






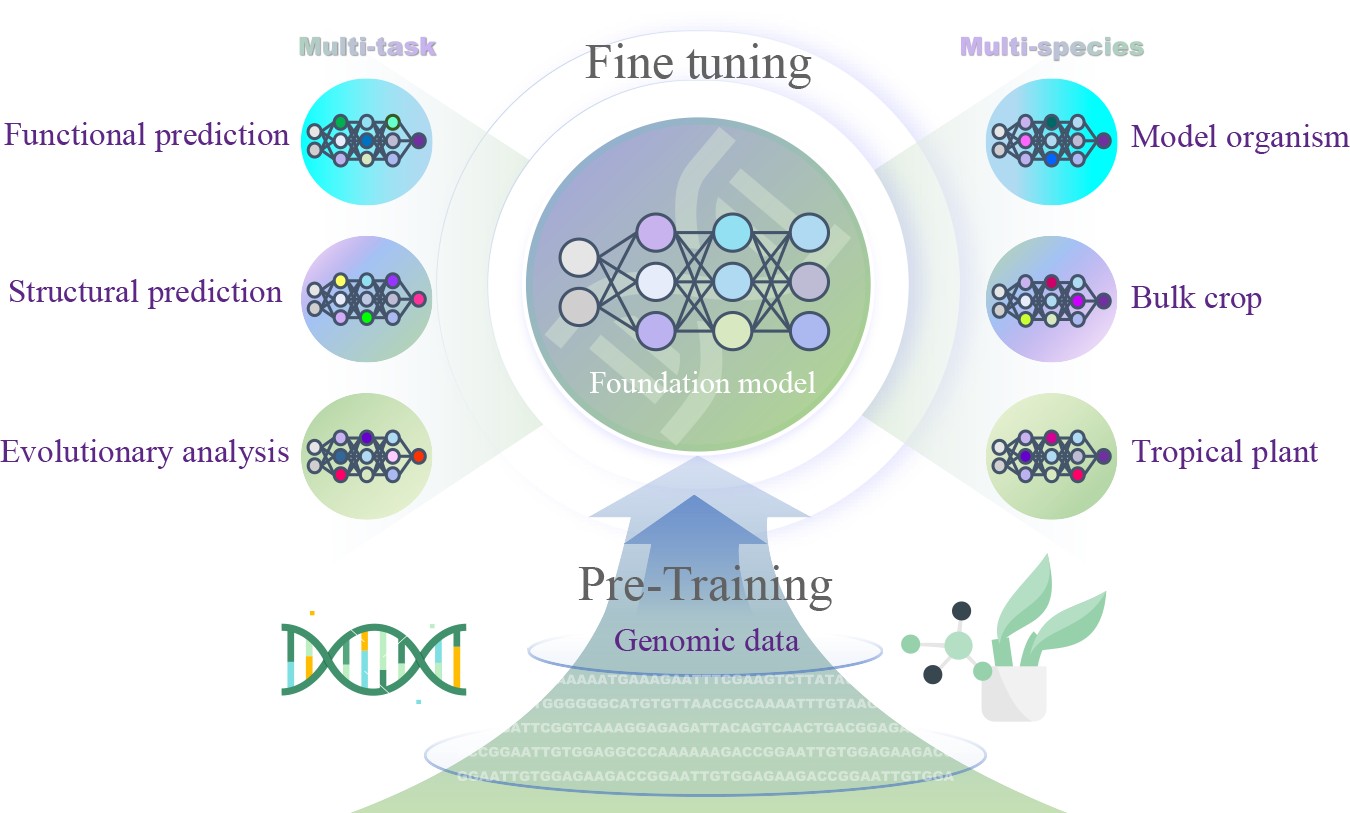


-
In the digital era, the rapid evolution of artificial intelligence (AI) technology has established large language models (LLMs) as a pivotal transformative force within the AI domain[1]. AI's inception dates to the mid-20th century Turing era, spurred by Alan Turing's concept of 'machine intelligence'[2]. This technology has undergone a qualitative leap, evolving from straightforward linear function algorithms to intricate neural networks[3]. Presently, LLMs, such as ChatGPT[4] and LLaMA[5], have emerged as seminal achievements in AI, inciting transformations across various sectors including academia, medicine[6], law[7], and finance[8]. These advancements have fueled researchers' aspirations towards the realization of Artificial General Intelligence (AGI).
In recent years, AI has made breakthrough progress in many fields. John J. Hopfield and Geoffrey E. Hinton were awarded the 2024 Nobel Prize in Physics for their pioneering contributions in neural network research[9]. Additionally, Demis Hassabis and John Jumper received the 2024 Nobel Prize in Chemistry for their significant contributions to protein structure prediction using the AI model AlphaFold2[10]. These recognitions have underscored the increasingly prominent role of AI in scientific research.
AI models are revolutionizing traditional biological research methods. In the realm of plant genomics, machine learning technologies and deep neural networks have fully demonstrated their immense potential in bioinformatics data analysis[11−13]. These technologies are widely applied in various aspects, including function prediction[14−17], gene expression regulatory network prediction[18,19], and protein-protein interaction prediction[20,21]. However, traditional machine learning models often exhibit limitations such as high data requirements, poor transferability, and weak generalization capabilities, which make it challenging to directly apply them to other tasks or species[22]. By adopting a two-step strategy involving pre-training LLMs on large volumes of unlabeled data and fine-tuning them on a small amount of labeled data, these limitations can be effectively overcome[23]. Currently, numerous studies based on LLMs have achieved more significant results compared to those based on traditional machine learning approaches[24−32]. Furthermore, LLMs specifically trained on plant data have started to emerge[33−36]. For example, AgroNT is a foundational LLM trained on the genomes of 48 plant species, which outperforms traditional analysis tools in regulatory annotation, promoter strength, and tissue-specific gene expression[35]. Furthermore, FloraBERT introduces BERT into the plant field and successfully predicts gene expression levels in various tissues of maize[36]; the Genomic Pre-trained Network (GPN) predicts gene functional elements in Arabidopsis thaliana[33].
However, despite the potential demonstrated by these LLMs in plant genomic data, they are currently primarily applied to research on animal genomic data[37]. How to effectively transfer these LLMs to the field of plant genomic research remains an urgent issue that needs to be addressed in the future. Meanwhile, due to the significant differences between natural language and biological genomic language, bridging this gap has also become a current research hotspot. This article aims to explore the application prospects and challenges of LLMs in plant genomic research.
Development of language models
-
Language models (LMs) are tools that learn from given language text data to acquire specific patterns and relationships within the language, with the aim of completing specified downstream language processing tasks. Based on algorithmic structure and task completion capabilities, LMs can be categorized into several types:
(1) Statistical Language Models (SLMs), which are predicated on the hypothesis of Markov chains, are designed to predict the next term in a sequence based on the preceding text. It is further subdivided into n-gram models according to the context coverage length. In the early stages, SLMs were primarily utilized for Information Retrieval (IR) and Natural Language Processing (NLP)[38].
(2) Neural Language Models (NLMs) utilize neural networks to simulate the sequential characteristics of language. Major network architectures include convolutional neural networks (CNNs)[39], recurrent neural networks (RNNs)[40], and long short-term memory networks (LSTMs)[41]. CNNs are specifically designed to process data with grid-like topology, with core features including convolutional layers and pooling layers[42]. RNNs, based on a cyclic connection architecture between network nodes, are specifically tailored for sequence data and are particularly effective at capturing dependencies in time series. Their cyclic connection mechanism enables them to effectively grasp temporal information in text[43]. CNNs excel at processing spatially structured data, such as images, while RNNs are adept at handling time series data, such as text and speech. Both architectures possess unique strengths in different task areas and can be combined to address more complex tasks.
