected for RNA-seq analysis in this study. Clean reads of the 12 samples range from 38.71 to 46.83 million. 96.90% to 98.04% clean reads were mapped to the reference genome, and 91.26% to 94.23% of the reads were uniquely mapped (Supplementary Table S2). These results confirm that both read quality and read counts were sufficient for quantitative gene expression analysis.
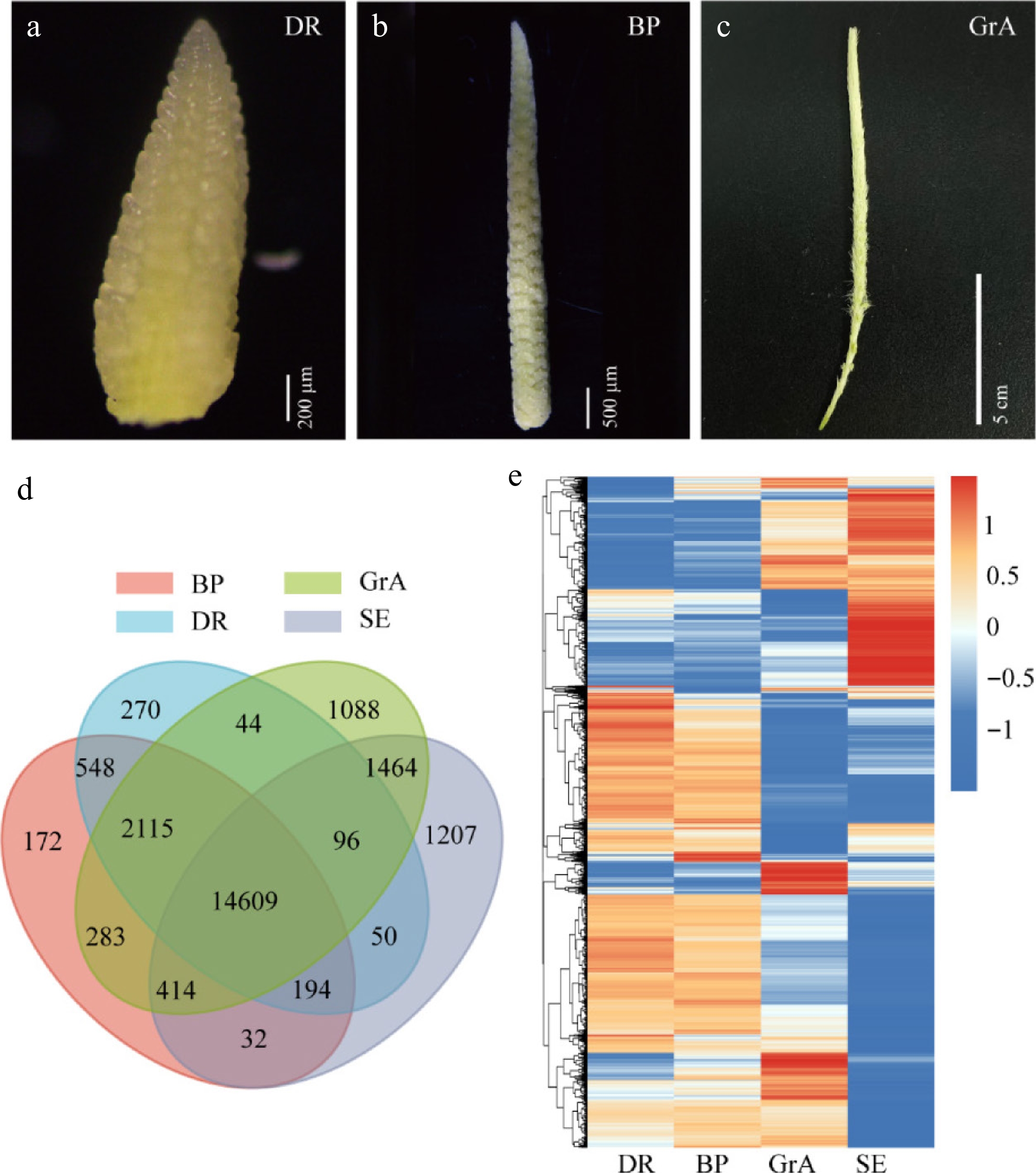
Figure 1.
Global gene expression profiling. (a)–(c) Phenotypic map of three periods of inflorescence development. (a) Scale bar = 200 µm. (b) Scale bar = 500 µm. (c) Scale bar = 5 cm. (d) Venn diagram of the four groups. (e) Hierarchical cluster analysis of genes expressed in four groups.
The principal component analysis (PCA) revealed that the three biological replicates within each sample group clustered closely, indicating high reproducibility (Supplementary Fig. S1a). RNA-seq results demonstrated that altogether 30,618 genes were detected in the four groups. The largest number of genes was measured in the GrA period, and relatively few during other groups (Supplementary Fig. S1b). A Venn diagram was created to understand the relationship between gene numbers in four groups. The findings indicated 14,609 genes were co-expressed, 270 genes specifically expressed in the DR stage, 172 genes specifically expressed in the BP stage, and 1,088 genes specifically expressed in the GrA stage (Fig. 1d). According to the gene expression levels, the genes were divided into six groups. The results revealed that a total of 22,587 genes were expressed (defined as FPKM > 1) (Supplementary Fig. S1c). Hierarchical cluster analysis was performed for all expressed genes (Fig. 1e). GO enrichment analysis and KEGG analysis were performed on all expressed genes (Supplementary Fig. S1d, S1e).
