-
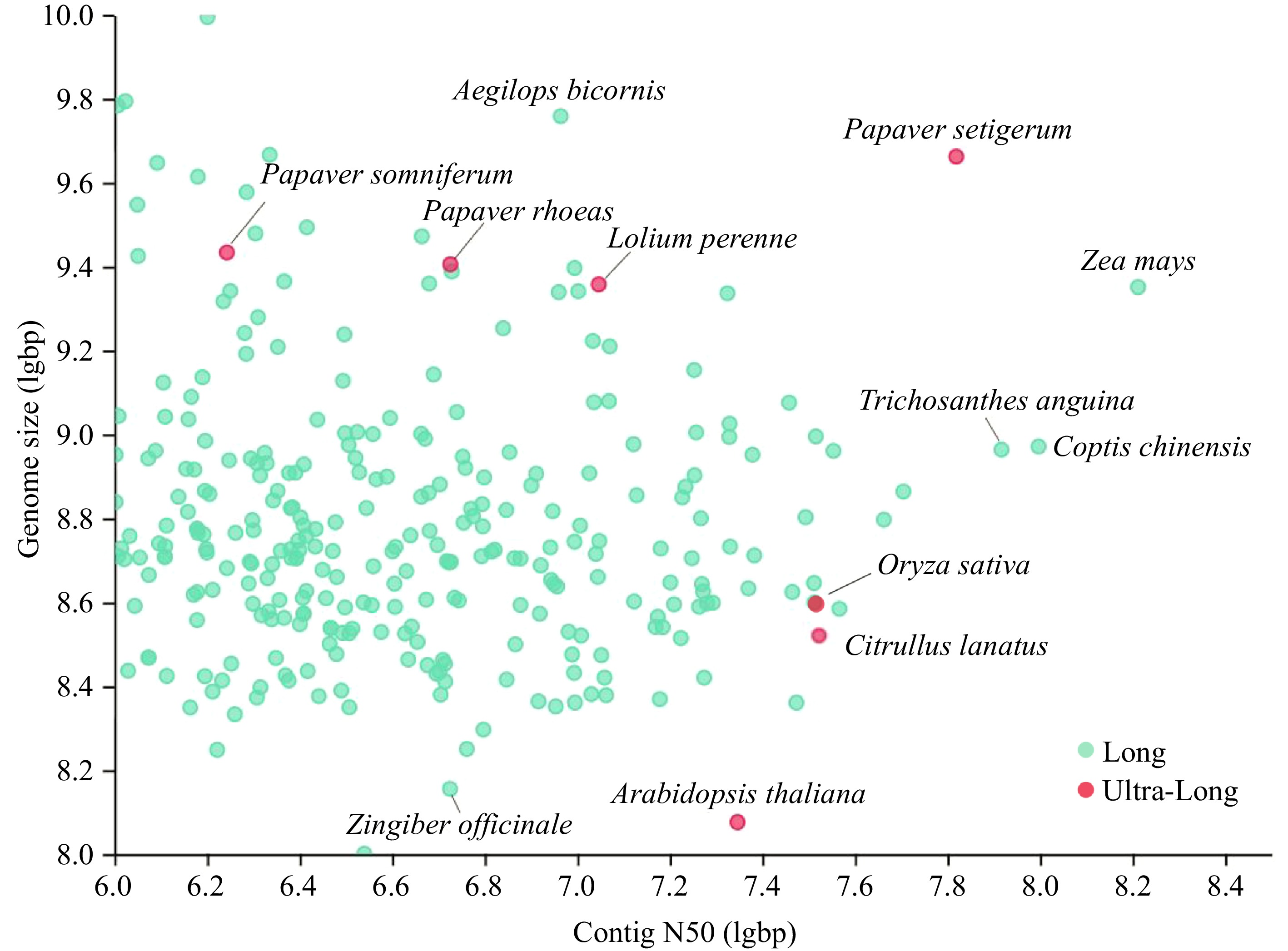
Figure 1.