(3) Pre-trained Language Models (PLMs)[44] have achieved notable success in computer vision and NLP tasks, significantly addressing the previous challenges of generalization and transferability faced by deep learning models (DLMs). These models have ushered in revolutionary breakthroughs in the development of AI technology (Fig. 1). Traditional DLMs are often constrained to specific tasks and datasets, with performance declines when applied to new environments. Utilizing the 'pre-training + fine-tuning' paradigm, PLMs first undergo pre-training on extensive unlabeled datasets, thoroughly extracting the rich feature rules of data through iterative learning. Subsequently, they are fine-tuned for different specific datasets. PLMs can rapidly adjust their parameters to accurately match and optimize for specific tasks. This approach not only enhances the model's transfer learning capabilities and facilitates the sharing and integration of cross-domain knowledge but also significantly reduces reliance on large-scale labeled datasets[45]. It performs effectively even in scenarios with limited data, thereby paving new avenues for AI applications in fields with scarce data resources.
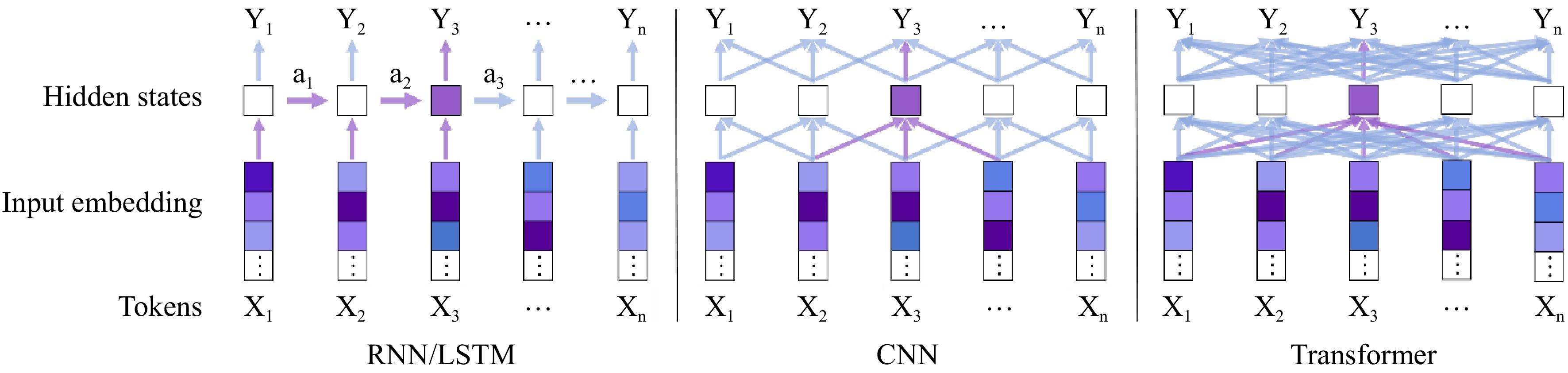
Figure 1.
Comparison of deep learning sequence models[49]: RNN / LSTM: iteratively transmitting sequence information based on hidden states. CNN: Converging the data of adjacent areas through the local perceptual field of view. Transformer: The self-attention mechanism is used to fully capture the pattern information of any span of the input sequence.
(4) Large Language Models (LLMs)[23], typically referring to large-scale neural networks with millions or even billions of parameters, are primarily based on the Transformer architecture. Researchers have observed that as model size increases, the performance in solving complex tasks significantly improves in ways that smaller models do not exhibit. Large pre-trained language models (e.g., GPT-3 with 175 billion parameters[46]) can execute downstream tasks using a small amount of sample data through 'In-Context Learning', a capability lacking in smaller pre-trained language models (e.g., GPT-2 with 1.5 billion parameters[47]). The unique capabilities of LLMs, termed 'Emergent Abilities', have led the academic community to collectively refer to these large pre-trained language models as 'LLMs'. As a quintessential example of LLMs, ChatGPT has been fine-tuned on human dialogue datasets using GPT series models, demonstrating remarkable human-machine dialogue capabilities. The launch of the initiative attracted extensive attention from diverse sectors of society[48].
Advantages of transformer architecture and large language models
-
The significant advancement in data processing capabilities of LLMs is primarily attributed to the Transformer neural network architecture, as introduced by Google in 2017[50]. The Transformer architecture comprises both encoder and decoder components and ingeniously integrates multi-head self-attention mechanism, masked pre-training task, and next sentence prediction task. These innovations significantly enhance the model's capacity to comprehend and generalize from the input data[51] (Fig. 2).
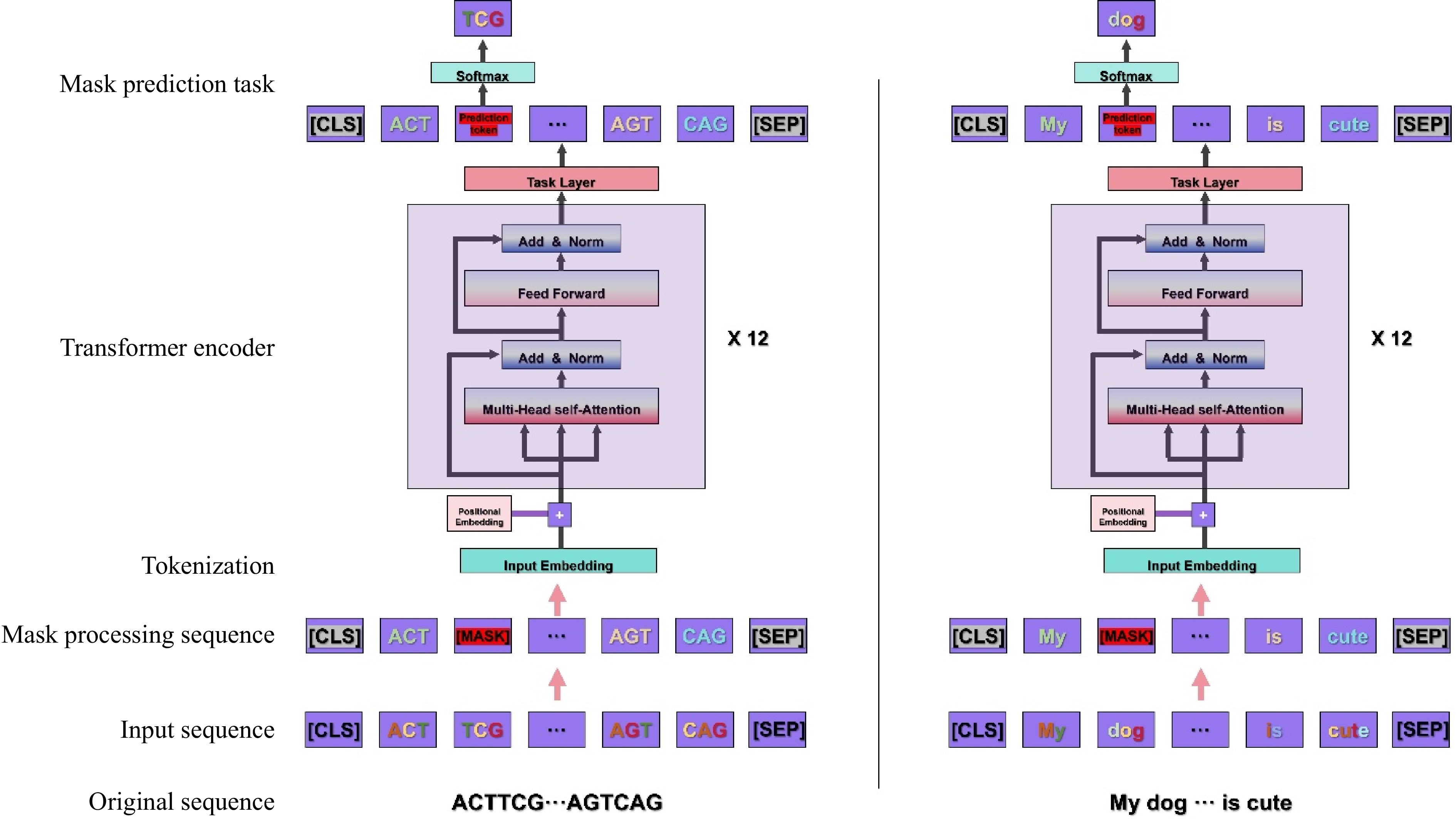
Figure 2.
