










-
Tea is one of the most popular beverages in the world. Currently, the demand for specific teas, such as purple tea[1], high gallotannin tea is on the rise. Among them, albino tea (white or yellow buds and leaves) is the most known[2−4]. The albino character is closely associated with the chlorophyll-deficiency, which results in physiological and biochemical changes[5]. Extensive studies had been carried out to identify the quality constituent changes[5−7]. As compared with normal green leaf teas, higher concentrations of amino acids, but lower catechin content were identified in albino teas which account for the umami taste. However, the underlying mechanism of the chlorophyll-deficient phenotype in albino tea still remains elusive.
Chlorophyll is a tetrapyrrole playing a central role in photosynthesis[8]. Active biosynthesis of chlorophyll and formation of chlorophyll–protein complexes are observed during leaf greening[9]. In contrast, the decrease of chlorophyll–protein complexes and degradation of chlorophyll are strictly coordinated with leaf senescence[10]. Chlorophyll metabolism is regulated by many internal or environmental factors, such as phytohormones[11], translation factors[12], feedback mechanisms associated with chlorophyll biosynthesis[13], light[14], redox state[15], temperature[16] as well as stresses[17].
With the development of RNA-sequencing technology, scientists tried to uncover the key genes associated with the chlorophyll-deficient phenotype of albino tea by identifying differentially expressed genes (DEGs) in green and albino leaves. For example, DEGs of the temperature-sensitive cultivar 'Anjibaicha' during different albescent stages were identified, which included genes involved in chlorophyll biosynthesis, chloroplast development and photosynthesis[18]. Moreover, the albino tea cultivar 'Baijiguan' showed 4,569 DEGs in leaves under control, 3 and 6 days' shading treatments. These DEGs were mainly correlated with reactive oxygen species (ROS) scavenging, photosynthesis, chloroplast development and secondary metabolism[19]. For another albino tea cultivar 'Yujinxiang', a total of 1,196 DEGs were identified between albino leaves under control and shading treatment, with 44 DEGs associating with chloroplast functions[20]. These studies offered valuable insights. However, several limitations do still exist which inhibit our understanding of albino tea. First, the lack of tea resources with similar genetic backgrounds inhibited us to focus on the key genes. Tea plant is a self-incompatible species. Although lots of DEGs were identified between the albino and normal tea cultivars, they might also be caused by the different genetic backgrounds rather than the key genes correlated with the albino character. Second, RNA-Seq analysis of tea leaves with different phenotypes (such as shading or different growth stages) in the same tea cultivars should also take the effects of environment or developmental stages into consideration. Finally, RNA-Seq analysis is a very useful method, but generally offers too much data. Since many DEGs might be just coincidently differentially expressed in the samples and not directly associated with the research target, further methods for identifying the key genes are still required.
Recently, a tea plant with both green and albino branches was identified in Haishun tea garden, Kaihua, China (Fig. 1). The identification of this plant enabled us to study the albino character without the interference of the genetic background between cultivars and the environmental effects. Furthermore, the publication of tea genomic data firstly provided a prerequisite for performing epigenetic study in tea plants[21]. In this study, both RNA sequencing and DNA methylation analysis of this special plant were conducted to narrow down the key genes associated with albino mechanism and chemical changes. The results will not only deepen our knowledge of the albino mechanism, but also provide key genes for further studies.
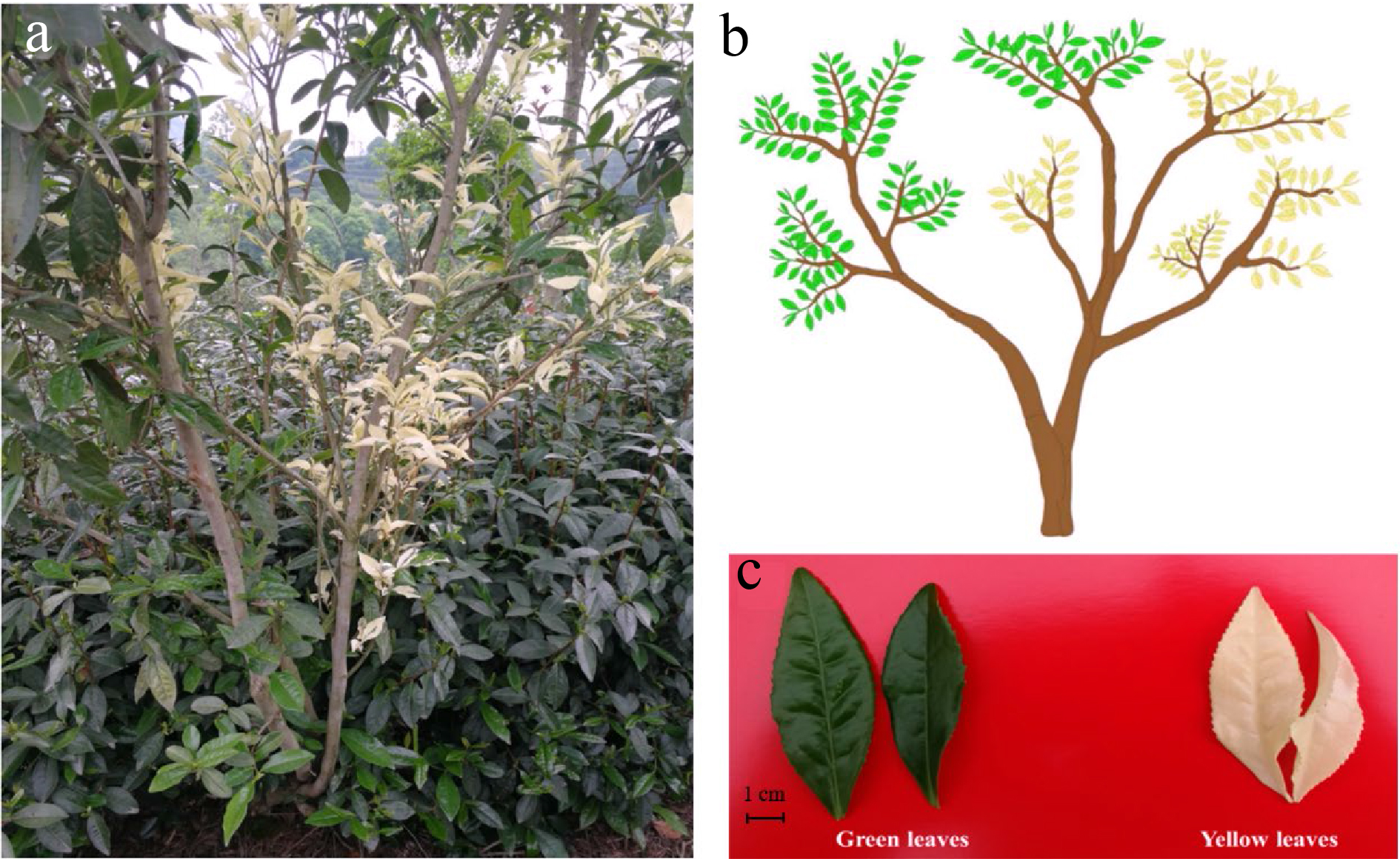
Figure 1. The phenotype of 'Haishun 2' tea mutant. (a) The original 'Haishun 2' tea plant; (b) Diagram of the 'Haishun 2' tea plant; (c) The green and yellow tea samples of 'Haishun 2' tea plant.