Analysis of genes highly expressed in each period.
-
Previous studies have shown that the gene expression patterns of DR and AP (the awn primordium meristem) period exhibited a relatively high degree of similarity in barley[41]. The results of the Correlation analysis indicated a strong developmental similarity between DR and BP (Supplementary Fig. S2a). A Venn diagram was created to understand the relationship between genes number in the DR and BP periods. The findings showed 17,466 genes were co-expressed, 480 genes specifically expressed in DR stage, 901 genes specifically expressed in BP stage (Supplementary Fig. S2b). Highly expressed genes in DR and BP developmental stages were analyzed (Fig. 2a). In the DR stage, the expression of 26 transcription factors (TFs) was higher than that in other groups. Compared with other groups, the expression of 1 TFs was higher in the BP period (Fig. 2b). KEGG and GO enrichment analysis were performed for highly expressed genes in DR and BP (Supplementary Fig. S2c, S2d). The result showed that high-expression genes were rich in cellulase activity and starch and sucrose metabolism.
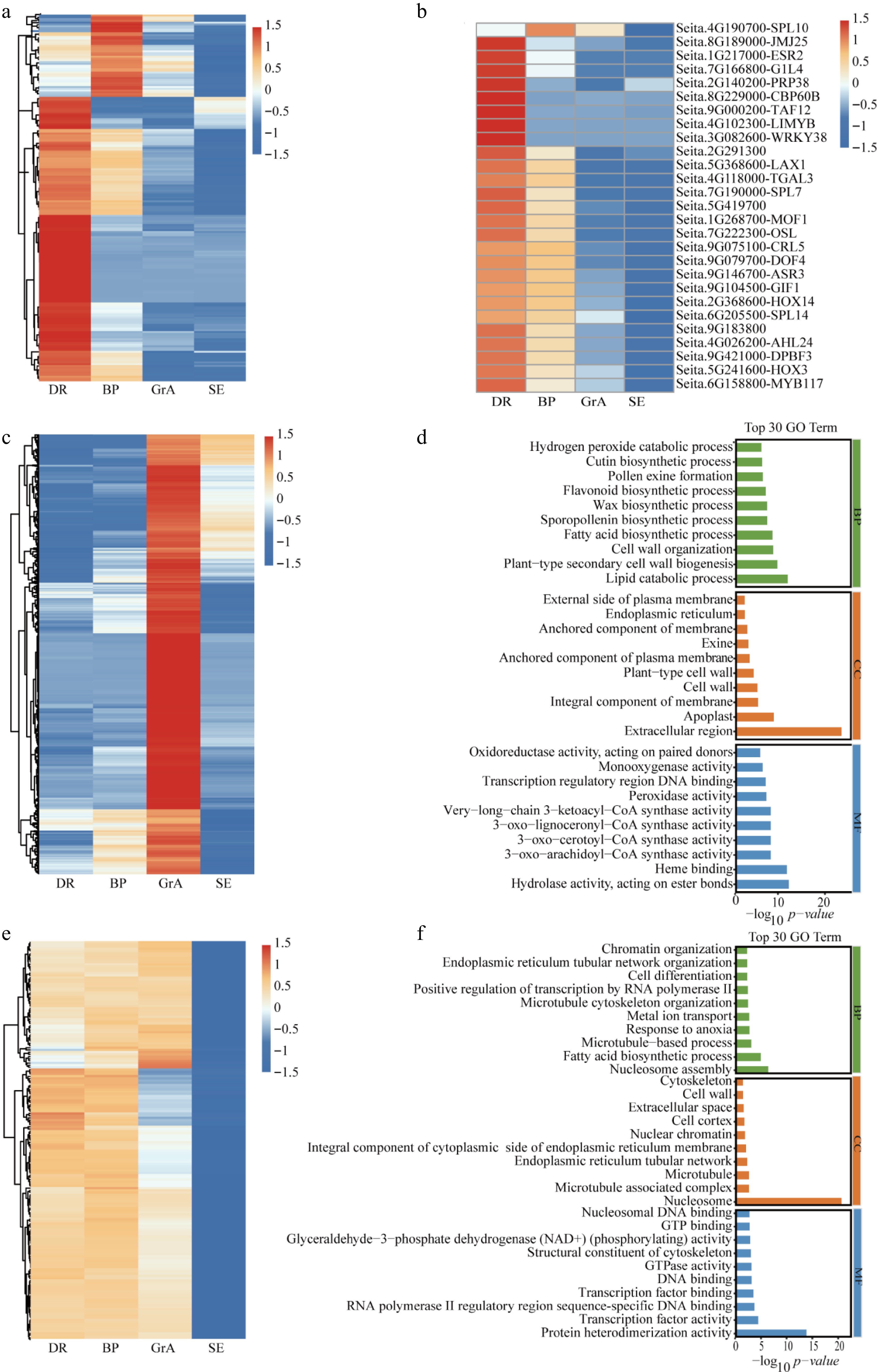
Figure 2.
Analysis of highly expressed genes in each period. (a) Hierarchical cluster analysis of highly expressed in the BP and DR periods. (b) Hierarchical cluster analysis of TFs highly expressed in the BP and DR periods. (c) Hierarchical cluster analysis of highly expressed genes in the GrA period. (d) GO enrichment analysis of the high-expression of genes at GrA stage. (e) Differential expression analysis of the three developmental stages. (f) GO enrichment analysis of the high-expression of genes in three developmental stages. BP: biological process; CC: cellular component; MF: molecular function.