Illustration of the masked pre-training task process for BERT models based on DNA and text sequences[49,51]. The process is as follows: 1) Input sequence preparation: One-dimensional character sequences serve as the initial input data. 2) Data preprocessing: The original character sequences undergo segmentation, trimming, and padding to standardize the sequence length, ensuring a consistent data format for subsequent processing. 3) Tokenization, masking, and special token insertion: The preprocessed sequences are tokenized, transforming character sequences into token sequences. Tokens within the sequence are randomly selected for masking, which involves replacing them with a special [MASK] token, randomly substituting them with other tokens, or leaving them unchanged. Special tokens [CLS] and [SEP] are inserted at the beginning and end of the sequence, respectively, to denote the start and end of the sentence, aiding the model in comprehending the overall structure of the sequence. Tokens, as the fundamental units for model learning, encode the semantic information of the sequence. 4) Embedding layer processing: The tokenized sequences are fed into the embedding layer, where each token is converted into a vector representation, thereby extracting and encoding the feature information of the data. 5) Transformer module training: The embedded vector sequences are input into the Transformer module for deep training. The Transformer module comprises multiple stacked encoder layers, each containing components such as self-attention mechanism, feedforward neural network, and normalization layer. These components collectively process the sequence to capture linguistic features and contextual information. 6) Model output and token prediction: The model outputs the vector representation of the masked tokens and attempts to predict the original tokens at the masked positions. 7) Similarity computation and parameter updating: The similarity between the predicted tokens and the original (pre-masked) tokens is computed (typically using the cross-entropy loss function). Through the backpropagation algorithm, the loss information is propagated back to the model, thereby updating the model parameters and gradually enhancing the model's ability to predict masked tokens. 8) Training iteration and optimization: The process is repeated over multiple training epochs to continuously optimize the model parameters, thus continuously improving the model's understanding and generation capabilities for the sequences.
(1) The multi-head self-attention mechanism directly captures complex interactions between sequences and is not theoretically limited by sequence length[52]. Compared to traditional RNNs, the Transformer architecture circumvents the challenge of gradient explosion or vanishing gradients when processing long sequences[53]. Furthermore, unlike CNNs, Transformer is not constrained by the window size of the convolutional kernel, enabling direct interactions among global tokens rather than being restricted to local contexts[52].
(2) The masked language model training task is to forecast the concealed words by relying on the visible segments of the input sequence. This approach encourages the model to delve deeper into the intrinsic patterns and contextual information within the sequence, thus further enhancing its generalization capabilities[50,51].
(3) The next sentence prediction task facilitates the model's ability to learn the coherence and logic between sentences. It does so by taking a pair of sentences as input and assessing whether the second sentence is a logical continuation of the first. This design not only enhances the model's capacity to understand textual structure but also provides robust support for handling complex linguistic tasks[50,51].
-
Language, as a medium for information exchange, whether it be human languages such as Chinese and English, programming languages like Python and Java, or even the genetic language of life, all adhere to specific vocabulary combinations and grammatical structures to precisely convey rich semantic content. Despite their varying forms, they exhibit similarities in information expression, structural characteristics, and logical rules.
In terms of information expression, all three employ specific element combinations to form meaningful information carriers. Genetic language[54] encodes genetic information via base sequences, natural language constructs sentences utilizing characters, vocabulary, and grammar, and programming languages manage computer operations[55] through variables, functions, and other syntactic structures. In terms of structural characteristics, they all display complex hierarchical structures and organizational patterns. Genetic language is composed of structures such as promoters, introns, and exons, which combine to form functional genes that regulate the expression and functional realization of organisms[56]; natural language forms texts through the specific combination of vocabulary, grammar, and sentences to facilitate information transmission and idea expression[57]; programming languages possess modular structures, such as functions and class modules, which form complex program architectures through invocation and nesting relationships, enabling efficient management of computer resources and the coordinated execution of tasks[58]. In terms of logical rules, they all follow specific rules and constraints to ensure the accuracy and comprehensibility of the language. Genetic language is subject to biochemical processes; for instance, the transcription process from DNA to mRNA is precisely regulated by enzymes such as RNA polymerase, and the translation process from mRNA to proteins depends on the synergistic action of ribosomes and tRNA. These processes are subject to strict biochemical regulation to ensure the accurate transmission of genetic information[59]; natural language must adhere to grammatical and contextual rules to convey information, otherwise, it may lead to misunderstandings or ambiguities; programming languages must be written in accordance with specific syntax rules, otherwise, they may result in compilation errors or program crashes.
In the enigma of life, the genome sequence serves as the language. It forms the foundation of genetic information that underpins life. Composed of the base sequence of DNA or RNA (A, T, G, C, U), it directs the gene expression and protein synthesis in organisms, thereby constructing a unique informational coding system[60]. Unraveling the genome's mysteries and elucidating its internal regulatory logic has consistently been at the forefront of biological research. Although the translation from DNA to protein follows universal biochemical principles, the regulatory mechanisms governing gene expression display considerable diversity across different cell types and organisms. Within a complex biochemical reaction network, a single gene may fulfill various physiological functions, and genes that are distant from each other may also share similar roles in regulating life processes[61]. The complexity, ambiguity, and long-range correlations inherent in the genomic language continue to challenge researchers, who have yet to develop a comprehensive grammatical system to analyze this language of life.
The application of advanced natural language processing (NLP) technologies, particularly LLMs, in AI, has unlocked unprecedented potential for the in-depth analysis of genetic information within DNA sequences[49], and for elucidating the intricate relationships between genes and biological phenotypes. As a significant milestone in deep learning, LLMs utilize deep neural networks to learn and comprehend the nuances of natural language. Initial language models, including Long Short-Term Memory (LSTM) architectures[41], encountered limitations due to their one-way processing approach and restricted memory capabilities when analyzing textual data[62]. However, with the rapid evolution of AI technology, LLMs have demonstrated a profound understanding of human language and a remarkable ability for creative generation. They can accurately capture grammatical, semantic, and contextual cues, and produce well-structured, semantically rich text[63]. Utilizing these models for interpreting the 'linguistic patterns' within DNA sequences is anticipated to markedly expedite the genetic code decryption process.
Throughout the trajectory of language acquisition, there are notable parallels between the learning mechanisms of extensive language models and those of biological entities in their linguistic development. In infancy, life learns language through imitation and repetition to master basic rules. With this growth, after mastering the basic logic, learning new information becomes more efficient, and a small amount of information can be quickly integrated into the knowledge system. LLMs have similarly evolved through a trajectory that begins with the pre-training on substantial datasets and proceeds to the fine-tuning on more constrained data sets, gradually enhancing their performance to enable precise language interpretation and creation.