-
A tea mutant 'Haishun 2' with both green and albino branches was identified in April 2018 in Haishun tea garden, Kaihua, China. The green and yellow leaves were collected on April 28, 2018. Parts of the tea samples were used for transmission electron microscopy (TEM) analysis. The green and yellow leaves of the 'Haishun 2' tea mutant were prepared for TEM of the ultrastructure of cells according to the method described by Wei et al.[1]. Parts of samples were dried using a freeze dryer at −5 °C for 72 h, and then subject to catechin analysis as described by Wei et al.[6]. The remaining samples were refrigerated (−70 °C) and subjected to transcriptome sequencing, quantitative RT-qPCR and DNA methylation analysis.
RNA Sequencing analysis was performed with three biological replicates according to the description by Wei et al.[21] and Trapnell et al.[22]. DNA methylation and RT-qPCR analysis were performed with three biological replicates according to the description by Park and Wu[23] and Zhang et al.[24].
-
The 'Haishun 2' tea mutant was first identified in Haishun tea garden, Kaihua, China. It has both green and albino branches on one plant (Fig. 1). Meanwhile, some of the albino branches could revert to green branches, indicating it may be caused by epigenetic changes.
To further confirm whether the albino phenotype change of 'Haishun 2' mutant is related to chlorophyll formation, the mature leaves of green and albino branches were collected for transmission electron microscopy (TEM) analysis (Fig. 2). More chloroplasts could be observed in green leaf cells than those in yellow leaf cells (Fig. 2a & b). Moreover, in the chloroplasts of green leaves, high numbers of thylakoids and tightly stacked grana were observed (Fig. 2a & c). However, the albino leaves exhibited abnormal chloroplasts with no grana stacking and thylakoids but some bubble-like structures (Fig. 2b & d). This result was similar to the previous reports on albino tea plants and indicates the albino phenotype of 'Haishun 2' is associated with chlorophyll formation problems.
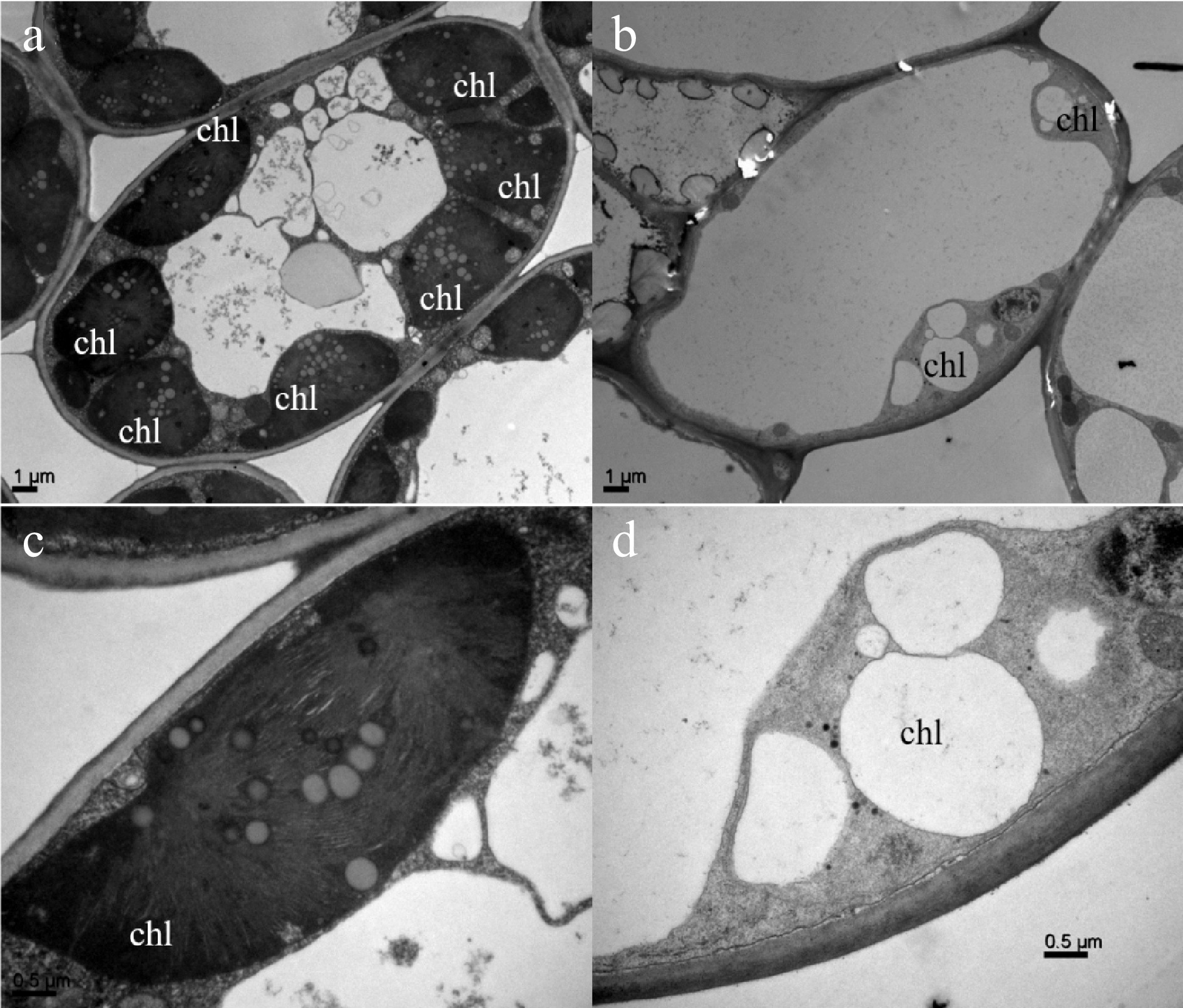
Figure 2. Comparison of subcellular structures of green and yellow leaves of 'Haishun 2' tea mutant.
Further HPLC analysis shows that in total, seven catechin components were detected, namely EGC, GC, ECG, C, EGCG, GCG and EC (Fig. 3). The content of most catechin components (EC, GC, C, EGCG, EGC and GCG) were reduced, whereas the content of ECG was significantly enhanced in albino tea leaves. In terms of the total catechins, the content was decreased by 42% in albino leaves when compared with normal green leaves of 'Haishun 2', which is consistent with previous studies.