The gene expression patterns of DR and BP exhibited a relatively high degree of similarity, while the transition to the GrA stage during development led to remarkable differences in many gene expressions. Highly expressed genes in the GrA stage were analyzed. In the GrA stage, 2,266 genes exhibited higher expression compared with the other groups (Fig. 2c). During the GrA stage, 233 TFs showed significantly higher expression compared to other groups. Additionally, 93 transporter-related genes, 27 auxin-related genes, 89 kinase-related genes, 63 E3 ligase-related genes, and 36 ROS-related genes were also markedly up-regulated in this stage (Supplementary Fig. S3a−S3f). GO enrichment analysis was performed on highly expressed genes in the GrA period. The result showed that high-expression genes were rich in lipid catabolic process, cell wall, and peroxidase activity (Fig. 2d).
Previous studies have shown that genes enriched in inflorescence development, including transporters, kinases, and transcription factors, exhibit comparatively steady expression patterns in early inflorescence development[41]. Therefore, genes that are highly expressed at all three developmental stages were analyzed. Firstly, a difference analysis was performed, which found 2,554 up-regulated genes (defined as genes that were highly expressed in all three periods) (Fig. 2e). Subsequently, genes exhibiting FPKM values greater than 100 across all three inflorescence developmental stages were selected for further analysis. In the three developmental periods, 10 TFs showed significantly higher expression compared to the two-week-old seedling leaves. Additionally, three transporter-related genes, two auxin-related genes, 10 kinase-related genes, two E3 ligase-related genes, and one ROS-related gene were also markedly up-regulated in the three developmental periods (Supplementary Fig. S4a−S4f). GO enrichment analysis demonstrated that high-expression genes were abundant in nucleosome assembly, the fatty acid biosynthetic process, protein heterodimerization activity, and the nucleosome (Fig. 2f).
Expression feature of the key genes in the foxtail millet developing inflorescence.
-
Within the ABCDE genetic model, which specifically governs floral organ identity, MADS-box homologous genes exhibit pleiotropic effects on plant reproductive growth and floral organ development[2]. Through transcriptome analysis, 30 MADS-box genes with altered expression were identified in this study (Fig. 3a). The results of RNA-seq revealed that only four genes were preferentially expressed in the two-week seedlings, and most of the MADS family genes were highly expressed in the three stages of inflorescence development. In early inflorescence development, the expression of most MADS family genes increased significantly in the GrA stage. The expression of SiMADS1 (Seita.9G490400) was the highest expressed in the GrA period, and its FPKM was as high as 1,000. These findings suggest that MADS-box could be of great importance in the development of foxtail millet inflorescence.
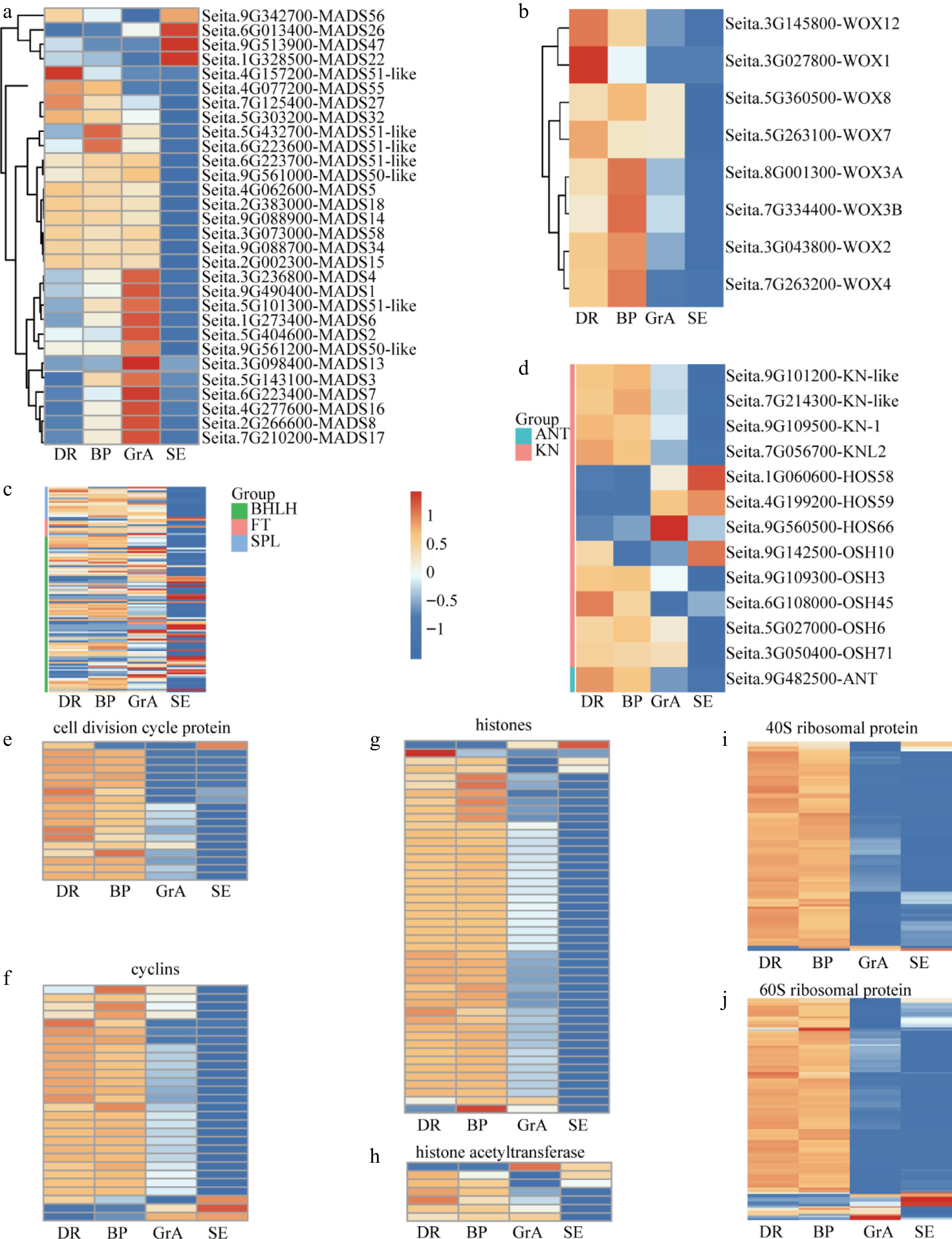
Figure 3.