The similarity between life language evolution and human language evolution
-
Biological entities, ranging from unicellular bacteria to intricate mammals, function as sophisticated systems for information retention[64]. These organisms encode hereditary traits within the nucleotide sequences of their genetic material, ensuring the perpetuation of biological traits and the propagation of their species. With each generation, the reproduction of offspring entails the conveyance of genetic information. Beyond genetic communication, organisms also engage in the transmission of knowledge through behaviors, social engagements, and cultural legacies[65]. The conveyance and accumulation of such information facilitates the adaptation to fluctuating environments and the evolution of more intricate and effective survival tactics. From a broad evolutionary perspective, DNA sequences, and human languages exhibit striking similarities in their evolutionary processes[54,66]. This observation offers a novel approach to analyzing DNA sequences by employing NLP technologies and methodologies (Fig. 3).
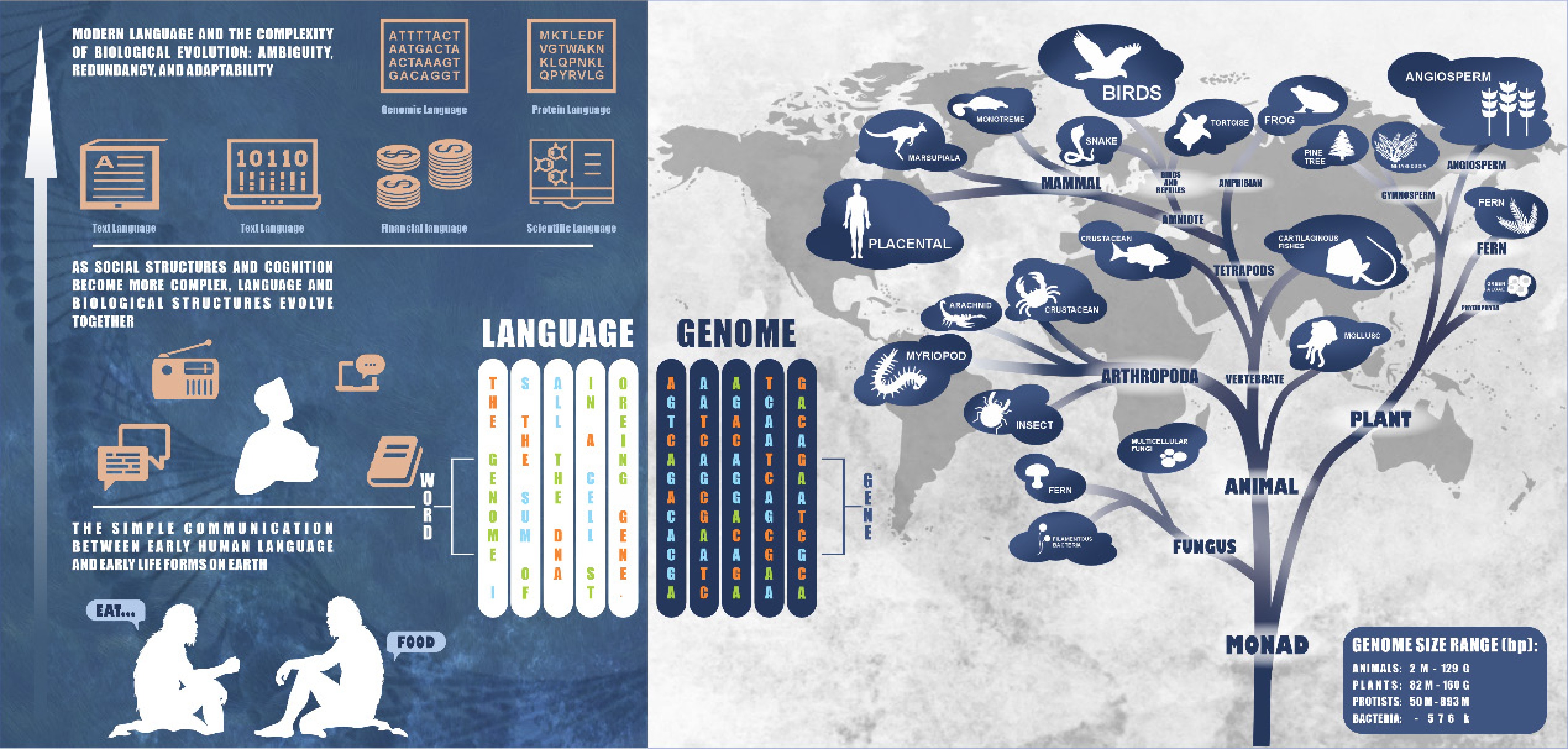
Figure 3.
Similarity between genome sequence and language sequence. The genome size data is derived from the NCBI genome database[82].
The transition from unicellular prokaryotes to multicellular eukaryotes is analogous to the evolution of early human language, evolving from simple and direct forms composed of basic syllables and simple words to meet basic survival needs (such as 'food' and 'danger') to more rigorous and complex linguistic rules and more specific vocabulary. Particularly in terms of the complexity of gene regulation, we initially seek to explore these similarities from the perspective of genome size, especially Whole-genome Duplication (WGD). Genome size often correlates with the time of species origin, and WGD is a significant driving force in biological evolution[67−70]. Studies have confirmed, by integrating extensive botanical cytogenetic and phylogenetic database information, that nearly one-third of contemporary vascular plant species have undergone chromosome doubling events[71]. The genomes of early prokaryotes, such as cyanobacteria, mycoplasmas, and bacteria, are characterized by their simplicity and directness. These organisms have relatively short gene sequences and mostly possess a single circular chromosome, with their structural genes generally arranged contiguously on the chromosome, and the transcription process does not require splicing or modification to meet basic survival and reproduction needs. In contrast, eukaryotic genomes are generally much larger than those of prokaryotes and contain multiple linear chromosomes[72]. Furthermore, prokaryotes have fewer repetitive and non-coding sequences, with the majority directly coding for proteins, with their gene functional regions being continuously distributed[73]. Eukaryotic genes, however, have a discontinuous distribution of expression regions, consisting of exons and introns, and the transcribed pre-mRNA must undergo splicing before being used for protein expression[74]. Eukaryotes possess many repetitive sequences; this diversification and redundancy in gene structure provide eukaryotes with a richer set of gene functions and enhance their robustness[75,76].
With the increasing complexity of ecological communities and the enhanced adaptability and responsiveness of organisms to environmental changes throughout biological evolution, the demand for gene regulation in organisms has steadily grown, leading to the evolution of a wider array of gene functions and regulatory pathways. This process mirrors the evolution of human language, from communicative speech to written language, scientific terminology, and other specialized domain-specific languages. Whole-genome duplication has brought about tremendous transformations in species' genomes, providing a potential genetic foundation for phenotypic evolution and innovation at the levels of functional genes and genetic regulatory networks[77]. Through processes such as gene loss, retention, and functional divergence following whole-genome duplication, genomes have expanded to encompass a wider range of gene functions[78] and have evolved more complex gene structures and regulatory networks. For example, approximately 88% of the duplicated genes in the yeast genome, stemming from a whole-genome duplication event that took place 100 million years ago, have been lost[79]. In multicellular organisms, the regulation of gene networks has become increasingly complex. For instance, there are marked differences in gene expression among different human tissues[80].