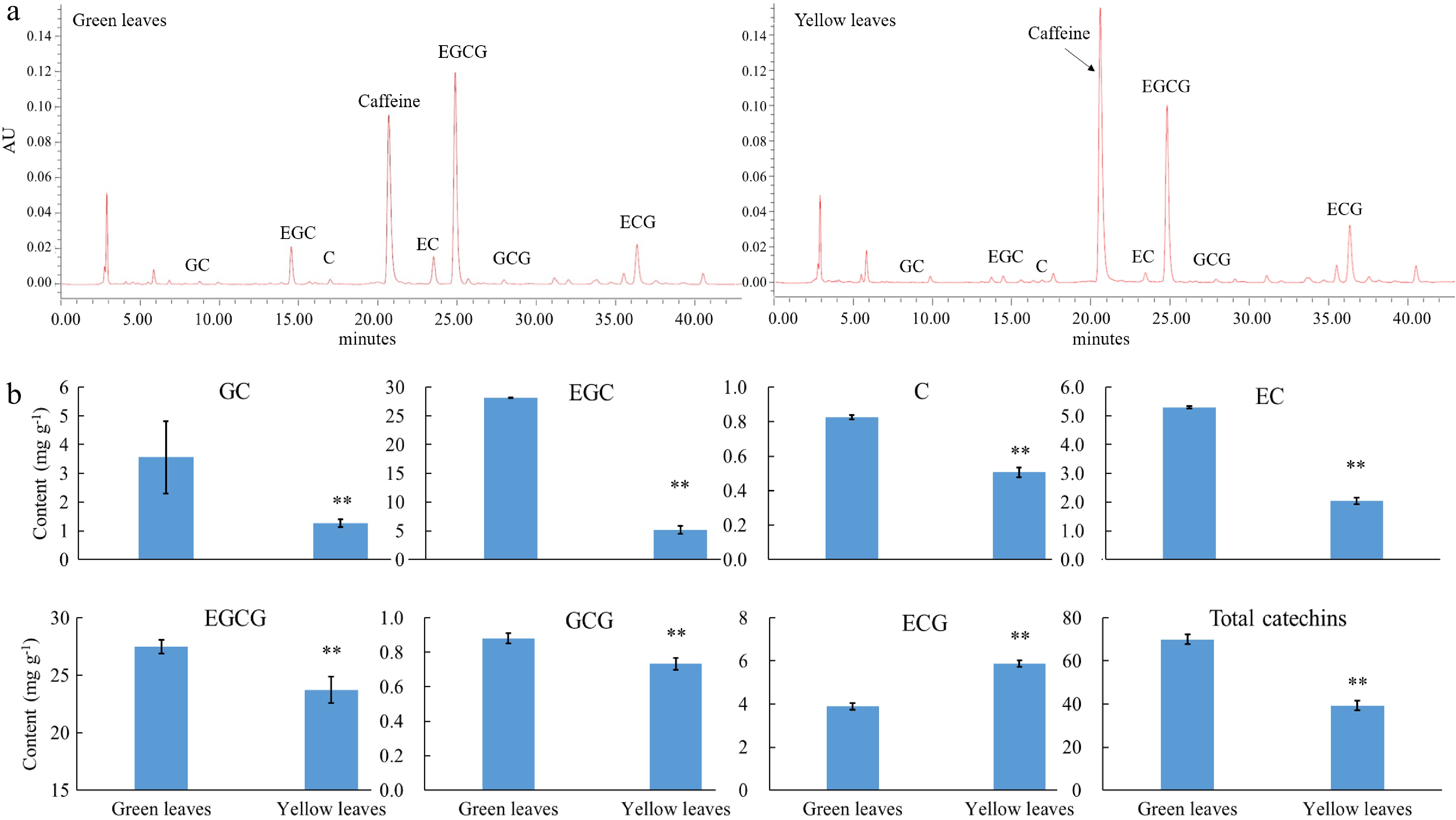
Figure 3. Catechin content in green and yellow leaves of 'Haishun 2'. (a) Chromatogram of the catechin component in green and yellow leaves of 'Haishun 2'. (b) The changes of catechin components and total catechins in green and yellow leaves of 'Haishun 2'.
Differentially expressed genes in green and albino tea samples identified by RNA-Seq analysis
-
To identify genes differentially expressed in green and yellow leaves of the 'Haishun 2' tea mutant, RNA-Seq analysis was performed. Within the entire samples, the clean reads and average numbers were 51.79 Gb and 8.63 Gb respectively. The rate of Q20 and Q30 to the clean reads were about 97% and 93% (Supplemental Table S2) showing high efficiency sequencing outcomes to the following analysis. Correlation analysis shows that the green and yellow samples were clustered into two groups, indicating the gene expressions were closely correlated with phenotype changes (Supplemental Fig. S1). All data were submitted to the NCBI database with the accession number PRJNAPRJNA624972 (www.ncbi.nlm.nih.gov/bioproject/PRJNA624972).
For differential expression analysis, we set a screening threshold (P < 0.05, fold change > 2 and maximum FPKM > 2) of DEGs to the green and yellow leaf samples. Quantitative profiling of RNA-Sequencing reveals that 3,747 genes were differentially expressed, with 1,725 genes up-regulated and 2,022 down-regulated in the yellow leaves.
To verify the RNA-Seq results, 19 DEGs were chosen from the data of RNA sequencing for RT-qPCR analysis (Fig. 4, Supplemental Table S1). Correlation analysis of the RT-qPCR and RNA-Seq results were also performed. RT-qPCR analysis shows that the expression patterns of these genes were highly consistent with the results of the RNA-Seq analysis. Furthermore, the correlation analysis also showed that the gene expressions obtained by both methods were highly significantly correlated (Fig. 4), revealing that the RNA-seq results are reliable.