Expression feature of the key genes in the foxtail millet developing inflorescence. (a) Hierarchical cluster analysis of the SiMADS gene family in foxtail millet developing inflorescence. Hierarchical cluster analysis of genes regulating (b) meristem signaling, (c) meristem identity, and (d) floral organ development, respectively. Hierarchical cluster analysis of (e) cell division cycle protein, (f) cyclins, (g) histones, (h) histone acetyltransferase, (i) 40S ribosomal protein and (j) 60S ribosomal protein during the development of foxtail millet inflorescence and seedlings.
Previous studies have shown that histones are indispensable proteins of the cell cycle. Core cell cycle regulators manipulate gene expression through modifying histone patterns[42]. In the transcriptome, it was found that many genes participating in cell cycle and cell division, for example, histones, cyclins, and cell division cycle protein (Fig. 3e–g), which may be required for the development of inflorescence. Most cyclins, cell division proteins, and histones are highly expressed in the BP and DR periods. Other genes associated with cell cycle regulation, for example, histone acetyltransferase, are also highly expressed in the DR and BP periods (Fig. 3h). Overall, hierarchical cluster analysis of cell cycle genes revealed relatively high cell cycle activity during the DR and BP stages of foxtail millet inflorescence development. Prior studies have demonstrated that 80S ribosomal proteins and 60S ribosomal proteins expression levels in plants decrease after WUS activation[43]. During transcriptome analysis, it was found that many 40S ribosomal proteins, and 60S ribosomal proteins were expressed at higher levels during BP and DR periods (Fig. 3i, j).
Stem apical meristem (SAM) homeostasis is critical for plant development, and the CLV-WUS negative feedback loop plays a crucial role in maintaining SAM homeostasis, and ensuring SAM homeostasis through regulating the amount of stem cells present in the central region[14,44]. Genes involved in stem apical meristem development were analyzed and categorized into three groups: those involved in meristem signaling; those related to meristem identity; and regulatory factors associated with inflorescence development. Firstly, genes associated with meristem signaling, for example, WUS and CLV, were analyzed. However, data analysis found that there were no CLV-related genes, only eight WUS-related genes, and they were highly expressed during the BP and DR periods (Fig. 3b). Secondly, genes associated with meristem identity were analyzed, for example KN1, ANT(AINTEGUMENTA). ANT is a transcription factor encoding the AP2 domain family that coordinates cell proliferation and cell growth by maintaining cell meristem capacity[45−47]. Most ANT was highly expressed in DR and BP stages (Fig. 3c). Finally, we analyzed that some regulatory factors, such as bHLH (basic helix-loophelix), FT (FLOWERING LOCUS T), and SPL (SQUAMOSA promoter-binding protein-like), also play important roles in inflorescence development. Transcripts of most bHLH, SPL, and FT genes were detected in developing inflorescences, and their expression levels generally decreased from the DR to the GrA stage (Fig. 3d).
RT-qPCR verification of RNA-seq data
-
To guarantee the dependability of the transcriptome data, six genes were chosen for RT-qPCR validation. Specifically, one gene (Seita.5G428100-MSP1) that was highly expressed during the DR period (Supplementary Fig. S5a) was analyzed. The rice MSP1 gene, which encodes a leucine-rich repeat receptor protein kinase, plays a critical role in initiating anther wall formation during flower development[48]. Two genes (Seita.8G251800-thionin-like peptide) were highly expressed during the BP period (Supplementary Fig. S5b, S5c). Four genes (Seita.9G490400-SiMADS1, Seita.6G134800-THI2, Seita.8G032600-Cellulase/Endoglucanase, and Seita.7G301000-LTP110-B) that were highly expressed during the GrA period (Supplementary Fig. S5d–S5f). Lipid transfer proteins (LTPs) have been proven to play an essential role in pollen development[49]. The RT-qPCR data showed that the changes in gene expression levels were consistent with the RNA-seq data. This shows that the present analysis results are reliable.
