








-
In recent years, more and more high-quality genomes of plants have been deciphered. As of August 1, 2022, we have collected a total of 300 plant genomes assembled at the chromosome level with contig N50 greater than 1 Mb from PLAZA (
https://plabipd.de/plant_genomes_pa.ep ), and classified them according to whether the sequencing technology are ultra-long reads (> 50 kb) (Fig. 1). Contig N50 increased from 99.5 ± 48.1 kb in 2010 to 3,395.2 ± 735.4 kb in 2020[1]. There are seven genomes assembled from ultra-long reads, including Arabidopsis thaliana[2], Lolium perenne[3], Papaver rhoeas, Papaver somniferum, Papaver setigerum[4], Citrullus lanatus[5] and Oryza sativa[6]. From Fig. 1, we can find that the genome size of most sequenced species is between 100 Mb and 10 Gb. The contig N50 of maize is the highest, about 162 Mb[7], and the Ginkgo biloba[8] genome is the largest, up to 9.87 GB. In the long reads sequencing, there are also many plant genomes with high sequencing and assembly levels, such as the high-quality reference genome HFTH1 of apple[9]. It has certain reference significance for identifying structural variation, integrating phenotypic and genotypic association, analyzing the pattern and speed of genome evolution and clarifying the genetic structure of important traits.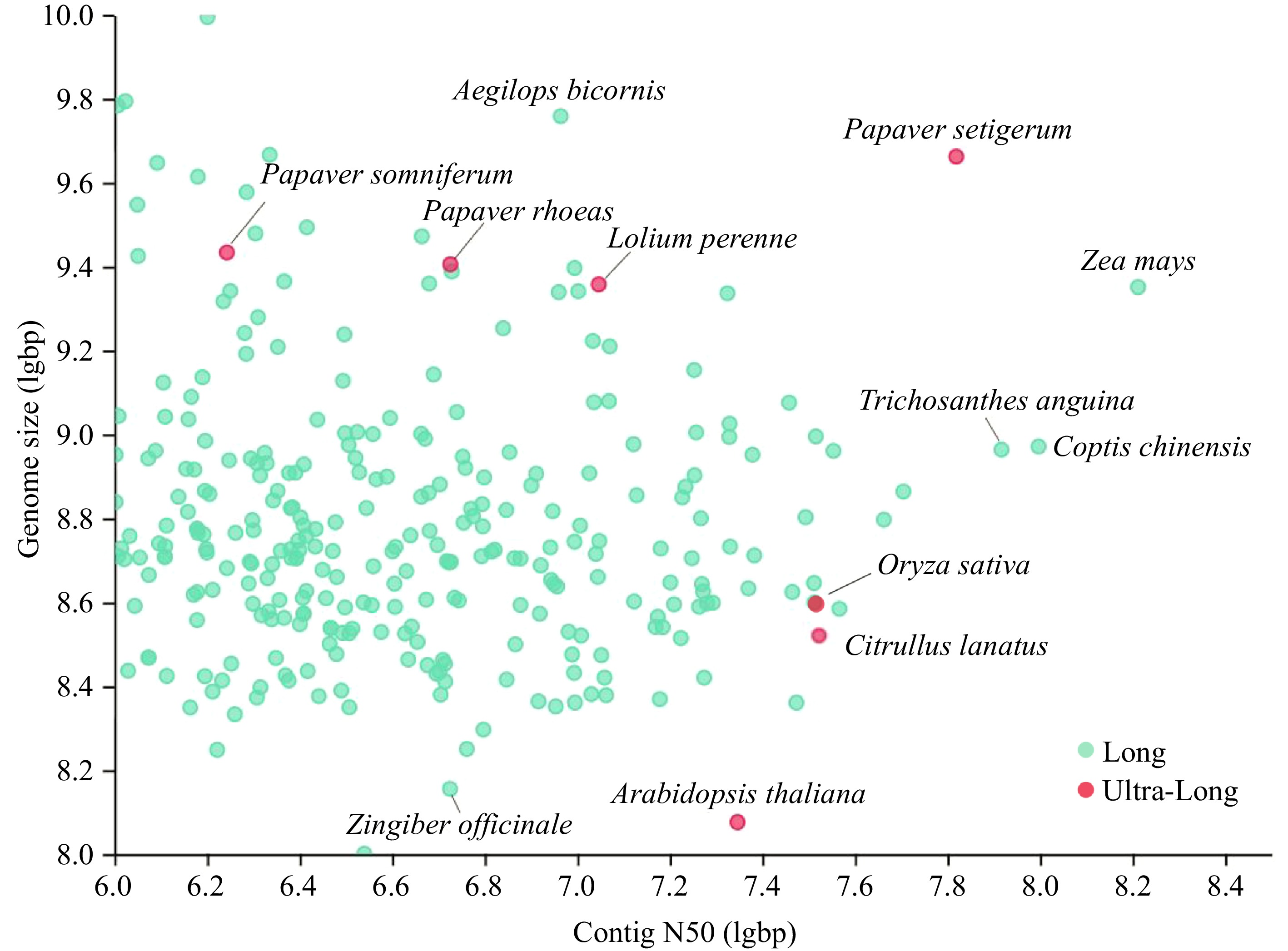
Figure 1.