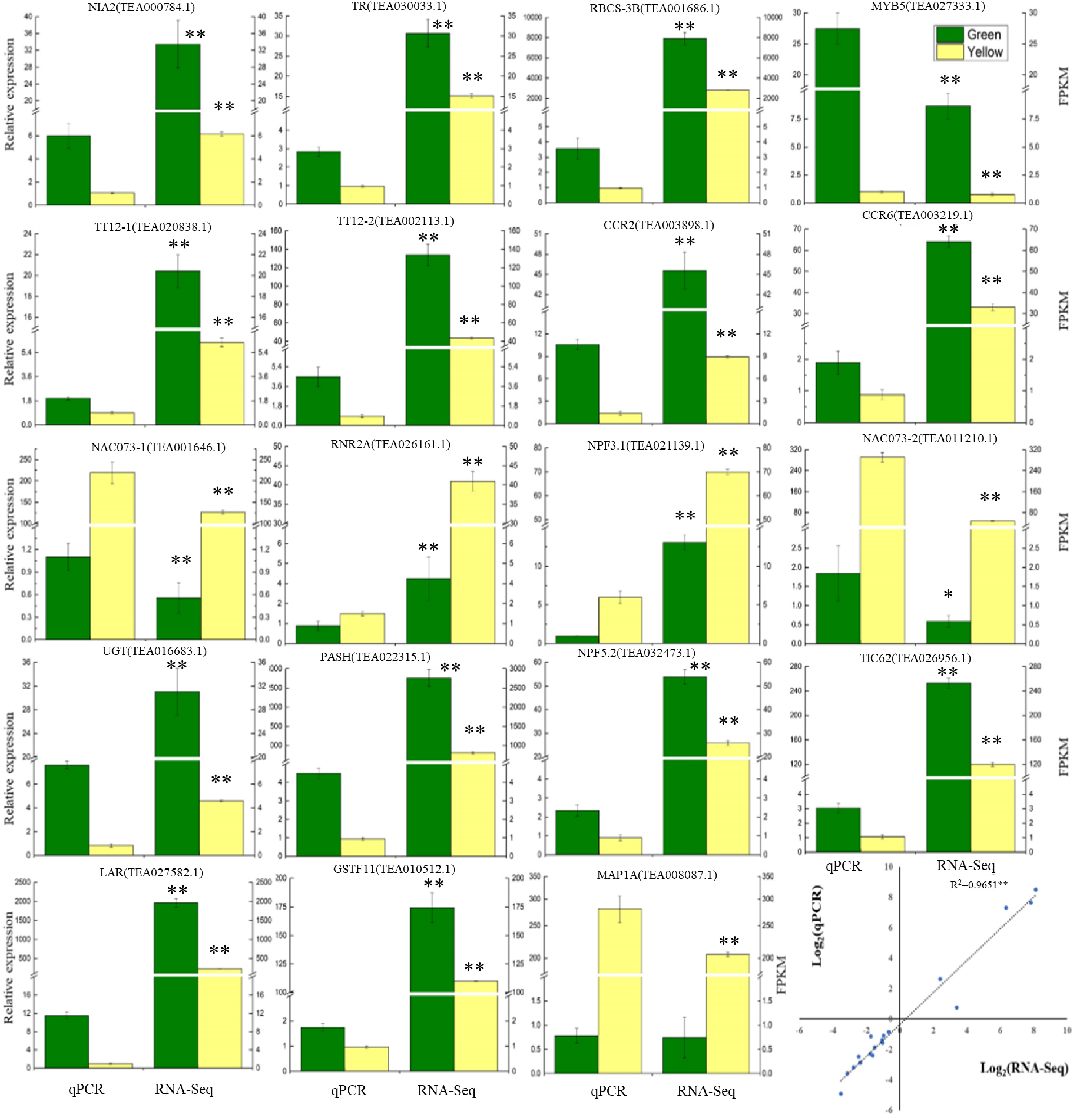
Figure 4. Verification of the gene expressions determined by RT-qPCR and RNA-Seq analysis. Data are shown as the means ± SD (n = 3).
DNA methylation analysis explored the overlap genes
-
To identify the potential epigenetic changes, we performed bisulphite sequencing (BS-seq) of green and yellow tea samples by an Illumina Hiseq 2500/4000 platform. A total of 673.87 Gb of sequencing data with an average of 112.31 Gb per sample were obtained for the whole genomebisulfite sequencing (Supplemental Table S3). The total numbers of raw and clean reads were 2.39 and 2.35 billion respectively, with an average of 398.2 and 391.8 million reads per sample. About 0.13% of the cytosines were read as Cinstead of T, revealing that the average bisulphite conversion rate is higher than 99.8% (Supplemental Table S3). Then the clean reads were mapped to the recent published tea reference genome, with an average mapping rate of 72.23% and duplication rate of 10.41% (Supplemental Table S4). This mapping rate is similar to those reported in other plant species, indicating the high quality of BS-seq in this study[25]. Based on the mapping results, it can be found that about 83.4%, 70.7% and 10.0% of cytosines in CpG, CHH and CHG contexts are methylated (Supplemental Table S5), which is similar to the recent finding in tea plant[25].
To identify the relationship between DNA methylation regions, transposable element (TE) density, gene density and RNA, a genome wide landscape was used with the first 20 scaffolds of 5mC/RNA as an example (Fig. 5a). From the landscape, we noticed that a large amount of methylation is present in TE-exuberant parts in the 'Haishun 2' genome. To further explore the correlation between DNA methylation and gene expression levels, these data in green and yellow tea leaves were compared in (Fig. 5b−g). In about 500 bp upstream of genes (promoter regions), the methylation levels in CpG and CHG contexts were negatively correlated with the gene expressions, which showed an opposite trend for that in CHH. On the other hand, the genebody methylation was negatively correlated with expression except for that in CpG. These results indicate that gene expressions controlled by DNA methylation in tea plant are quite complex and need further research.