Collection of the high-quality genome of 300 plants from PLAZA (
https://plabipd.de/plant_genomes_pa.ep ). Ultra-long sequencing dots are shown in red, and the rest are shown in green. -
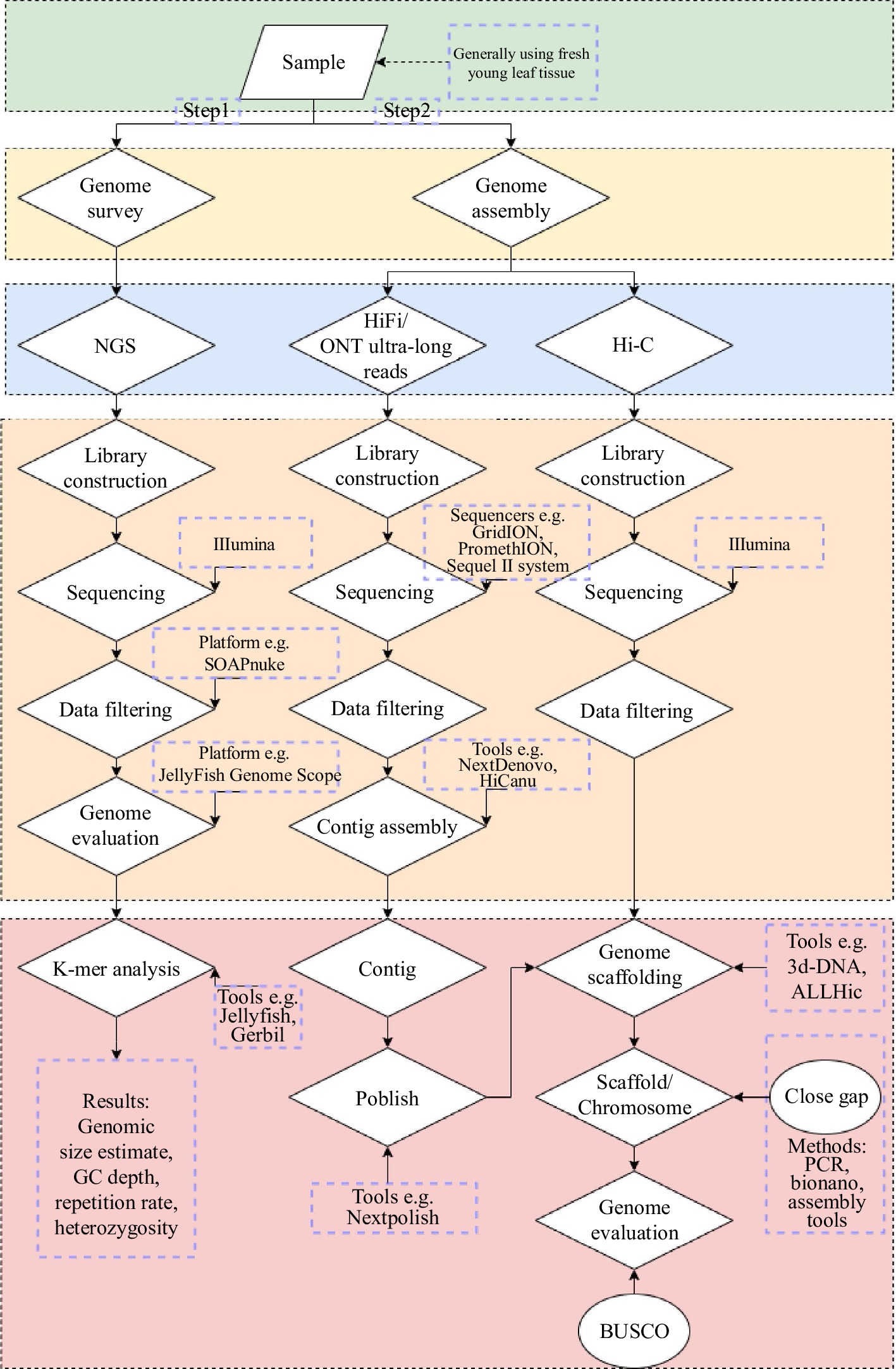
Figure 2.
Process of de novo assembly of the complete plant genomes. Sequencing platform, software, precautions, etc are noted in the figure alongside the steps.