Collection of the high-quality genome of 300 plants from PLAZA (
https://plabipd.de/plant_genomes_pa.ep ). Ultra-long sequencing dots are shown in red, and the rest are shown in green. -
Nowadays, based on different shearing methods and Nanopore sequencing kits, there are two ways to construct ultra-long DNA libraries: one is based on mechanical shearing, and the other is based on transposase. Mechanical fragmentation refers to the physical method for breaking DNA molecules into varying sizes which is considered the gold standard, including ultrasonic, spraying and hydrodynamic shearing methods. The N50 produced by the former is between 50 and 70 kb, and the construction of the library takes about 8 h. The N50 produced by the latter is between 90 and 100 kb, and it only takes 90 min to build the library[10]. However, the scheme of inputting the same DNA mechanical shearing can make it possible to obtain more productive fragments.
Oxford Nanopore and Circulomicsde Ultra-long DNA Sequencing Kit have supported the reading of sequences up to 4.2 Mb to maximize the number of ultra-long fragments. The kit is based on transposase chemistry: the transposase simultaneously cleaves template molecules and attaches tags to the cleaved ends. Its consumption of fuel during a sequencing run is reduced significantly. Combined with the Nanobind Kit from Circulomics, the Oxford Nanopore Ultra-long DNA Sequencing Kit can maximize the number of ultra-long read lengths, and has supported the continuous sequencing of single DNA fragments up to 3+ Mb (user data) and up to 4+ Mb (internal data)[11]. At first, the transposase of the Ultra-long DNA Sequencing Kit will simultaneously cleave the template molecule and attach the molecular marker to the cleaved end, then add the rapid sequencing linker to the labeled end, and finally elute the DNA library overnight with a Circulomics Nanobind disk (5 mm) to remove the free linker and short DNA fragments. With this method, it is possible for users to obtain more than 100 read-length sequences larger than 1 Mb by running a PromethION sequencing chip. For example, the sequencing reading length N50 of Lolium perenne by this method is as long as 62 kb[3].
-
Next-generation sequencing including Roche 454 and Illumina is a technology of sequencing while synthesizing, but its reading length is less than 200 bp, and time consuming. ONT (Oxford Nanopore Technologies) single molecule sequencing and Pacbio (Pacific Biosciences) HiFi sequencing are two current mainstream technologies. First of all, it is necessary to complete the preliminary assembly of the genome: survey, PacBio HiFi and ONT ultra-long sequencing of the genome to be tested are completed by using DNBSEQ short-length sequencing technology. And ONT PacBio single molecule real-time sequencing (SMRT) makes the single molecule read length exceed 10 kb, which is beyond the length of most simple sequence repeats.