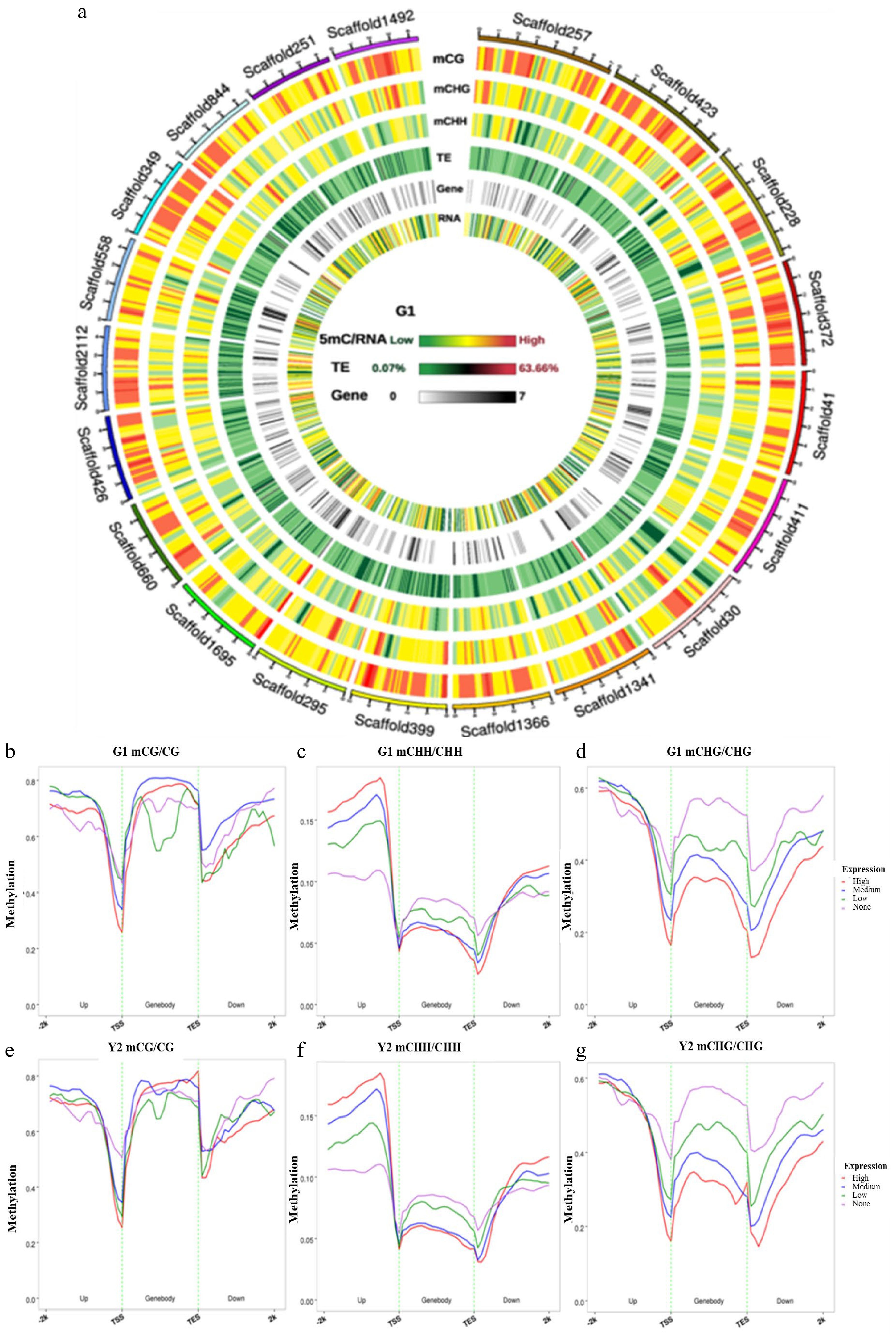
Figure 5. Circos plots of scaffolds in tea genome and the association between DNA methylation and gene expression. (a) Circos plots of scaffolds in tea genome. Track order: density plot of 5mC in CpG, CHG and CHH contexts; density of transposable elements (TEs); gene density of each scaffold. (b−g) Distributions of methylation levels within gene bodies partitioned by different expression levels: 1st_quintile is the lowest and 4th_quintile is the highest; genes with FPKM value < 0.1 were considered non-expressed (none). (b−d) The association between CpG, CHG and CHH methylation and gene expression in green leaves; (e−g) The association between CpG, CHG and CHH methylation and gene expression in yellow leaves; Genes were divided into four groups according to their expression levels. Association between gene expression and DNA methylation in the CpG, CHG and CHH sequence contexts. Genes with FPKM value groups by their gene expression levels. This means that the first group of genes contained the highest expressed genes. Each upstream 2 kb, gene body, and downstream 2 kb region was divided proportionally into 20 bins, and average methylation levels were estimated for each bin.
A total of 3,250 sites in gene bodies and 820 sites in promoter regions were found to be differentially methylated between green and yellow leaves of tea mutant 'Haishun 2' (Supplemental Fig. S2). Among them, CHG methylation is a major characteristic of DMRs in gene bodies, with 2,137 DMR genes. While both CHH and CHG methylations are the major characteristics of DMRs in promoter regions, with 363 and 346 DMR genes. The DNA methylation data suggests that DMRs in 'Haishun 2' mainly occur in gene bodies rather than promoter regions (about 4 fold). Then an integrative analysis of DMRs and DEGs were performed to identify the overlapped genes. A total of 311 and 97 overlapped genes were identified between DEGs vs DMRs in gene bodies and DEGs vs DMRs in promoters (Fig. 6). It could be found that most of these overlapped genes were higher methylated and lower expressed in albino leaves, indicating DNA methylation in tea plants mainly inhibits the expressions of genes.
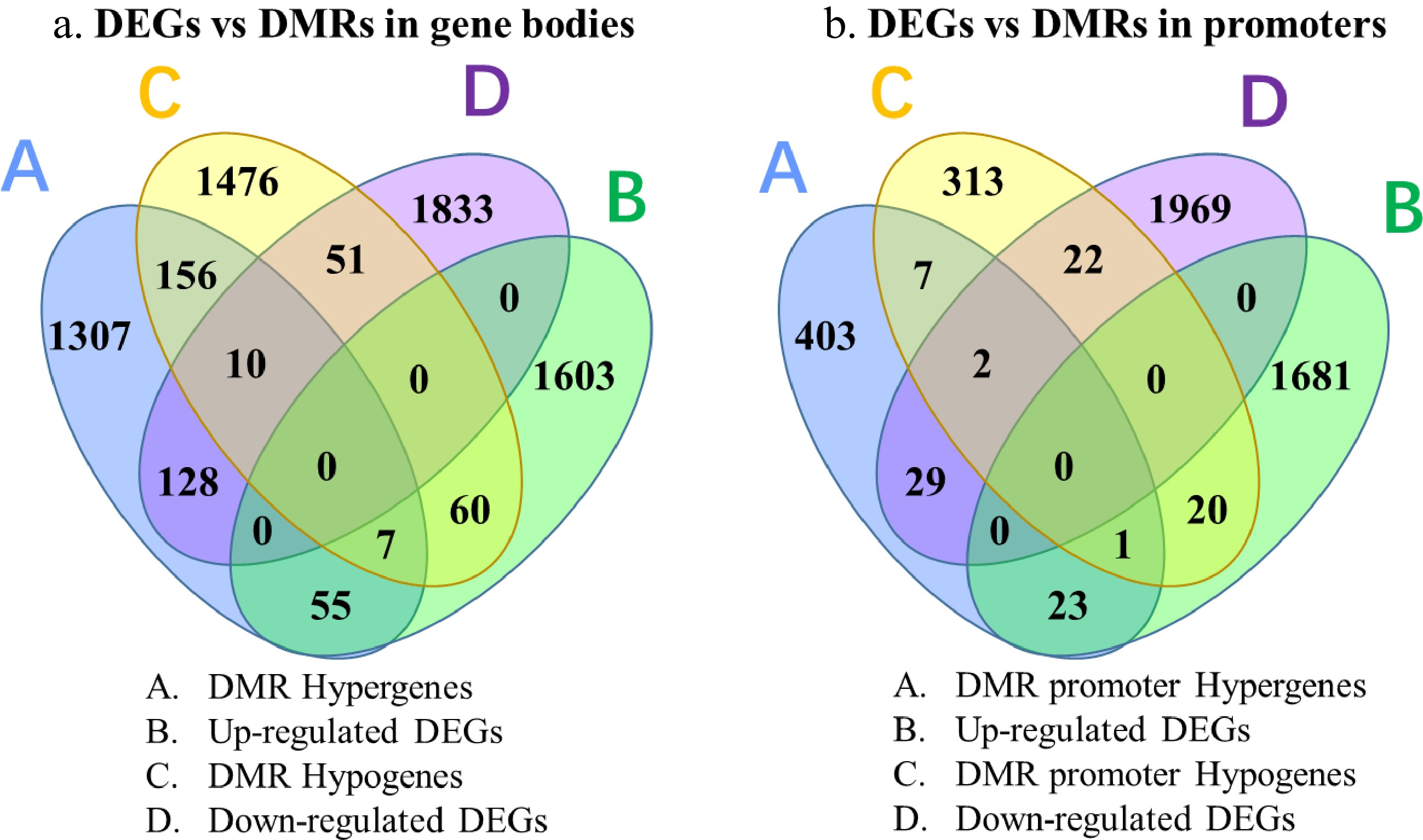
Figure 6. Venn diagrams of genes identified by RNA-Seq and BS-Seq analysis.