Functional verification of SiMADS1 by the CRISPR-Cas9 system
-
The analysis of the transcriptome revealed that the expression of SiMADS1 was very high in the GrA period. MADS1 has been previously reported to regulate inflorescence development in barley and rice[50,51], but has not been reported in foxtail millet. To confirm the function of SiMADS1 in foxtail millet, SiMADS1 was knocked out in the background of Ci846 by means of the CRISPR-Cas9 system. The distance between the two targets is 7 bp, and both are located in the first exon of SiMADS1. Altogether 25 positive transgenic plants with six editing types were identified (Fig. 4a). Two lines were selected (simads1-4 and simads1-7) for plant phenotype analysis. The analysis showed that, compared to the wild type (WT), the knockout line had a lower seed setting rate (Fig. 5a–h). Grain size and hundred-grain weight were also measured. The results showed that there were no significant differences in grain length, grain width, and hundred-grain weight (Fig. 5i–r). One of them, (simads1-4), was selected for phenotypic analysis. Five distinct floral phenotypes were identified: Type I (5.44%, n = 441), characterized by twin flowers per panicle; Type II (57.37%, n = 441), exhibiting the absence of inner floral organs; Type III (8.17%, n = 441), showing stigma dysplasia; Type IV (23.58%, n = 441), featuring anther deletion; and Type V (5.44%, n = 441), demonstrating ovule deletion (Fig. 4b, c). These findings imply that SiMADS1 plays an essential role in determining flower organ identity.

Figure 4.
Gene structure diagram and phenotype analysis of WT and simads1 knockout line. (a) Gene structure map and identification of mutation site. (b) Phenotype analysis of simads1 and WT flower. Type I: double flower (Scale bar = 1 mm); Type II: empty shell (Scale bar = 1 mm); Type III: stigma dysplasia (Scale bar = 1 mm); Type IV: anther deletion (Scale bar = 500 μm); Type V: Ovule deletion (Scale bar = 1 mm); WT (Scale bar = 500 μm). (c) The number and percentage of five types in simads1 flowers.
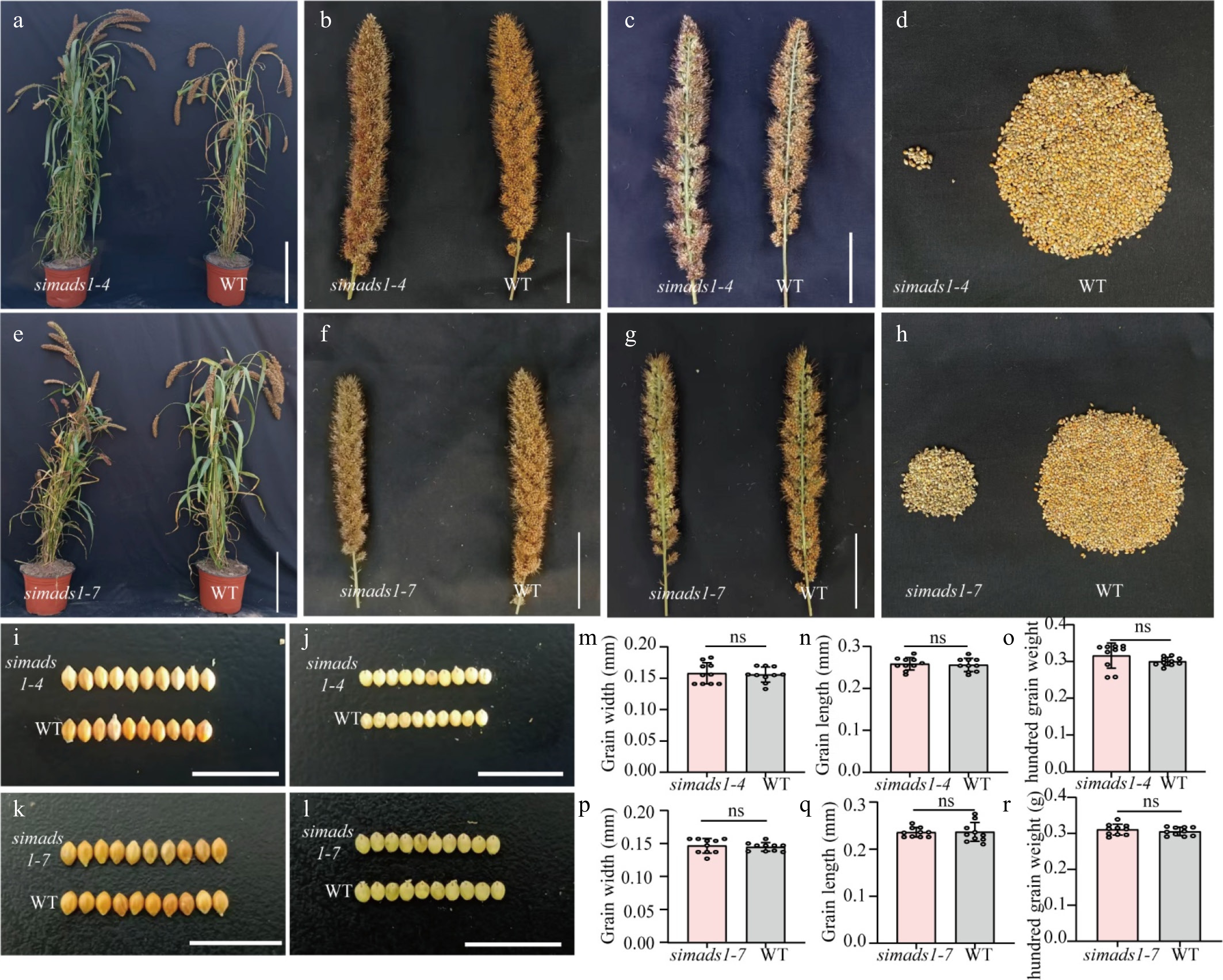
Figure 5.