-
Tools Features URL PacBio and ONT Assemblage relevant NextDenovo[33] (V2.5.0) String graph-based de novo assembler for long reads which uses
a 'correct-then-assemble' strategy, but requires significantly less computing resources and storage.https://github.com/Nextomics/NextDenovo Hifiasm[26] (V0.16.1) Fast haplotype-resolved de novo assembler for PacBio HiFi reads. https://github.com/chhylp123/hifiasm SMARTdenovo[34] A de novo assembler for PacBio and Oxford Nanopore (ONT) data.
It produces an assembly from all-vs-all raw read alignments
without an error correction stage.https://github.com/ruanjue/smartdenovo NGMLR[35] (V0.2.7) Long-read mapper designed to align PacBio or Oxford Nanopore (standard and ultra-long) to a reference genome with a focus on reads that span structural variations. https://github.com/philres/ngmlr CentroFlye[36] (V0.8.3) Algorithm for centromere assembly using long error-prone reads. https://github.com/seryrzu/centroFlye_paper_scripts Canu[21] (V2.2) Fork of the Celera Assembler, designed for high-noise single-molecule sequencing (such as the PacBio RS II/Sequel or Oxford Nanopore MinION). https://github.com/marbl/canu Wtdbg2[22] (V2.5) De novo sequence assembler for long noisy reads produced by
PacBio or ONT which assembles raw reads without error correction and then builds the consensus from intermediate assembly output.https://github.com/ruanjue/wtdbg2 HiCanu[25] Modification of the Canu assembler designed to leverage the full potential of HiFi reads via homopolymer compression, overlap-
based error correction, and aggressive false overlap filtering.https://github.com/marbl/canu HINGE[20] Long read assembler based on OLC (Overlap-Layout-Consensus). https://github.com/HingeAssembler/HINGE Peregrine Fast genome assembler for accurate long reads (length > 10 kb, accuracy > 99%). https://github.com/cschin/Peregrine Flye[19] (V2.9) De novo assembler for single-molecule sequencing reads, such as those produced by PacBio and Oxford Nanopore Technologies designed for a wide range of datasets. https://github.com/fenderglass/Flye Shasta[23] (V0.9) The goal of the Shasta long read assembler is to rapidly produce accurate assembled sequence using DNA reads generated by
Oxford Nanopore flow cells as input.https://github.com/chanzuckerberg/shasta NECAT[37] (V0.01) Error correction and de novo assembly tool for Nanopore long
noisy reads.https://github.com/xiaochuanle/NECAT Wengan[24] (V0.2) Wengan avoids entirely the all-vs-all read comparison. The key
idea behind Wengan is that long-read alignments can be inferred by building paths on a sequence graph.https://github.com/adigenova/wengan NextPolish[38] (V1.4) NextPolish is used to fix base errors (SNV/Indel) in the genome generated by noisy long reads, it can be used with short read
data only or long read data only or a combination of both.https://github.com/Nextomics/NextPolish Miniasm[18] (V0.3) Very fast OLC-based de novo assembler for noisy long reads. https://github.com/lh3/miniasm Falcon[15−17] (V0.3) Experimental PacBio diploid assembler https://github.com/PacificBiosciences/FALCON Raven[39] (V1.7) De novo genome assembler for long uncorrected reads. https://github.com/lbcb-sci/raven HERA[40] Local assembly tool using assembled contigs and self-corrected
long reads as input which can generate ultra-long, even chromosome-scale, contigs.https://github.com/liangclab/HERA Hi-C scaffolding relevant HiC-Pro[41] (V3.1) HiC-Pro was designed to process Hi-C data, from raw fastq files (paired-end Illumina data) to normalized contact maps. https://github.com/nservant/HiC-Pro SALSA[42] (V2.3) A tool to scaffold long read assemblies with Hi-C. https://github.com/marbl/SALSA 3D-DNA[28] (V201008) 3D de novo assembly (3D DNA) pipeline. https://github.com/aidenlab/3d-dna ALLHiC[29, 30] Phasing and scaffolding polyploid genomes based on Hi-C data. https://github.com/tangerzhang/ALLHiC LACHESIS[27] First tool to use Hi-C data to assist genome assembly. https://github.com/shendurelab/LACHESIS Table 1.
Tools for assembling long reads and Hi-C reads.
Figures
(2)
Tables
(1)