To have a global view of the potential metabolisms involved in the albino phenotype in 'Haishun 2', the overlapped genes were mapped to reference canonical pathways according to KEGG database. For DMRs in gene bodies, the most representative pathway was biosynthesis of secondary metabolites (12 overlapped genes, P-value = 0.030), followed by pantothenate and CoA biosynthesis (two overlapped genes, P-value = 0.087; Supplemental Table S6). For DMRs in promoter regions, the most representative pathway was photosynthesis (two overlapped genes, P-value = 0.191; Supplemental Table S7). These pathways are already recognized to be closely related with chloroplasts in previous reports. Therefore, these overlapped genes might also play key roles in the albino phenotype of the 'Haishun 2' mutant, which are worthy of further study.
-
DNA methylation is a major epigenetic mechanism which interferes with transcription factor binding to DNA or the recruitment of inhibitory proteins, thereby affecting normal development and cellular function in plants and animals. In this study, DNA methylation analysis shows that CpG was the major methylation pattern in the 'Haishun 2' mutant (Supplemental Table S5). However, most of DMRs occurred in the CHG context, suggesting CHG methylation might also play a dominant role in gene regulation. Wang et al.[25] reported that CHG methylation in tea was significantly higher than that in potato or tomato. Our results were consistent with that report, indicating CHG methylation should be especially emphasized in future tea epigenetic studies.
An integrative analysis of DNA methylation and RNA-Seq data in tea plants was first reported here and a close correlation between the global DNA methylation changes and albinism was identified, which was similar to the findings in Agave angustifolia Haw[26,27]. We identified 385 overlapped genes which potentially affect albinism in 'Haishun 2' tea. These genes include 288 genes only differentially methylated in gene body regions, 74 genes only differentially methylated in promoters and 23 genes differentially methylated in both regions. Most of the DMRs occurred in gene body regions rather than promoters. As Wang et al.[25] also found the lowest expressed genes possessed the highest methylation levels in gene body regions, we hypothesized that changes in methylation levels in gene body regions might be a key epigenetic pattern for gene regulation in tea. Compared with the previous studies on albino tea by RNA-Seq method alone, the number of candidate genes identified here was largely narrowed down by the use of integrative analysis in a specific tea mutant. Therefore, it would be helpful for us to avoid the interference of thousands of unrelated genes and deepen our understanding of the albino mechanism.
From the overlapped genes, two NAC transcription factors (TEA001646.1 and TEA011210.1) were found to be clearly differently expressed. The CpG methylation in the promoter of TEA001646.1 and the exon of TEA011210.1 were significantly less in the albino leaves compared with green leaves of 'Haishun 2' (Fig. 7). Their expressions were all enhanced, from almost none expression in the green leaves, to 127.2 (TEA001646.1) and 48.1 (TEA011210.1) in the albino leaves, which were closely correlated with the less CpG methylation. Previous studies showed that the transcription factor of the NAC family is involved in leaf senescence[28]. Oda-Yamamizo et al.[29] reported that an Arabidopsis NAC transcription factor (ANAC046) is a positive regulator of leaf senescence and exerts its effect by controlling chlorophyll degradation. Tak et al.[30] found that overexpressing a stress associated NAC transcription factor (MpSNAC67) showed highly senesced phenotype including yellowing and de-greening of leaves. Therefore, the up regulation of TEA001646.1 and TEA011210.1 should be especially emphasized in future studies, which might also participate in chlorophyll degradation and be the key reasons for the albino phenotype in 'Haishun 2'.
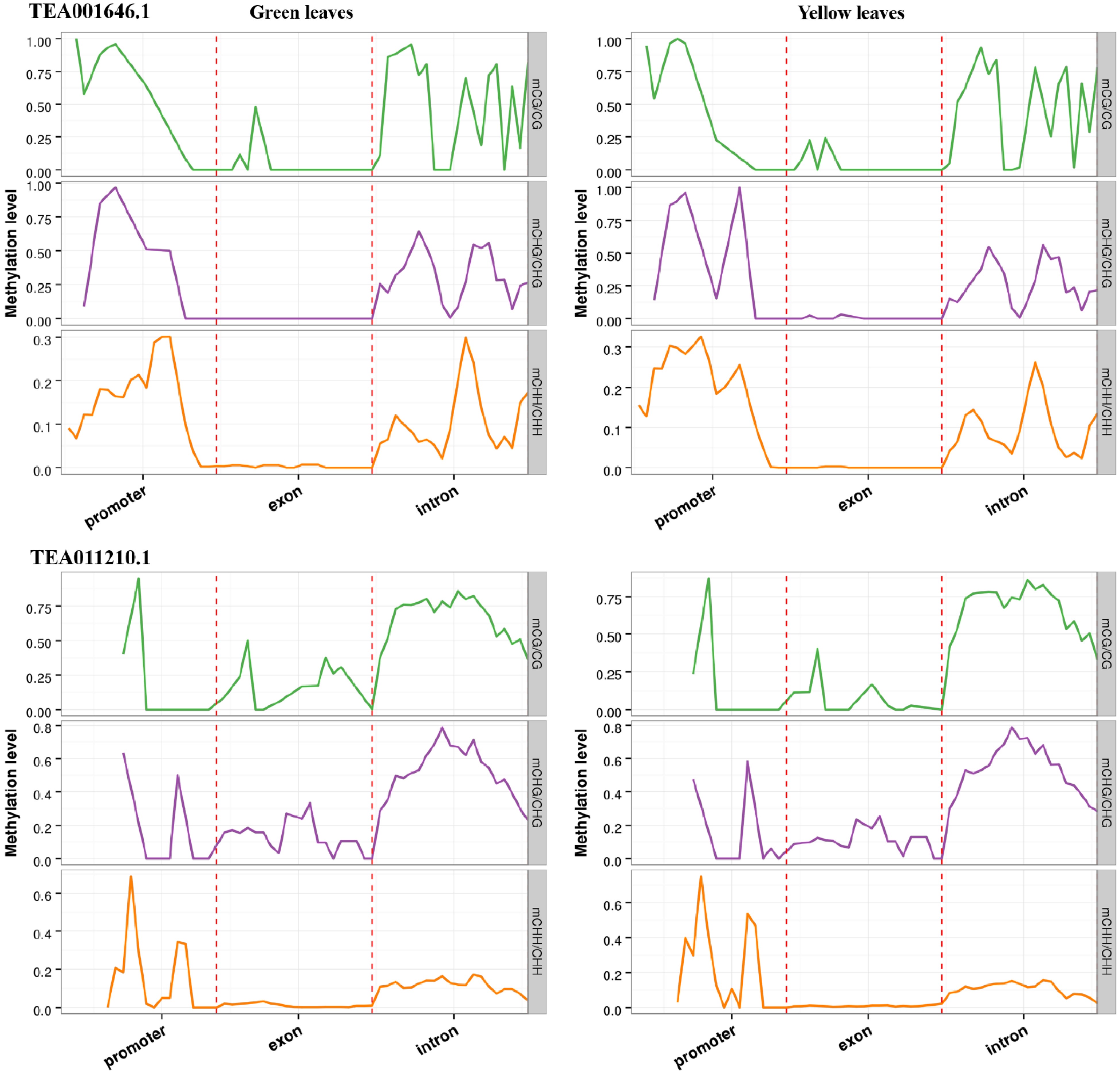
Figure 7. DNA methylation patterns of two NAC transcription factors (TEA001646.1 and TEA011210.1). In all cases, promoter indicates the upstream 2,000 bp of transcriptional start sites, exon and intron are the corresponding parts of gene bodies.