Both of the two sequencing technologies have their own merits. With the comparison of HiFi, ONT ultra-long reads delivered higher contiguity. However, the ultra-long fragment obtained by ONT still has a relatively high base error rate before error correction. PacBio HiFi sequencing is a sequencing technology based on circular consensus sequencing (CCS). Its accuracy is as high as 99.8%, and the average length of generated HiFi reads is as long as 13.5 kb[12]. The quality of HiFi data is relatively stable in regions with different GC content and the repeatability is better. In this sequencing mode, it still has the same or even longer enzyme reading length (over 100 kb) as CLR (Continuous long reads) sequencing mode, but the inserted fragment is only 10−20 kb[13], which is far less than the reading length of the enzyme. Therefore, when sequencing, the enzyme will perform rolling circle sequencing around the DNA template, that is, the insert will be sequenced many times. In this way, random sequencing errors caused by single sequencing can be self-corrected by the algorithm, and finally high-accuracy HiFi Reads can be obtained. Because the amount of data used for assembly is small, and there is no need for data self-correction, the required computing resources in the assembly process are less than those in the traditional CLR mode, and the assembly cost is saved.
Combining ONT with HiFi can realize ultra-long sequencing. Finally, by combining Hi-C technology to obtain the relative position information of genes on chromosomes, the genome chromosome level assembly is completed, and the complex region is manually adjusted, and the T2T reference genome sequence is obtained (Fig. 2). PacBio HiFi sequencing depth is about 60×, and ONT ultra-long sequencing depth is about 30−200×.
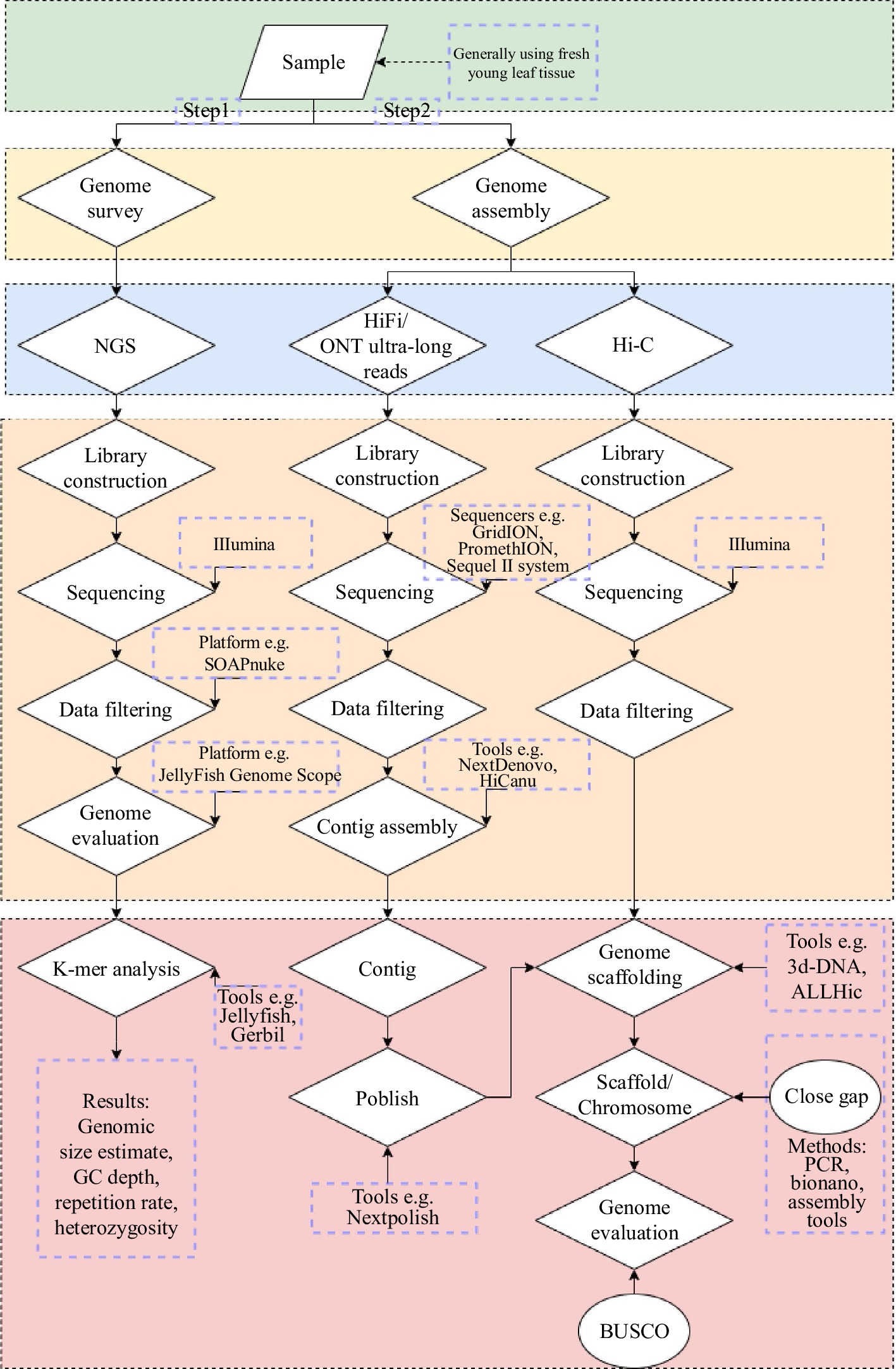
Figure 2.