The genomes of modern organisms have evolved into a highly complex and multifaceted system, displaying characteristics akin to 'polysemy' and 'redundancy' in language. The significant expansion of non-coding genes has played a crucial role in the evolution of life. By enhancing the complexity and flexibility of gene sequences, they empower organisms to respond to environmental challenges in various ways, thereby markedly improving the stability and adaptability of biological information[77]. The genome's 'long-distance correlation' feature, in which many well-studied regulatory elements, such as enhancers, repressors, and insulators, can influence gene expression over distances exceeding 20 kb[81], enables different genes to establish functional connections across the linear regions of chromosomes, forming complex gene regulatory networks. This characteristic is analogous to how words in human language can establish semantic connections across sentences or paragraphs, forming complex networks of thought and expression.
-
Currently, the majority of mainstream LLMs are based on the Transformer architecture. In the domains of botany and biology, these models can be categorized into three structural types: encoder, decoder, and encoder-decoder. The pre-training and fine-tuning paradigm are extensively employed in these fields[83]. Given their proficiency in analyzing word sequences, LLMs can utilize various types of sequence data as training inputs. In biological research, BERT[51], and GPT[46] models have been widely applied to study genome, proteome, and gene expression data, as illustrated in Table 1.
Table 1. Summary table of genomic LLMs.
Model Time Base model Model architecture Parameters Pre-trained data species Capability Tokenizer type Tokens length (bp) Ref. DNABERT 2021.02 BERT Transformer Encoder 110 M Animal Function Prediction K-mer (3−6) 512 [49] Enformer 2021.10 Transformer + CNN Transformer Encoder 240 M Animal Function prediction Single-base 196,608 [81] MoDNA 2022.08 BERT Transformer encoder - Animal Function prediction K-mer (6) 512 [84] GPN 2022.08 BERT + CNN Transformer encoder − Plant Variant prediction Single-base 512 [33] iEnhance-BERT 2022.08 BERT(DNABERT) +
CNNTransformer encoder − − Enhancer prediction K-mer (3−6) 512 [85] FloraBERT 2022.08 BERT Transformer encoder − Plant Function prediction BPE 256 [36] TFBert 2022.09 BERT(DNABERT) Transformer Encoder 110 M Animal Epigenomics prediction K-mer (3−6) 512 [86] iDNA-ABF 2022.10 BERT(DNABERT) Transformer encoder 110 M Animal Function prediction K-mer (3−6) 512 [24] iEnhancer-ELM 2022.12 BERT(DNABERT) Transformer encoder 110 M Animal Enhancer prediction K-mer (3−6) 512 [87] Species-aware DNALM 2023.01 BERT(DNABERT) Transformer encoder 110 M Fungus Function prediction K-mer (6) 512 [25] Nucleotide Transformer 2023.01 BERT Transformer encoder 50 M−2.5 B Animal Function Prediction K-mer (6) 1,000−2,048 [88] GPN-MSA 2023.01 BERT Transformer encoder 86 M Animal Variant prediction Single-base 128 [89] SpliceBERT 2023.02 BERT Transformer encoder 19.4 M Animal Splicing prediction Single-base 1,024 [28] miProBERT 2023.03 BERT(DNABERT) Transformer encoder 110 M − Promoter prediction K-mer (6) 512 [90] RNA-MSM 2023.03 BERT+CNN Transformer encoder − − Structure prediction Single-base 512 [31] MRM-BERT 2023.06 BERT(DNABERT) Transformer encoder 110M Animal + Microorganism RNA modifications prediction K-mer (6) 512 [26] GENA-LM 2023.06 BERT/BigBird Transformer encoder 110−360 M Animal Function prediction BPE 512−4,096 [91] DNABERT-2 2023.06 BERT Transformer encoder 117 M Animal Function prediction BPE 128 [92] Geneformer 2023.06 BERT Transformer encoder − Animal Function prediction Custom tokenizer 2,048 [93] PLPMpro 2023.07 BERT(DNABERT) Transformer encoder − Eucaryon Promoter prediction K-mer (6) 512 [94] EpiGePT 2023.07 BERT + CNN Transformer encoder − Animal Epigenomics prediction Single-base 128 [29] Uni-RNA 2023.07 BERT Transformer encoder 25−400 M − Function prediction Single-base 512−1,280 [32] AgroNT 2023.10 BERT Transformer encoder 1B Plant Function prediction K-mer (6) 1,024 [35] FGBERT 2024.02 BERT Transformer encoder 954.73 M Metagenomics Functional prediction − − [27] RiNALMo 2024.02 BERT Transformer encoder 135−650 M − Functional prediction Single-base 1,024 [95] gLM 2024.04 BERT Transformer encoder 1B Metagenomics Functional prediction − − [96] RNAErnie 2024.05 BERT Transformer encoder 105 M − Functional prediction Single-base 512 [30] GenSLMs 2022.10 Diffusion + GPT Transformer decoder 25−250 M Virus Variant prediction K-mer (3) 2,048 [97] DNAGPT 2023.08 GPT Transformer decoder 100 M−3 B Animal Function prediction + Sequence generation K-mer (6) 512−4,096 [98] ENBED 2023.11 Transformer Transformer encoder−decoder 1.2 B Animal + Plant + Insect + Bacteria Function prediction + Sequence generation Single-base 16,384 [99] HyenaDNA 2023.06 Hyena Hyena 0.44−6.6 M Animal Function prediction + Species classfication Single-base 64 K−1 M [100] Evo 2024.02 StripedHyena StripedHyena 7B Bacteria + archaea Function prediction + Sequence generation Single-base 131 K [101] PDLLM 2024.12 Mamba Mamba 130 M Plant Function prediction Single-base/
K-mer (2−6)/BPE512 [34] Encoder architecture for genomic large language models
-
The encoder model constitutes a neural network architecture that encompasses solely the encoder component of the Transformer. It specializes in the deep feature extraction of input sequences, condensing key information within the model's hidden layers to generate a feature representation of a fixed size or within a specific range. In the domain of NLP, the BERT model stands as an exemplary representative of such architectures, excelling at generating meaningful data representations. Numerous Genomic LLMs, trained based on the BERT framework, have demonstrated robust performance in tasks related to gene sequence analysis. DNABERT[49] represents a pioneering initiative that integrates biological sequences with pre-trained language models. Leveraging the pre-trained bidirectional encoder representation from BERT, it has been successfully deployed in the analysis of genomic DNA sequences. Through fine-tuning on targeted datasets, DNABERT has achieved significant breakthroughs in gene data analysis, including the prediction of promoters, splicing sites, and transcription factor binding sites. To improve the model's capability for recognizing long sequences, DNABERT incorporates k-mer of varying lengths (3−6-mers) to segment the genome sequence during training, significantly expanding the model's vocabulary. However, subsequent studies, such as DNABERT-2[92] and HyenaDNA[100], have indicated that the k-mer lexical approach may impede the model's ability to delve deeper into the underlying patterns of biological sequences. These studies advocate the use of single-nucleotide resolution models to facilitate a more nuanced exploration. Recently, several studies have successfully extended the application of genomic LLMs to plant genomic data[34−36]. Among them, PDLLM adapted models that were trained on animal data, such as DNABERT[49] and NT[88], to various plant genomic-related datasets (e.g., core promoters, sequence conservation, histone modifications, open chromatin, long non-coding RNA (lncRNA), and promoter strength) for fine-tuning, achieving accuracy rates exceeding 77% in promoter prediction tasks[34].