Several key genes involved in chlorophyll formation or function were found to be down regulated and differentially methylated in albino leaves of 'Haishun 2', such as chlorophyll a-b binding proteins (TEA017256.1, TEA008963.1, TEA021966.1), photosystem II 22 kDa protein (TEA011923.1), curvature thylakoid 1B (TEA010578.1), photosystem I reaction center subunit VI (TEA022315.1) and chloroplast stem-loop binding protein (TEA006794.1). Previous reports of their homolog genes showed a direct correlation with leaf color changes. For example, chlorophyll a-b binding proteins, which collect and transfer light energy to photosynthetic reaction centers were widely identified to be down regulated in albino tea cultivars[31]. In barley, single nucleotide polymorphisms in a chlorophyll a/b-binding protein were identified to be significantly associated with leaf color[32]. While the chloroplast stem-loop-binding protein is a key protein associates with pre-ribosomal particles in chloroplasts. It was found that a rice mutant in a chloroplast stem-loop-binding protein gene (CSP41b) showed altered leaf color[33]. However, as their expressions were also high in albino leaves and fold changes were only slightly higher than 2, they were considered to be controlled by NAC transcription factors rather than the direct reason for the albino phenotype.
Flavonoids, especially catechin contents are widely observed to be lower in albino tea leaves than those in normal green leaves. Recent transcriptomic analysis also identified that the expression levels of many flavonoid biosynthetic genes were significantly reduced in albino tea leaves, which was consistent with the metabolism data[7,34,35]. However, whether the decrease of flavonoid biosynthesis is the cause or result of tea albinism is a question waiting to be answered. Generally, flavonoids are considered to be biosynthesized in endoplasmic reticulum and then transported into vacuoles, which don't seem to be correlated with chlorophyll formation[36]. However, recently, Brillouet et al.[37,38] found the formation of catechins deposit in the chlorophyllous organs of Tracheophyta. This suggests the chloroplasts might also be an important organ for catechin biosynthesis or accumulation. If the same mechanism occurs in tea plants, the lower catechin content in albino leaves should be caused by less chlorophyll content and thereby less factories for catechin biosynthesis. On the other hand, Ma et al.[34] considered that the color change of an albino tea cultivar 'Huabai 1' might be a combined effect of phenylpropanoid biosynthesis pathway and other metabolic pathways including flavonoid biosynthesis. In this study, many key genes involved in flavonoid biosynthesis, such as 4-coumarate-CoA ligase, flavonol synthase and leucoanthocyanidin reductasegenes were found to be both differentially expressed and methylated in albino and normal leaves, which is consistent with the finding of Ma et al.[34]. Therefore, whether their expression changes result in, or are caused by, the reduction of chlorophyll content is still a question to answer.
It is also interesting that both CsMYB5 (TEA027333.1) and CsGSTF11 (TEA010512.1) were found to be differentially methylated and expressed in albino and green leaves in the 'Haishun 2' mutant. CsMYB5 was found to participate in the regulation of flavonoid biosynthesis[39]. While, CsGSTF11 was found to be significantly and positively correlated with catechin content in our previous study[24]. The homolog of CsGSTF11 in poinsettia was also reported to be closely correlated with flavonoid accumulation[40]. Our previous research found a coupled role for CsMYB75 and CsGSTF1 in anthocyanin hyperaccumulation in purple tea[1]. The CsGSTF1 is only involved in anthocyanin transportation and is controlled by CsMYB75. Their expressions control the anthocyanin accumulation in purple tea. Here, whether CsMYB5 and CsGSTF11 play similar roles in catechin accumulation is an interesting question for future studies.
-
In summary, we conducted an integrative analysis of metabolomics, transcriptome and DNA methylation analysis of the green and albino leaves in a tea mutant 'Haishun 2' and identified 385 overlapped genes. Two NAC transcription factors were less methylated and highly up regulated in the albino leaves of 'Haishun 2', which was the first identification of the key roles of NAC transcription factors in albino tea and worthy of further study. Many genes involved in catechin biosynthesis were down regulated in albino leaves, such as 4-coumarate-CoA ligase, flavonol synthase and leucoanthocyanidin reductase genes, suggesting chlorophyll formation is important for catechin biosynthesis. Furthermore, CsMYB5 and CsGSTF11 were found to be differentially methylated and expressed in albino and green leaves, indicating their potential roles in catechin accumulation (Fig. 8). Overall, the identification of these key genes deepened our understanding of the albino mechanism and catechin accumulation, which will facilitate molecular breeding of albino tea cultivars. Further exploration of these genes will also largely improve relative studies.