Process of de novo assembly of the complete plant genomes. Sequencing platform, software, precautions, etc are noted in the figure alongside the steps.
-
The huge amounts of data of the second-generation sequencing increases the computational complexity of the assembly. And it is difficult to distinguish repetitive sequences of the genome which will produce incomplete assembly. Then, with the development of the third-generation sequencing technology, researchers combined the second and third-generation sequencing technology, plus optical mapping, genetic mapping or Hi-C technology to assemble the genome, which greatly improved the quality of assembly. Recent advances in optical mapping have allowed the genome comparison and identification of large-scale structural variations which also enables construction of improved genome assemblies with greater contiguity[14]. There are a large number of repetitive sequencing in the genome, which interferes with assembling, so genetic maps are required. These maps have their own advantages and disadvantages, and they need to be integrated and corrected with each other.
To achieve the complete genome, firstly, researchers use DNBSEQ short-read sequencing technology to complete Survey. Then, using ONT and HiFi makes the assembling and polishing process simpler and the results more accurate. Finally, obtaining the relative position information of genes on chromosomes by combining Hi-C technology could accomplish the chromosomal level of the genome. Subsequently, haplotype genomes were constructed by combining Hi-C data and short-read sequencing data from the parents. Tools for assembling long reads are constantly emerging and updated, and the assembly efficiency and accuracy are getting higher and higher, such as Falcon[15−17], miniasm[18], Flye[19], Hinge[20], CANU[21], wtdbg[22], Shasta[23] and Wengan[24], etc (Table 1). But with the emergence of HiFi reads, HiCanu[25] and hifiasm[26] have become the most important assembly tools chosen by researchers. Most assembly results are based on multiple software, and it is necessary to try different sorts of assembly software constantly, so that the reliability of the results obtained is often the highest, and few assemblies only use a single specific assembly software for assembly. In the assembly of Hi-C reads, the LACHESIS[27] tool has not been updated, and 3D-DNA[28] and ALLHiC[29, 30] gradually take its place. ALLHiC uses signal density to remove the link between alleles, which makes it easier for homologous chromosomes to be separated, so it can be used to solve the problem of plant polyploid assembly.
Table 1. Tools for assembling long reads and Hi-C reads.
Tools Features URL PacBio and ONT Assemblage relevant NextDenovo[33] (V2.5.0) String graph-based de novo assembler for long reads which uses
a 'correct-then-assemble' strategy, but requires significantly less computing resources and storage.https://github.com/Nextomics/NextDenovo Hifiasm[26] (V0.16.1) Fast haplotype-resolved de novo assembler for PacBio HiFi reads. https://github.com/chhylp123/hifiasm SMARTdenovo[34] A de novo assembler for PacBio and Oxford Nanopore (ONT) data.