Researchers have discovered that the integration of encoder models with CNNs significantly enhances the capability to analyze super-long DNA sequences. This synergy improves pattern recognition in long-fragment DNA sequences, as exemplified by models such as Enformer[81], GPN[33], and IEnhancer-BERT[85]. Notably, Enformer[81] leverages the strengths of the Basenji2 and ExPecto models, and innovatively combines a Transformer module with traditional convolutional layers to forge a novel deep learning architecture. This architectural innovation not only substantially broadens the model's receptive field, allowing it to capture sequence information up to 196,608 bp, but also significantly enhances the precision of gene expression prediction from DNA sequences.
Decoder architecture for genomic large language models
-
The decoder model is based on the Transformer decoder, which comprises solely the decoder component of the Transformer architecture[23]. Its core function is to generate or expand relevant information based on the input sequence, thereby completing sequence generation tasks and summaries. In the field of NLP, the GPT model is an exemplary representative of such architectures. In the context of genome data, models such as DNAGPT[98] and GenSLMs[97] have achieved notable results. DNAGPT[98] addresses complex tasks related to DNA sequence analysis through multi-task pre-training and an innovative markup language design, performing exceptionally well across various domains and offering robust support for biological research. GenSLMs[97] innovatively combines a Transformer-based generation model with a Stable Diffusion-based diffusion model to capture contextual and long-range interactions within the genome, which is particularly useful for virus mutation identification.
Encoder-decoder architecture for genomic large language models
-
The encoder-decoder model serves as an architecture for information conversion from one sequence to another. The encoder's role is to extract features from the input data and compress them into a tensor-based representation that is rich in contextual information. The decoder then receives this compressed feature information and produces the corresponding output sequence[23]. The separation of encoding and decoding processes allows the model to excel in understanding and generating sequences, making it suitable for a variety of downstream tasks, such as sequence generation and feature extraction. In the context of genomic data applications, the encoder-decoder model has demonstrated significant potential. It can efficiently analyze and interpret complex genomic sequences, capturing essential features and patterns within DNA sequences. For instance, the Ensemble Nucleotide Byte-level Encoder-Decoder (ENBED) model[99] utilizes byte-level tokenization and an encoder-decoder architecture to conduct in-depth analyses of nucleotide sequences. Pre-trained on reference genome sequences such as those of E. coli, Drosophila, mouse, maize, and human, ENBED has achieved notable results in a range of downstream tasks. These include the identification of enhancers, promoters, and splicing sites, as well as annotating the biological functions of genome sequences. Furthermore, the AlphaFold series of models, also based on the encoder-decoder framework, can predict the three-dimensional structure of proteins from amino acid sequences[102].
Non-transformer architecture for genomic large language models
-
In the domain of AI, numerous model architectures have been developed that innovate upon the Transformer architecture in terms of input word length, model architecture compression, and data feature extraction. Consequently, not all LLMs are based on the Transformer architecture[34,100,101]. A case in point is Evo[101], which employs the StripedHyena architecture with 7 billion parameters to achieve single nucleotide resolution modeling at a context length of 131 K. Researchers have determined that the StripedHyena architecture outperforms various baseline architectures at every scale, including the conventional Transformer architecture. Evo excels in a range of prediction tasks, including zero-sample predictions, and can identify key genes. The model is capable of learning to regulate DNA and other sequence information within the central dogma, as well as understanding the common variations and regulatory elements of multiple genes, thus underscoring the significance of DNA as the foundational layer of biological information.
Through research, it is found that the core research method of Genomic LLMs mainly focuses on the encoder using Transformer architecture. In comparison, decoders, encoder-decoder models, and other innovative model architectures still have relatively limited application practices in the complex and unique field of biological language[83]. Currently, the interdisciplinary convergence of fields like life sciences and AI is in its nascent phase. During this initial stage, investigators from diverse scholarly backgrounds exhibit notable heterogeneity and divergent focuses in the deployment of AI within their research endeavors. Given the nascent and complex nature of these interdisciplinary interactions, experts from various fields must move beyond their traditional disciplinary boundaries and immerse themselves in acquiring a more extensive and profound body of specialized knowledge[103]. This is to seamlessly incorporate the adept and potent analytical instruments from the AI sector into the profound exploration of life's enigmas with greater adaptability and innovation. Such interdisciplinary collaboration and innovation are poised to furnish genetic researchers with formidable technological backing, empowering them to dissect and analyze vast genomic datasets with an unprecedented level of efficacy and precision.
-
Currently, the majority of Genomic LLMs are primarily trained on DNA sequence data from humans and animals, or on short sequence biological data from viruses and prokaryotes. For instance, the Nucleotide Transformer[88] utilized a dataset from the 1000 Genomes Project, which encompassed 3,202 human genomes with genetic diversity. The EVO[101], on the other hand, was trained using a comprehensive collection of over 80,000 bacterial and archaeal genome datasets.
It is noteworthy that, although plant sequence data holds significant value for life science research, its utilization in the training of genomic LLMs remains relatively constrained. To date, only a limited number of models have incorporated plant genome data into their training processes. For instance, the GPN[33] utilizes eight reference genomic DNA sequences of Brassicas, a factor that inevitably narrows the scope and depth of the model's applicability within the plant domain.
With the growing proliferation of openly accessible plant databases, such as the comprehensive database Phytozome[104], which spans multiple taxa from algae to higher plants, Gramene[105] specializing in genomic information of grasses, Sol Genomics Network (SGN)[106] dedicated to genomic research on Solanaceae species, CottonGen[107] offering a platform for cotton genomic data, Rice Genome Annotation Project database (RGAP)[108] containing pan-genome and annotation data for 3,000 rice varieties, and The Arabidopsis Information Resource (TAIR)[109] serving as the definitive source for Arabidopsis genomic information, these databases collectively form a rich repository of plant bioinformatics data. They hold immense potential for the training and application of genomic LLMs in the plant domain. These datasets not only furnish basic genomic sequence information but also encompass multi-omics data, including transcriptomics, proteomics, and metabolomics, along with multidimensional information on plant phenotypes, ecological niches, evolutionary trajectories, etc., providing comprehensive training materials for genomic LLMs. Through pre-training on vast amounts of data, LLMs can more precisely capture unstructured information within plant genomic data[36]. Leveraging pre-trained base models and employing transfer learning strategies, various downstream tasks can be accomplished with merely a small quantity of domain-specific data on consumer-grade graphics processing unit (GPU)[34]. In contrast to traditional machine learning, which demands large volumes of domain-specific data and encounters difficulties in transferability, particularly when training predictive models for non-model plants, the advent of LLMs presents new avenues of opportunity.
Tropical plants, often growing in environments characterized by high environmental variability and species richness, may contain unique genes and mutations in their genomes that facilitate adaptation to these unique environments. However, research on tropical plants remains relatively scarce, with genomic data for many species still absent or sparse. Taking tropical plants such as macadamia[110] and passion fruits[111] as examples, relevant open-source datasets are often lacking. In such cases, the cross-species transfer learning capabilities of LLMs become particularly crucial. By leveraging genomic data from related or phylogenetically close species and employing transfer learning strategies with genomic LLMs[93], it is possible to predict and analyze the genomic structure and function of tropical plants. This approach not only saves considerable time and resources but also uncovers the unique genetic characteristics and adaptation mechanisms of non-model plants. Given that many tropical plants are endangered or rare species, studying their genomes is crucial for species conservation. LLMs can rapidly analyze the genomic data of these species, revealing their genetic diversity and evolutionary history, thereby providing a scientific basis for formulating effective conservation measures. Furthermore, in-depth research on the genomes of tropical plants can aid in the discovery of new germplasm resources, opening new avenues for crop improvement and biotechnological breeding.