Phenotypic analysis of simads1 and WT. (a)–(d) Phenotypic analysis of WT and simads1-4. (e)–(h) Phenotypic analysis of WT and simads1-7. (a) and (e) Scale bar = 25 cm. (b), (c), (f) and (g) Scale bar = 5 cm. (i) and (j) Grain size of WT and simads1-4. (k) and (l) Grain size of WT and simads1-7. Scale bar = 1 cm. (m)–(o) Grain width, grain length and hundred grain weight of WT and simads1-4. (p)–(r) Grain width, grain length, and hundred grain weight of WT and simads1-7. Statistically significant differences between samples are represented by the number of asterisks on the column (* p-value < 0.05, ns: not significant).
Combined RNA-seq and DAP-seq to identify downstream-regulated genes of SiMADS1
-
To elucidate the molecular mechanism of SiMADS1 in regulating foxtail millet inflorescence development, transcriptome sequencing of both WT and simads1 during GrA and panicle stages was performed, followed by differential gene expression analysis (Supplementary Fig. S6a–S6d). To better search for the downstream regulatory genes of SiMADS1, DAP-seq of SiMADS1 was performed. A total of 20,792 peaks were identified, of which 3.38% were in the exon region, 9.45% in the intron region, 66.81% in the intergenic region, and 20.36% in the promoter region (Fig. 6a). Next, the 7,283 genes identified in the DAP-seq analysis were overlapped with DEGs from each of the two periods to try to identify the direct target genes regulated by SiMADS1. The Venn diagram revealed that there were 155 overlapping genes in the GrA stage, and 215 overlapping genes in the panicle stage (Fig. 6b, c). The results of GO enrichment analysis revealed that the target genes in the GrA period were more concentrated in carpel development, floral organ morphogenesis, plant-type ovary development, and floral meristem determinacy (Fig. 6d). The target genes in the panicle stage are rich in the regulation of gene expression, stamen development, stamen formation, and plant-type ovary development (Fig. 6e).
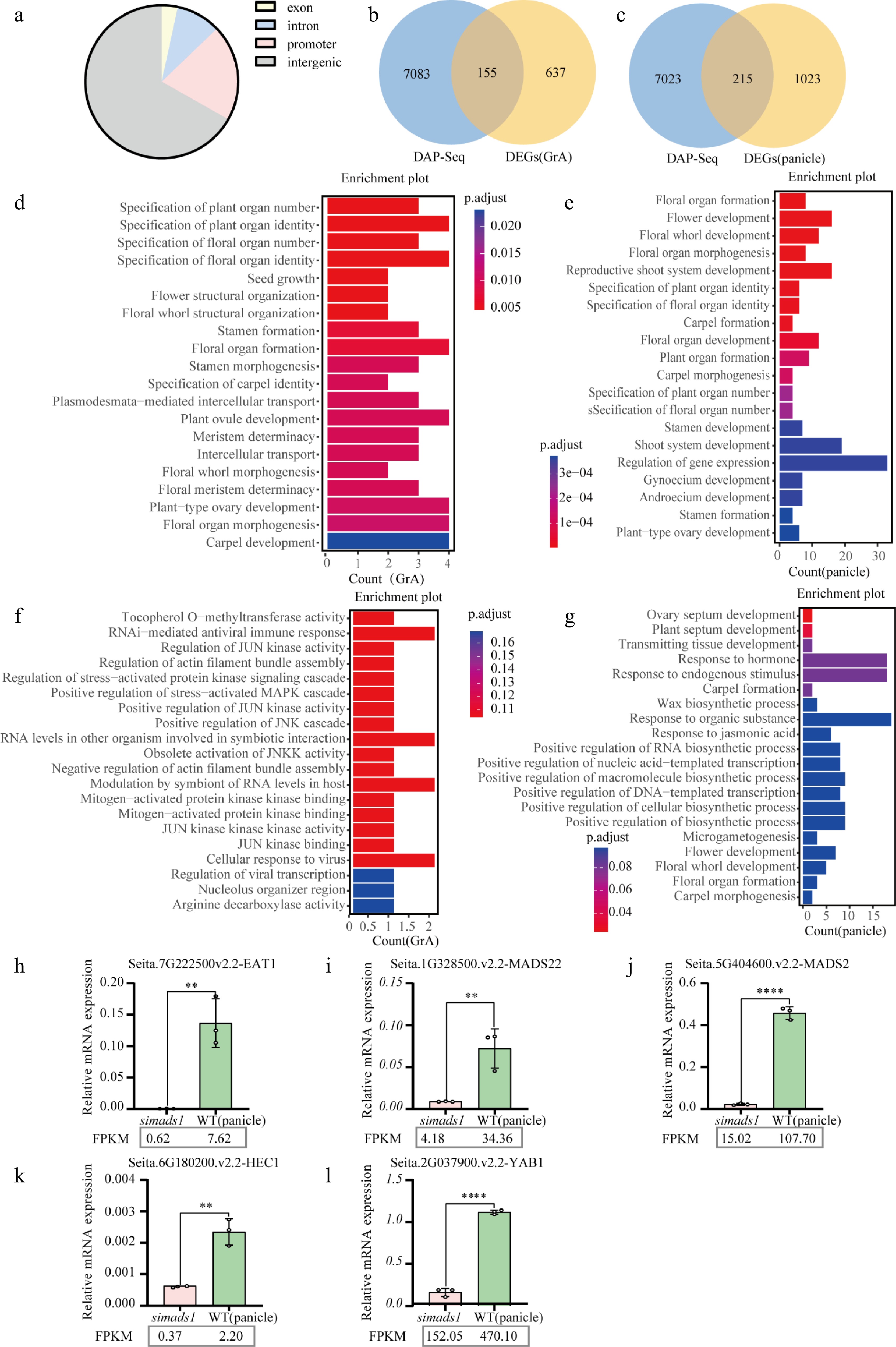
Figure 6.