It produces an assembly from all-vs-all raw read alignments
without an error correction stage.https://github.com/ruanjue/smartdenovo NGMLR[35] (V0.2.7) Long-read mapper designed to align PacBio or Oxford Nanopore (standard and ultra-long) to a reference genome with a focus on reads that span structural variations. https://github.com/philres/ngmlr CentroFlye[36] (V0.8.3) Algorithm for centromere assembly using long error-prone reads. https://github.com/seryrzu/centroFlye_paper_scripts Canu[21] (V2.2) Fork of the Celera Assembler, designed for high-noise single-molecule sequencing (such as the PacBio RS II/Sequel or Oxford Nanopore MinION). https://github.com/marbl/canu Wtdbg2[22] (V2.5) De novo sequence assembler for long noisy reads produced by
PacBio or ONT which assembles raw reads without error correction and then builds the consensus from intermediate assembly output.https://github.com/ruanjue/wtdbg2 HiCanu[25] Modification of the Canu assembler designed to leverage the full potential of HiFi reads via homopolymer compression, overlap-
based error correction, and aggressive false overlap filtering.https://github.com/marbl/canu HINGE[20] Long read assembler based on OLC (Overlap-Layout-Consensus). https://github.com/HingeAssembler/HINGE Peregrine Fast genome assembler for accurate long reads (length > 10 kb, accuracy > 99%). https://github.com/cschin/Peregrine Flye[19] (V2.9) De novo assembler for single-molecule sequencing reads, such as those produced by PacBio and Oxford Nanopore Technologies designed for a wide range of datasets. https://github.com/fenderglass/Flye Shasta[23] (V0.9) The goal of the Shasta long read assembler is to rapidly produce accurate assembled sequence using DNA reads generated by
Oxford Nanopore flow cells as input.https://github.com/chanzuckerberg/shasta NECAT[37] (V0.01) Error correction and de novo assembly tool for Nanopore long
noisy reads.https://github.com/xiaochuanle/NECAT Wengan[24] (V0.2) Wengan avoids entirely the all-vs-all read comparison. The key
idea behind Wengan is that long-read alignments can be inferred by building paths on a sequence graph.https://github.com/adigenova/wengan NextPolish[38] (V1.4) NextPolish is used to fix base errors (SNV/Indel) in the genome generated by noisy long reads, it can be used with short read
data only or long read data only or a combination of both.https://github.com/Nextomics/NextPolish Miniasm[18] (V0.3) Very fast OLC-based de novo assembler for noisy long reads. https://github.com/lh3/miniasm Falcon[15−17] (V0.3) Experimental PacBio diploid assembler https://github.com/PacificBiosciences/FALCON Raven[39] (V1.7) De novo genome assembler for long uncorrected reads. https://github.com/lbcb-sci/raven HERA[40] Local assembly tool using assembled contigs and self-corrected
long reads as input which can generate ultra-long, even chromosome-scale, contigs.https://github.com/liangclab/HERA Hi-C scaffolding relevant HiC-Pro[41] (V3.1) HiC-Pro was designed to process Hi-C data, from raw fastq files (paired-end Illumina data) to normalized contact maps. https://github.com/nservant/HiC-Pro SALSA[42] (V2.3) A tool to scaffold long read assemblies with Hi-C. https://github.com/marbl/SALSA 3D-DNA[28] (V201008) 3D de novo assembly (3D DNA) pipeline. https://github.com/aidenlab/3d-dna ALLHiC[29, 30] Phasing and scaffolding polyploid genomes based on Hi-C data. https://github.com/tangerzhang/ALLHiC LACHESIS[27] First tool to use Hi-C data to assist genome assembly. https://github.com/shendurelab/LACHESIS Some scientists are interested in reference-guided genome assembly, that is, how to mount newly assembled genomes to the homology of genome chromosomes of related or the same species. RaGOO[31] is a fast and reliable reference-guided scaffolding method.
After obtaining the genome sequence, it is of great importance to assess assembly quality. There are always some conserved sequences among similar species, and BUSCO (Benchmarking Universal Single-copy Orthologs) uses these conserved sequences to compare with the assembly results[32]. To identify whether the assembly results contains these sequences, so the integrity of the assembly can be obtained.