In the future, as more plant data becomes accessible and integrated, genomic models will be able to more comprehensively cover the plant field and delve deeper into the intricacies of plant genomics. This advancement will not only significantly accelerate the progress of plant biology research but also furnish the sectors of agricultural production, ecological protection, and biodiversity conservation with more robust tools and support. Consequently, these efforts will aid in a better comprehension and utilization of the Earth's invaluable plant life resources.
-
Although substantial progress has been achieved in the investigation of genomic large language models, the absence of standardized benchmarks remains a significant challenge[92]. The validation data employed by various models exhibit differences, necessitating the use of benchmark data for testing when assessing AI tools[112]. However, as the training datasets in the majority of studies are not publicly accessible, researchers face challenges in ascertaining whether the model has been exposed to the test dataset during training when comparing multiple existing models. This situation may introduce bias into the evaluation results. Consequently, devising methods to more transparently compare the performance of different genomic models has become a pressing issue that requires resolution.
Currently, comparisons of model performance primarily rely on the Nucleotide Transformer Benchmark[88], the Plants Genomic Benchmark (PGB)[35], and the Genome Understanding Evaluation (GUE)[92] benchmark datasets, supplemented by datasets from models that demonstrate superior performance. The Nucleotide Transformer Benchmark constitutes a comprehensive evaluation framework, engineered to assess the performance of foundational genomics models. This benchmark includes 18 distinct genomics analysis tasks[88]. GUE, on the other hand, incorporates 36 diverse datasets from nine genome analysis tasks, with input lengths varying from 70 to 10,000 nucleotides. This range provides a rich and diverse dataset, which supports a thorough evaluation of model performance[92]. PGB is a benchmark dataset based on edible plants[35].
-
Biological large language models have demonstrated robust capabilities in genomics research, primarily across four pivotal areas: functional prediction, structural prediction, sequence generation, and analysis of sequence variation and evolution.
Function prediction
-
Gene function prediction has consistently been a central focus within genomics research. Traditional methodologies are constrained by their reliance on the direct training of task-specific sequences and are heavily dependent on manually annotated data. Nonetheless, the advent of LLMs has revolutionized the prediction of gene function regions. In recent years, LLMs, including GENA-LM[91], DNABERT[49], MoDNA[84], PLPMpro[94], and miProBERT[90], have achieved notable success in promoter prediction. Notably, the MoDNA[84] has adeptly mastered promoter prediction through pre-training on human genomic data, thereby learning common genomic features, and subsequent fine-tuning using the Eukaryotic Promoter Database (EPDnew) dataset. Furthermore, the PLPMpro[94] has enhanced the performance of promoter sequence prediction by integrating prompt learning with pre-training models. Additionally, iEnhancer-ELM[87] has demonstrated strong performance in enhancer recognition by effectively extracting location-related multi-scale contextual information.
Structure prediction
-
Structural prediction is a pivotal area in genomics research, encompassing the anticipation of the spatial conformation of biological macromolecules, such as DNA, RNA, and proteins. The deployment of LLMs has markedly enhanced the precision and efficiency of these predictions. Notably, HyenaDNA[100] has achieved a significant breakthrough in the prediction of chromatin structure, enabling the forecasting of chromatin maps and epigenetic markers, and subsequently quantifying the functional impacts of non-coding variations. TFBert[86], leveraging 690 ChIP-seq datasets for pre-training, has effectively predicted DNA-protein binding sites. Building on the Transformer architecture of RoBERTa, gLM[96] has adeptly discerned potential functions and regulatory interplays among genes through training on the MGnify database, which comprises seven million metagenomic contigs. This model is capable of concurrently capturing genomic context and intrinsic protein sequence information, demonstrating substantial potential in a variety of downstream applications, including enzyme function prediction, operon prediction, homologous protein alignment, and contig classification. Furthermore, MoDNA[84] concentrates on predicting transcription factor binding sites, offering valuable support for the analysis of transcriptional regulatory networks.
Sequence generation
-
Sequence generation is a critical component of bioinformatics, with the core objective being the creation of artificial sequences that mimic authentic biological sequences. This technology has exhibited considerable potential, particularly in the artificial generation of human genomes. It serves not only to construct a protective shield for genetic privacy but also significantly diminishes the costs associated with the collection of genetic samples. DNAGPT[98] has successfully synthesized 5,000 human genomes encompassing 10,000 single nucleotide polymorphism (SNP) regions, thereby showcasing its exceptional capability in sequence generation.
Analysis of sequence variation and evolution
-
Sequence variation and evolutionary analysis are pivotal in examining DNA sequence variation and delineating its evolutionary trajectory. The emergence of genome-wide association studies (GWAS) has significantly expanded our comprehension of the genetic underpinnings of complex plant traits and diseases[113]. Within this domain, tools predicated on LLMs, such as GPN[33], GenSLMs[97], GPN-MSA[89], and Evo[101], are increasingly demonstrating robust predictive and analytical prowess. Notably, GPN[33] anticipates genome-wide variation effects via unsupervised pre-training, outperforming conventional methodologies. Analyses utilizing Arabidopsis data indicate that GPN's predictive accuracy exceeds that of traditional conservative scores, and it is not reliant on genome-wide alignments or functional genomics data. This advancement lays the groundwork for the development of a cross-species genome-wide variation effect predictor, which is instrumental for genetic disease diagnosis, GWAS fine mapping, and the computation of multi-gene risk scores. The predictive outcomes of GPN can be visualized using the UCSC Genome Browser, facilitating researchers' interpretation of the results. GenSLMs[97] was successfully trained by pre-training on over 110 million prokaryotic gene sequences and fine-tuning on 1.5 million SARS-CoV-2 genomes, thereby enabling the identification of salient variations. Concurrently, to enhance the model's interpretability, GenSLMs incorporates an integrated visualization tool designed to graphically represent genomic relationships and model attention mechanisms. Furthermore, Evo[101] was trained on a comprehensive dataset comprising 300 billion nucleotides of prokaryotic genome-wide DNA. Evo has adeptly learned the information encoded within regulatory DNA and other modal sequences in the central dogma, capturing common variations involving multiple genes and regulatory elements across the evolutionary diversity of prokaryotes.
-
Although Genomic LLMs have achieved significant milestones in the domains of structural prediction, sequence generation, and sequence variation and evolutionary analysis — thus expanding the horizons of genomics and bioinformatics research — they still confront a myriad of substantial challenges. These challenges pertain not only to technical aspects but also to the model's interpretability, the constraints on sequence length, and issues surrounding word segmentation.
At the technical level, Genomic LLMs must manage vast amounts of data, high dimensionality, and strong complexity, imposing unprecedented demands on computing power and storage resources. The exponential growth of biological data presents the challenge of designing a more efficient model architecture to achieve faster processing speeds and higher prediction accuracy within limited computing resources[114]. This requires not only enhanced data compression and feature extraction capabilities within the model but also algorithmic innovations to simultaneously improve computational efficiency and predictive performance.
The interpretability of models is also a significant bottleneck hindering the widespread application of Genomic LLMs. Despite these models' remarkable accuracy in various predictive tasks, their decision-making processes are often regarded as 'black boxes'[115], which are challenging for researchers to understand and interpret. This lack of transparency not only restricts the model's in-depth application in scientific research but also impacts its credibility and acceptance in practical applications. Therefore, enhancing model interpretability to allow researchers to comprehend the decision logic and internal mechanisms is crucial for enhancing the model's application value and advancing research progress in related fields.