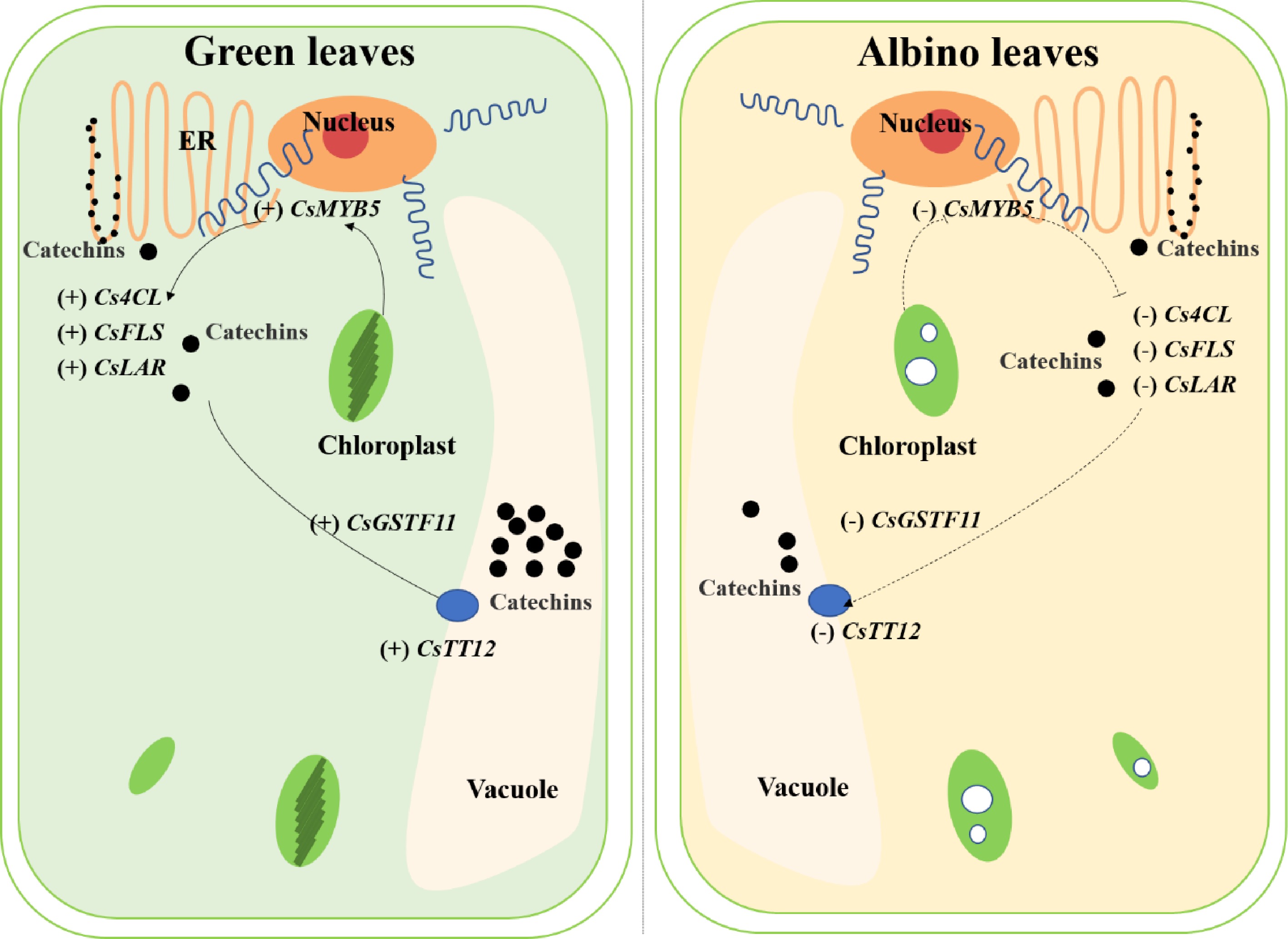
Figure 8. The potential mechanisms involved in catechin accumulation in green and albino leaves of 'Haishun 2'. 4CL, 4-coumaroyl CoA ligase; FLS, flavonol synthase; GST, glutathione S-transferase; LAR, leucoanthocyanidin reductase; TT12, transparent testa 12.
- We appreciate Mr. Haishun Guo for the use of tea plant 'Haishun 2'. This work was supported by the Central Public-interest Scientific Institution Basal Research Fund (1610212020002), the Major Project of Agricultural Science and Technology in Breeding of Tea Plant Variety in Zhejiang Province (2021C02067) and Earmarked Fund for China Agriculture Research System (CARS-19).
- The authors declare that they have no conflict of interest.
- Supplemental Table S1 Primers used for quantitative real time PCR analysis.
- Supplemental Table S2 Summary of RNA-Seq datasets of green and yellow leaves of Haishun2 tea mutant.
- Supplemental Table S3 Summary of BS-seq datasets of green and yellow leaves of Haishun2 tea mutant.
- Supplemental Table S4 Summary of mapping results to the published tea genome.
- Supplemental Table S5 Summary of cytosine methylation in C, CpG, CHG and CHH contexts.
- Supplemental Table S6 KEGG analysis of the overlap genes of DEGs and DMRs in gene bodies in ‘Haishun2’ tea mutant.
- Supplemental Table S7 KEGG analysis of the overlap genes of DEGs and DMRs in promoters in ‘Haishun2’ tea mutant.
- Supplemental Fig. S1 Correlation analysis of the RNA-Seq results.
- Supplemental Fig. S2 Venn diagrams of DMR genes.
- Copyright: © 2022 by the author(s). Exclusive Licensee Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Wei K, Yu S, Quan Q, Aktar S, He M, et al. 2022. An integrative analysis of metabolomics, DNA methylation and RNA-Seq data reveals key genes involved in albino tea 'Haishun 2'. Beverage Plant Research 2: 2 doi: 10.48130/BPR-2022-0002

An integrative analysis of metabolomics, DNA methylation and RNA-Seq data reveals key genes involved in albino tea 'Haishun 2'
- Received: 01 December 2021
- Accepted: 20 December 2021
- Published online: 10 January 2022
Abstract: Albino tea, a type of tea closely associated with the chlorophyll-deficient phenotype is of great interest due to its multiple benefits to human health. To explore the potential mechanisms involved in albino tea, we performed metabolomics, DNA methylation and RNA-Seq analysis of green and albino leaves in a special tea mutant 'Haishun 2'. The albino leaves accumulated significantly less catechins compared with the green leaves, which is closely associated with their difference in chlorophyll formation. A total of 385 candidate genes were identified by the integrative analysis. Two NAC transcription factors were less methylated and highly up regulated in the albino leaves of 'Haishun 2', which was the first identification of the key roles of NAC transcription factors in albino tea and worth further study. Many genes involved in catechin biosynthesis were down regulated in albino leaves, such as 4-coumarate-CoA ligase, flavonol synthase and leucoanthocyanidin reductase genes, suggesting chlorophyll formation is important for catechin biosynthesis. Furthermore, CsMYB5 and CsGSTF11 were found to be differentially methylated and expressed in albino and green leaves, indicating their potential roles in catechin accumulation. This was the first report of the integrative analysis of transcriptome and DNA methylation data in tea plants and the results suggest that the integrative analysis is useful for exploring key genes associated with epigenetic changes in tea plants.
-
Key words:
- Albino tea /
- DNA methylation /
- Transcriptome analysis /
- Catechin biosynthesis /
- Chlorophyll degradation