-
In view of the availability of whole genome assembly from scratch, especially those from long-reads sequencing data, gap closure sequences can be determined. There will be more gaps in the middle of the scaffold. In order to make the assembled sequence more complete, it is necessary to connect contigs again by using the pairing relationship between the sequenced double-ended data, and fill the holes between contigs by using the covering relationship between the sequenced data and the assembled contigs, so as to extend the contigs. The length of contigs after filling holes is generally increased by 2−7 times compared with that before filling holes. GapFiller is the software for filling holes[43], TGS-GapCloser[44], GAPPadder[45], PBJelly[46] and so on. Similarly, it is necessary to try different software and keep trying to choose the best solution.
Among them, Bionano can make complete single DNA molecules arranged in parallel in nanochannels through its unique chip technology, and take photos and images, so that the genome structure can be fully displayed[47]. Therefore, it can assist genome assembly. Five contigs were obtained from the human MHC region through NGS. Through Bionano, their positions in the genome and the size of gaps can be accurately determined. By comparing the genome map with sequencing fragments, it is easy to determine the positions of gaps and two contigs separated by 36.4 kb on the chromosome[48]. It can also read through repetitive sequence information. There are many repetitive fragments in the human genome. The copy number of repetitive fragments can be accurately determined through the Bionano system, which can read through long-chain single-molecule DNA fragments, so that this result can be clearly presented.
Researchers also introduced BAC (bacterial artificial chromosome)-anchor strategy to fill the remaining gaps in ONT-HiC assemblies which used ONT-generated ultra-long reads[49]. In short, for each gap, BAC sequences used to replace ONT contigs with HiFi contigs because HiFi contigs enjoy better continuity. All the BACs used share more than 99.9% sequence identity with their target contigs.
-
The genome assembled by the second-generation sequencing technology can only be regarded as a draft genome. With the continuous development of sequencing technology, the third-generation sequencing technology takes into account the advantages of the first-generation sequencing technology and the second-generation sequencing technology in terms of length and high-throughput, and can obtain a longer sequence, thus obtaining a reference-level genome. Therefore, more genetic information can be obtained through the third-generation sequencing technology[50].
However, due to the highly repetitive sequences such as telomeres and centromeres in the genome, almost all the genomes obtained today have a relatively high number of gaps, which are usually expressed by N or n. There are two reasons for the gap. One is that the gap is generated because of the restriction of the reading length. For example, if sequencing only has a reading length of 150 bp at both ends, and one fragment has 350 bp, the remaining 50 bp will not be known, so the longer the reading length, the smaller the gap will be. Second, assembly technology restricts the generation of gaps, such as comparing the sequenced reads with the contigs, and assembling contigs into scaffolding by using the connection relationship between the reads and the size information of inserted fragments (Mate-Pair), in which the undetermined region in scaffolding sequence. At present, many complete chromosomes have been sequenced and assembled in human beings[51], rice[6], Arabidopsis thaliana[2], banana (Musa acuminata)[52]. T2T genome refers to the high-precision, high-continuity, high-integrity genome assembly from telomere to telomere. It is realized by combining a variety of sequencing technologies, which is helpful to clarify the complex structure of highly repetitive regions in the genome, such as the detailed study of the variation characteristics and evolution patterns of centromeres and telomeres. T2T Alliance researchers have assembled and published the first completed picture of the human X chromosome[53]. Autosomal completion diagram[54] is a complete map of the human genome. Therefore, the satellite array in the centromere region, telomeres, large genome duplication and important genetic information in the rRNA region have been uncovered, among which there are many genes related to human aging and diseases. In human X chromosome sequencing, more than half of the reads obtained are over 70 kb, and the longest one is 1.04 Mb. The centromere contains a highly repetitive DNA region, which spans 3.1 million base pairs. In 2021, on the occasion of the 20th anniversary of the release of the draft human genome sequence, T2T Alliance released the latest complete human genome sequence CHM13 v1.1, which not only contains all the unresolved data, but also corrects the original assembly errors. In this completed picture of the human genome, researchers newly added or corrected 238 Mb of sequences, of which 182 Mb is a brand new sequence, and annotated 2,226 new genes, which is the most complete human genome ever. In the future, scientists will perform pan-genome sequencing on individuals of different races, so as to understand the genetic diversity of different races and individuals, and provide greater help for the future goal of precision medicine.