Combined analysis of RNA-seq and DAP-seq. (a) Distribution of SiMADS1 binding regions in the foxtail millet genome. (b) and (c) The Venn diagram shows the overlapping genes of SiMADS1 binding genes and DEGs. (d) and (e) Gene ontology (GO) enrichment analysis was performed for genes with overlap between SiMADS1 binding genes and DEGs. (f) and (g) Gene ontology (GO) enrichment was used to analyze the overlapping genes between SiMADS1 binding genes, and DEGs in the promoter region. (h)–(l) The representative genes related to inflorescence development at spikelet period were quantitatively confirmed by RT-qPCR. The ACTIN gene served as the internal control in this study. The error bars represent the standard deviation of three biological replicates. Statistically significant differences between samples are represented by the number of asterisks on the column (** p < 0.01, **** p < 0.0001).
To further identify the downstream regulatory genes of SiMADS1, the genes in the promoter region were separately enriched. The results showed that there were no targets related to floral development in the GrA stage, while the targets in the panicle stage were rich in floral development, floral whorl development, floral organ formation, and carpel morphogenesis (Fig. 6f, g). Therefore, genes (EAT1, HEC1, MADS2, MADS22, YAB1) related to flower development at the panicle stage were selected for RT-qPCR. These genes play important roles in inflorescence development. The RT-qPCR results showed that these genes were down-regulated compared with WT, indicating that these genes may be positively regulated by SiMADS1 (Fig. 6h–l).
-
Inflorescence development is closely related to crop yield. Although some genes have been previously reported to regulate inflorescence development[2,5], the process of foxtail millet inflorescence development has been less studied. The transcriptome at different stages of foxtail millet inflorescence development was therefore analyzed and compared with the transcriptome of two-week seedlings. This study provides new genetic resources for further analysis of the molecular mechanism of spike development in foxtail millet. Employing CRISPR-Cas9 genome editing, a functional validation of SiMADS1 was conducted. Integrated analysis of RNA-seq and DAP-seq data further identified its downstream target genes. These findings enhance our understanding of the molecular mechanisms governing inflorescence development and provide a valuable resource for future functional studies of related genes.
In the early stages of inflorescence development, the cell division and cell cycle plays a crucial role[41]. In the DR and BP phases of foxtail millet, the expression of cell cycle-related regulators, cyclin, and histone, are preferentially expressed, indicating active cell proliferation and differentiation, resulting in the formation of flower organs. Furthermore, genes highly expressed during the GrA period were enriched in cell wall components, which also reveals the active growth of floral organs in the late developmental stages of foxtail millet. Previous studies have indicated that after WUS activation in Arabidopsis thaliana, the expression level of 80S ribosomal proteins and 60S ribosomal proteins expression levels decreased[43]. In the present study, it was found that 40S ribosomal protein and 60S ribosomal protein were all highly expressed in the DR and BP periods, indicating that their expression patterns may be different in foxtail millet, but more specific studies are needed.
Previous studies have indicated that the expression of WUS is highest in barley during the DR period[41], and higher in foxtail millet during the DR and BP periods, but no CLV homologous gene were found in foxtail millet. A class of genes were found which encoded thionin-like peptides that were very highly expressed (Supplementary Fig. S7, Supplementary Table S3). Therefore, it is speculated that these genes may be a specific class of genes in foxtail millet and have similar functions to CLV, but further research is needed. KNOX family members are further partitioned into two groups based on sequence and expression patterns: Class I genes are essential for the formation and maintenance of the stem apical meristem, while Class II genes exhibit various expression patterns depending on the gene[52,53]. The high expression of KN1 homologues in foxtail millet during DR and BP periods indicates their conserved function in the development of foxtail millet inflorescence.
Previous studies have indicated that non-specific lipid transfer proteins plays a crucial role in anther development[49]. Many non-specific lipid transfer proteins were found in the transcriptome of simads1, the expression of these genes decreased significantly compared with WT (Supplementary Fig. S8), which showed that non-specific lipid transfer proteins may be regulated by SiMADS1, but the specific regulatory mechanism needs further gene research.