The limitation on DNA sequence length poses a significant challenge for Genomic LLMs. The intricacy of the attention mechanism leads to a computational complexity that scales with the square of the sequence's length, indicating that the computational requirements escalate exponentially alongside an increase in sequence length[116]. Consequently, under current technological constraints, most genomic LLMs can only be pre-trained within a relatively short context range, namely 512 to 4,096 tokens, representing an extremely small fraction, less than 0.001%, of the human genome[49,87]. While preprocessing or compression algorithms, such as EVO, which extends the maximum input genome length to 131 K[101], and Enformer which reaches 196 K[81], have been developed, these methods often sacrifice the single-nucleotide resolution of DNA sequences. For example, Enformer reduces the input sequence from 196,608 to 1,536 bp using seven convolutional blocks with pooling, which is akin to training the transformer module with token inputs at a resolution of 128 bp[81], significantly affecting the accuracy and interpretability of gene sequence analysis. Due to the limitations on the length of model input sequences, input sequences are typically split into fixed-length segments. However, plant genome sequences often exhibit high heterozygosity and complex structures, with some plant genomes reaching billions of bases[117], such as the 160 Gb genome of Tmesipteris oblanceolata[118]. This immense scale suggests that even with the maximum sequence length that current Genomic LLMs can handle, only a tiny fraction of the plant genome can be analyzed. This fragmentation process may lead to the loss or distortion of critical information, since genes and regulatory elements often span across multiple segments, and their interactions and regulatory networks may be disrupted during the fragmentation process. In addition, repetitive sequences and transposable elements comprise a significant portion of plant genomes, playing pivotal roles in evolution yet also complicating genome interpretation[75]. However, the inability to input extensive genomic contexts into the models may cause confusion when predicting gene functions or structures. Polyploidy is widespread in plants, indicating the existence of multiple similar copies of the genome within a single plant, further adding to the complexity of the genome. Models need to distinguish and integrate information from these different copies, particularly when handling complex gene structures or long-distance regulatory relationships, since they may not be accurately captured, thereby affecting their application in plant genomics research. This limitation on the length of input DNA sequences significantly impedes the model's ability to gain a deep understanding and conduct a comprehensive analysis of complete genomes or longer DNA sequence fragments, thus limiting the widespread application and potential of the model in genomics research. Striking a balance between extending the length of input DNA sequences and maintaining high resolution remains a pressing challenge that needs to be addressed urgently.
The issue of tokenizer limitations is highlighted. To transform continuous DNA sequences into discrete units manageable by computational models, Genomic LLMs typically employ fixed-length k-mers. K-mer, which are n-gram sequences analogous to words in human language, serve as the fundamental 'words' of DNA[92]. Additionally, tokenizer are utilized to aggregate these meaningful DNA units. However, this method of word segmentation often overlooks the significant impact of single nucleotide variations, such as single nucleotide polymorphisms (SNPs), on biological traits[92]. As a crucial source of biodiversity and a pivotal factor in the etiology of diseases, single nucleotide variations play a critical role in the genetic information and functional regulation within organisms. Yet, due to the rigidity and constraints of the k-mer approach, this key variation information may be overlooked or misinterpreted during model processing, thereby compromising the model's predictive accuracy and interpretive capacity.
In the context of the current wave of AI sweeping genomic research, LLMs have powerful data processing and analysis capabilities[37]. However, there are still many challenges in the combination of LLMs in plant genome research. With the rapid development of multi-omics technology[119], the field of plant research has ushered in the era of data explosion, however, existing technologies find it challenging to effectively manage, store, and analyze these vast amounts of data. LLMs have shown significant advantages in this context: they can deeply mine the inherent patterns of genomic data through pre-training on vast amounts of genomic sequence data. Subsequently, they can be fine-tuned using only a small amount of labeled data[47], allowing them to quickly adapt and be applied to various downstream research tasks, achieving high-precision data analysis and prediction across the entire genome. Particularly in the realm of non-model plants, such as tropical species, where the diversity of species and complexity of data result in a scarcity of labeled data, traditional machine learning algorithms are inadequate for analysis tasks. For example, the AgroNT model[35] utilizes reference genomes from 48 plant species for pre-training to achieve accurate predictions of various regulatory features. By fine-tuning the model specifically for the tropical crop 'cassava', it successfully predicts enhancer elements and gene expression in multiple tissues of cassava, further demonstrating the feasibility of using LLMs in non-model plant research. This approach not only significantly enhances data processing efficiency, but also ensures excellent performance on limited datasets, providing robust technical support for advancements in research areas such as tropical plants and other non-model plants.
Looking ahead, as deep learning technology continues to advance and genomic data continues to expand, there is an expectation that the fusion of AI with genomic data will yield more precise and efficient methods for genomic data analysis. Notably, the utilization of LLMs, such as GPT, in genomics research is still in its nascent stages. Nevertheless, these models have exhibited substantial potential. Through ongoing research and innovation, they may transcend the limitations of current models, integrate multidisciplinary knowledge, and explore the convergence of AI-driven approaches with plant genomics. The role of AI in advancing plant genomics research is likely to become increasingly significant. This paper encourages more botanists to engage in the research and application of LLMs. It is anticipated that a collaborative effort will elevate the development of both AI technology and plant science to unprecedented levels.
The research was supported by Biological Breeding-National Science and Technology Major Project (2023ZD04073), the Project of Sanya Yazhou Bay Science and Technology City (SCKJ-JYRC-2022-57), and the High-performance Computing Platform of YZBSTCACC.
-
The authors confirm contribution to the paper as follows: study conception and design: Yang L, Zou M, Chai H, Xia Z; data collection: Yang L, Wang H; analysis and interpretation of results: Yang L; draft manuscript preparation: Yang L. All authors reviewed the results and approved the final version of the manuscript.
-
There are no original data associated with this article. Referenced data are available in the literature.
-
The authors declare that they have no conflict of interest.
-
Received 25 November 2024; Accepted 18 February 2025; Published online 14 April 2025
-
Artificial intelligence models are revolutionizing traditional biological research methods.
LLMs have not yet been effectively applied to the field of plant science.
New opportunities arise from combining non-model species such as tropical plants with LLMs.
Genomic language shares similarities with natural language.
- Copyright: © 2025 by the author(s). Published by Maximum Academic Press on behalf of Hainan University. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Yang L, Wang H, Zou M, Chai H, Xia Z. 2025. Artificial intelligence-driven plant bio-genomics research: a new era. Tropical Plants 4: e015 doi: 10.48130/tp-0025-0008 

Artificial intelligence-driven plant bio-genomics research: a new era
- Received: 25 November 2024
- Revised: 20 January 2025
- Accepted: 18 February 2025
- Published online: 14 April 2025
Abstract: With the rapid development of artificial intelligence (AI) technology, particularly the emergence of large language models (LLMs) such as the GPT series, AI has been increasingly integrated into scientific research. These models exhibit robust cross-domain applicability by assimilating vast repositories of world knowledge and demonstrating proficiency in understanding and generating natural language. Leveraging the inherent similarities between genome sequences and natural language, this paper examines the recent advancements of AI in genomics. It elucidates the foundational principles of LLMs and their latest research developments in architectural design and functional analysis within the context of genomic data analysis. The paper also thoroughly explores the current challenges and prospective research directions. Despite the preliminary successes of LLMs in genomic research, significant obstacles remain in the integration of plant genomics with these models. This study highlights that LLMs offer innovative tools and perspectives for genomics research, extending to the fields of biology, agriculture, and even the study of tropical plants. Consequently, the effective utilization of AI technology by biologists to advance plant science has become a critical area of inquiry.
-
Key words:
- Artificial intelligence /
- Transformer /
- Large language model /
- Plants /
- Genomics