Due to the high similarity of homologous chromosomes in diploid species, genome assembly usually does not distinguish homologous chromosomes, and only assembles genomes with mixed parental genetic information. However, this assembly method will lead to inaccurate genetic annotation, which is not conducive to some biological studies that distinguish the genetic information of parents. Therefore, obtaining two haplotype genomes from parents provides important reference information for further study of allele mutation and understanding of species genetic relationship and evolutionary history. Furthermore, the sex chromosomes of a species usually carry important sex-determining genetic information, determine the development of reproductive organs, and show many completely different genomic characteristics and evolutionary patterns from them. However, the highly repetitive sequence and heterochromatin of sex chromosomes make them difficult to assemble. By sequencing and assembling the complete sex chromosomes, we can deeply analyze the specific differences of different sex individuals in species.
For plants, as early as 1999, the American Genome Research Institute (TIGR) assembled chromosome 2 of Arabidopsis thaliana without a gap, and the chromosome length obtained was 19.6 Mb[55]. It includes centromere and nucleolar organizer regions, but the function of nearly half of the genes on this chromosome is unknown. Presently, three high-quality assemblies of A. thaliana, Col-CEN[56], Col-XJTU[49] and Col-PEK[2], were deciphered and their quality was progressively improved. The rice[6] genome is the first complete gap-free plant genome published so far, and it fills the gap of 223 (ZS97RS1) and 167 (MH63RS1) between the two genomes.
In the past 100 years or so, 60% of the plants on the earth have been wiped out[57]. There are many wild varieties with excellent genes and traits, which is a great loss for those engaged in agricultural production and scientific research. The goal of Vertebrate Genome Project (VGP)[58], is to collect, sequence and assemble at least one high-quality, error-free, nearly gap-free, haplotype staged and annotated reference genome of all existing vertebrates, and to use these genomes to solve basic problems in biology, diseases and ecological protection. The minimum expected measurement values are contig N50 > 1 Mb, scaffold N50 > 10 Mb[59], and 90% of the assembly is located at the chromosome level. Tropical plants include 2/3 of higher plant species which have extremely rich genetic diversities. To protect endangered tropical wild plants, we could launch a tropical plant genome project and we hope that researchers can make use of the latest genome sequencing and assembly technology to assemble as complete and gapless as possible, which is more conducive to us to identify and classify a large amount of genetic information contained in excellent species.
High-definition genome provides complete gene sequences and complete repetitive sequences, which can help us understand the composition of centrioles and telomeres, promote the development of comparative genomics and evolutionary biology, and better modify the genome[60], providing genome data for genetic breeding and domestication, which in turn further promotes the development of the three major omics.
-
Nowadays, for plants, the complete genome can only be assembled at the level of a single chromosome or simple species. Sequencing and assembling of the large genome (5 GB ≤ genome size < 10 GB) or very large genome (Genome Size ≥ 10 Gb) is still a worldwide problem. For example, the genome size of Fritillaria pallida exceeds 40 Gb[61]. Therefore, it is even more difficult to obtain its complete genome. The amount of data required for the assembly of very large genomes often reaches the Tb level. To obtain sequencing data quickly, the sequencing platform must have ultra-high throughput, and its computing cost and occupied server resources are huge. However, species with very large genomes often have a large number of repetitive regions, and short reading and long sequencing technology are difficult to span. At the same time, a large number of short clips lead to extremely complicated genome assembly, and it is difficult to get ideal results. For example, Paris japonica[62] has the largest genome of any plant yet assayed, about 150 Gb which is 50 times larger than that of a human haploid genome. The genome size of Paris polyphylla[63], the same genus as former, is about 82.55 Gb, making it the largest genome draft to date. The difference in genome size of plants belonging to the family Nigellaceae is as high as 230 times, which is an ideal model for studying the change of genome size. Deciphering its genome is of great significance for studying the evolution in genome size and biosynthesis pathway of Paris polyphylla saponins. In this study, 10.25 Tb of sequenced reads were assembled by using SOAPdenovo2[64] to get a genome sketch of 70.18 Gb, but it was not assembled by sequencing with the third generation technology, and it was not mounted on the chromosome with Hi-C. The genome of gymnosperm Pinus tabulaeformis was assembled to the chromosome level, reaching 25.4 Gb[65]. The genome contains a large number of super-long introns, the average length of which is 10 kb. It takes 1.3 million CPU hours to assemble the whole genome by WDL (Workflow Description Language)-Canu. Therefore, an ultra-high throughput sequencer, a method of obtaining ultra-long fragments, a set of resource-saving assembly algorithms and a powerful CPU are urgently needed to overcome the last field of genome sequencing.