MADS-box plays a crucial role in the development of plants. So far, a series of MADS-box genes have been researched in Arabidopsis, rice, and barley[41,54,55]. Previous studies have shown that SiMADS34 can affect grain yield by regulating inflorescence development, but its seed size does not change[25]. SEP subfamily members are key regulators of inflorescence development in angiosperms[56−58]. OsMADS1 is known to be a member of the SEP-like gene[59]. The present study shows that the SiMADS1 knockout line can lead to the loss of flower organs and abnormal development, such as the loss of ovule and stigma development. This agrees with the results reported in rice[51]. It has been previously reported that HvMADS1 can not only regulate the development of inflorescence but also affect the grain size[50]. However, it does not affect grain size in foxtail millet (Fig. 5i–p), indicating that it has different functions in the development of foxtail millet inflorescence.
The rice genome contains three SVP MADS-box genes (OsMADS22, OsMADS47, and OsMADS55), and their role in regulating meristem identity is conserved[60]. Previous studies have indicated that OsMADS2 plays a crucial role in floret opening and stamen emergence[61]. The HECATE (HEC) basic helix-loop-helix transcription factor regulates SAM's cellular behavior and is critical for maintaining SAM integrity, and its loss of function also contributing to stigma dysplasia[62−64]. In this study, multiple direct target genes of SiMADS1 were identified, such as EAT1, MADS22, MADS2, HEC1, and YAB1. However, the functions and molecular mechanisms of these genes in foxtail millet are still poorly characterized.
-
This study revealed the phased gene expression pattern during the development of foxtail millet inflorescence. It also revealed that SiMADS1 plays an essential role in determining the identity of flower organs. Multiple direct target genes of SiMADS1 were identified. It offers precious genetic resources for a deeper understanding of the development process of foxtail millet inflorescence. It expanded our knowledge of the molecular mechanism of inflorescence development, and contributed to the future functional study of genes associated with inflorescence development.
This work was supported by the National Key Research and Development Program of China (2023YFD1202702-6), the National Natural Science Foundation (32400217, and 32200222), and the Start-up Fund of Shanxi Agricultural University (2021BQ84).
-
The authors confirm their contributions to the paper as follows: experiment execution: Zhang JJ, Zhang MH, Wu HJ, Hu JX, Hao JH, Zhang LQ, Dong S, Li X, Gao L, Yang G, Yuan X; data analysis: Zhang JJ, Zhang MH, Wu HJ, Hu JX, Hao JH, Zhang LQ, Dong S, Li X, Gao L, Yang G, Yuan X, Chu X, Wang JG; draft manuscript preparation: Zhang JJ, Chu X, Wang JG; funding: Chu X, Wang JG. All authors reviewed the results and approved the final version of the manuscript.
-
The RNA-seq and DAP-seq data can be found in the NCBI database. The transcriptome of different inflorescence development period: PRJNA1285567. The transcriptome of Ci846 and simads1: PRJNA1332245. The DAP-seq of SiMADS1: GSE309337.
-
The authors declare that they have no conflict of interest.
- Supplementary Table S1 The sequences of the primers used in this study.
- Supplementary Table S2 Summary of RNA-seq read mapping result.
- Supplementary Table S3 The expression of thionin-like peptide.
- Supplementary Fig. S1 Study of global gene expression in different periods.
- Supplementary Fig. S2 Gene analysis of high-volume expression in BP and DR periods.
- Supplementary Fig. S3 Classification of highly expressed genes in GrA period.
- Supplementary Fig. S4 Classification of highly expressed genes in all three periods.
- Supplementary Fig. S5 The representative genes in inflorescence development were quantitatively confirmed by RT-qPCR.
- Supplementary Fig. S6 Transcriptome sequencing analysis of simads1.
- Supplementary Fig. S7 Expression characteristics of the thionin-like peptide in the foxtail millet developing inflorescence.
- Supplementary Fig. S8 Expression feature of the non-specific lipid transfer proteins (LTP) simads1 and WT in panicle period.
- Copyright: © 2025 by the author(s). Published by Maximum Academic Press on behalf of Hainan Yazhou Bay Seed Laboratory. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Zhang JJ, Zhang MH, Wu HJ, Hu JX, Hao JH, et al. 2025. Transcriptome profiling of developing inflorescences reveals SiMADS1 as a key regulator of floral development in foxtail millet. Seed Biology 4: e024 doi: 10.48130/seedbio-0025-0025 

Transcriptome profiling of developing inflorescences reveals SiMADS1 as a key regulator of floral development in foxtail millet
- Received: 21 August 2025
- Revised: 28 September 2025
- Accepted: 24 October 2025
- Published online: 17 December 2025
Abstract: Inflorescence development of foxtail millet (Setaria italica L.) is important for its yield and quality. The molecular basis of foxtail millet inflorescence development is poorly understood. In this study, the transcriptome landscape of three developing inflorescence stages in foxtail millet was analyzed and compared them with two-week seedlings. The transcriptome revealed the expression model of genes that may be engaged in inflorescence development, such as kinase, transporter, E3 ubiquitin ligase, reactive oxygen species (ROS) related proteins, MADS-box family, shoot apical meristem pathway related proteins, cell cycle associated proteins, ribosomal proteins, etc. The SiMADS1 knockout line exhibits a phenotype of defective floral organs, resulting in greatly reduced yield. The transcriptomes of simads1 at the green anther and panicle stages were further analyzed. By integrating transcriptome data with DAP-seq analysis, multiple downstream regulatory genes of SiMADS1 were identified. This study provides new genetic resources for further analysis of the molecular mechanism of inflorescence development in foxtail millet.
-
Key words:
- Foxtail millet /
- Inflorescence development /
- Transcriptome analysis /
- SiMADS1 /
- DAP-seq