Sequencing and assembling the genome of polyploid plants are also a future trend. Polyploidy mainly occurs in angiosperms, and many polyploid plants are of great value in agricultural production. It can be divided into two types: autopolyploidy from whole genome doubling, like Medicago sativa[66]; allopolyploidy whose chromosome doubles after interspecific or intraspecific hybridization, such as allohexaploid Triticum aestivum[67] (AABBDD, 2n = 6x = 42) and allotetraploid Arachis hypogaea[68] (AABB,2n = 4x = 40). The relationship between phenotypes and genotypes of polyploids is more complicated. For example, they need complicated regulation to ensure the consistent expression of homologous genes. In genome assembly, autopolyploid is more difficult than allopolyploid, because the whole genome doubling event will result in highly similar segments. Therefore, one of the challenges in genome assembly is that similar fragments in two subgenomes can't be assembled by mistake.
The whole genome of rice gap-free was evaluated by gene BUSCO. ZS97RS3 and MH63RS3 both covered 99.88% of the reference gene sets, but there was no gap in the reference genome, but BUSCO still didn't reach 100%. BUSCO may not be the best method to evaluate genome integrity in the future, and whether the genome is gap-free may replace BUSCO to evaluate gene integrity. In the near future, we can predict that simple diploid plants will be sequenced and assembled into gap-free genomes, and the data quality of large plant genomes and polyploid plant genomes will be greatly optimized.
The authors acknowledge the funding from National Natural Science Foundation of China (32172614) and a startup fund from Hainan University to Fei Chen.
-
The authors declare that they have no conflict of interest.
-
Received 1 August 2022; Accepted 13 September 2022; Published online 26 September 2022
- Copyright: © 2022 by the author(s). Published by Maximum Academic Press on behalf of Hainan University. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Zhou Y, Zhang J, Xiong X, Cheng Z, Chen F. 2022. De novo assembly of plant complete genomes. Tropical Plants 1:7 doi: 10.48130/TP-2022-0007 

De novo assembly of plant complete genomes
- Received: 01 August 2022
- Accepted: 13 September 2022
- Published online: 26 September 2022
Abstract: Plant genomes encode the mysteries of how plants cope with complex environments over long evolutionary histories. Over the past 20 years, rapidly developing technologies have allowed the decoding of hundreds of plant draft or reference genomes. The diversity, polyploidy and heterozygosity of plants make it technically challenging and time-consuming to generate high-quality plant genome assemblies. Recently invented ultra-long read sequencing technologies have achieved a milestone where several plant genomes have been gapless and assembled into telomere to telomere. Telomere-to-telomere (T2T) genome refers to a high-quality complete genome with high genomic accuracy, high continuity, and high integrity. With the release of the completed human genome and Arabidopsis thaliana genome, the era of complete T2T species genome has arrived. In this review, we summarize the history leading up to the gap free plant genomes based on emerging ultra-long read sequencing technologies. We discuss to close gaps relying on targeted genome sequencing and assembling technologies. However, there are still quite a lot of challenges in super large, polyploidy, and unstable genomes. Nevertheless, these complete genomes have already provided unprecedented information, which will certainly deepen our understanding of plant genomes and the exploration of more functional sequences. By taking advantage of the complete genomes, a series of important genes could be annotated, which will help achieve the goal of genome design in crop species.
-
Key words:
- plant genome /
- assembly /
- long-read sequencing /
- tools