









-
Ornamental plants, including annual herbs, perennial herbs, ornamental shrubs, and trees[1], play an important role in the construction of human settlements by promoting urban and rural greening, regulating air humidity, absorbing harmful gases and dust, and preventing soil erosion[2,3]. The extracts of ornamental plants can also be used for cosmetics, foods, fine chemicals and other industries[4−6]. The breeding objectives of ornamental plants are focused on new shapes, new colors and new fragrances. Conventional breeding techniques, such as cross and mutation breeding, have produced a large number of new cultivars of ornamental plants. However, these traditional methods are time-consuming and laborious, the compatibility of distant hybridization is poor, and the results are unpredictable. Genomics is a new branch of biology, which takes DNA sequencing as its core technology, combined with genetics, molecular biology, and bioinformatics to accurately interpret genome information. The rapid development of genomics has pushed the breeding work to new heights. Through the precise location of functional genes and genetic transformation, we can potentially accelerate the breeding process and obtain new cultivars with favorable features.
The completion of the whole genome sequence of Prunus mume[7] fuels the development of ornamental plant genomics. Currently, more than 72 species of ornamental plants have been sequenced, including Rosa chinensis[8,9], Chrysanthemum nankingense[10], Phalaenopsis aphrodite[11], Paeonia suffruticosa[12] and other important ornamental plants, thanks to the rapid development of sequencing technology and the reduction in cost. Through the mapping and cloning of functional genes, and the study of gene expression and regulatory networks at the genome-wide level, researchers can systematically analyze the genetic mechanisms underlying important traits of ornamental plants. By analyzing genomic variation in plant populations, we can infer the population structure, population history, and environmental adaptability of ornamental plants.
In this paper, we review the recent progress in genome sequencing of important ornamental plants, including the sharing of genomic data and its application in ornamental character mapping and variety classification, and discuss the application prospects of new technologies. We pinpoint several challenges faced by ornamental plant breeding. We limit our review to the genomics of floral traits.
-
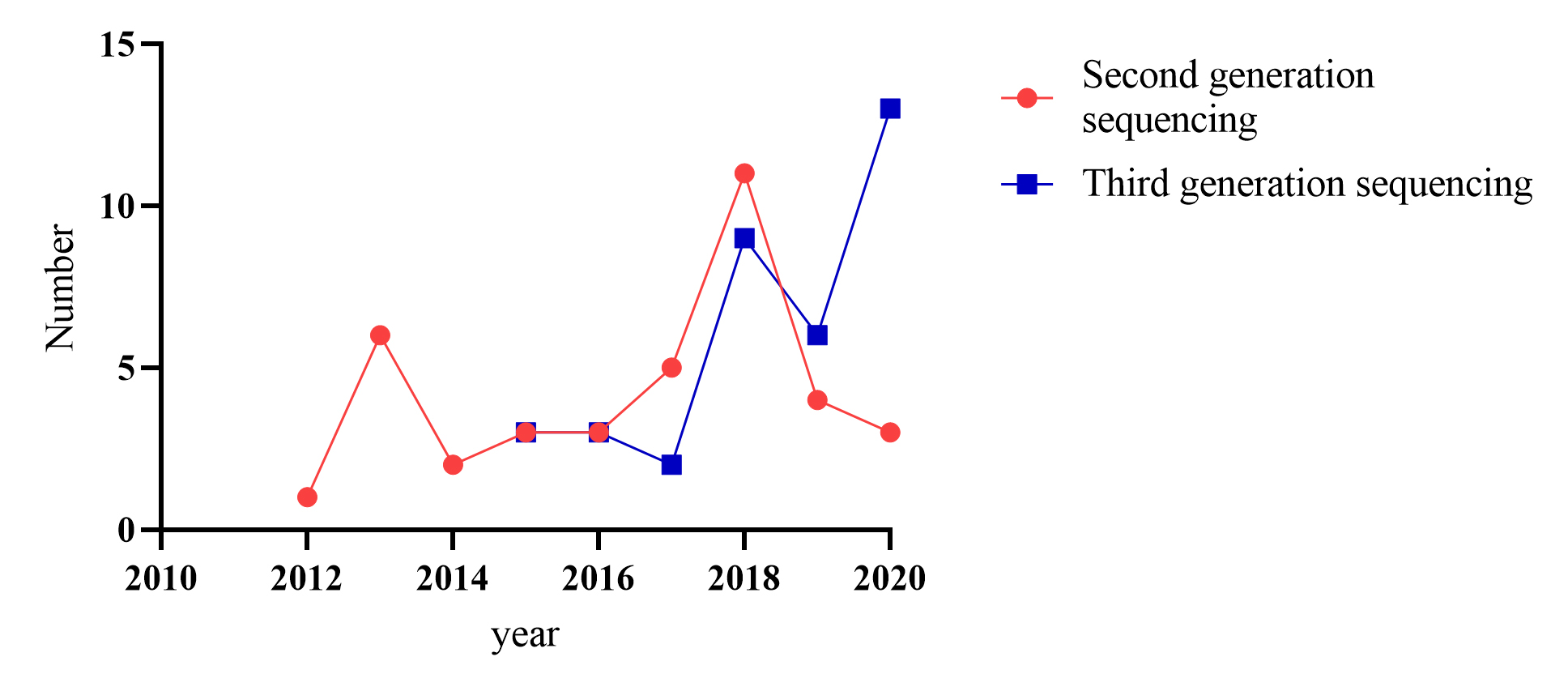
To date, at least 72 species of ornamental plants have been sequenced (Supplemental Table S1, Table S2), among which perennial herbs, shrubs, trees, and annual herbs account for 36%, 24%, 24%, and 17% (Table 1, Fig. 1), respectively. Sequenced species include 10 from Rosaceae, five from Orchidaceae, five from Asteraceae and six from Fabaceae (Table 2, Supplemental Table S3, Fig 2). Illumina sequencing (second-generation) has been the main sequencing technique of ornamental plants over the past decade. Yet, since 2015, third-generation sequencing technology has been applied, which markably reduces sequencing cost and improves sequencing quality (Supplemental Fig S1).
Table 1. Representative species of different tpye of ornamental plants.
Representative species Perennial herbaceous flowers Catharanthus roseus, Chrysanthemum nankingense, Nelumbo nucifera Annual herbaceous flowers Helianthus annuus, Ipomoea nil Flowering trees Osmanthus fragrans, Prunus mume, Prunus yedoensis Flowering shrubs Lavandula angustifolia, Paeonia suffruticosa, Rosa chinensis 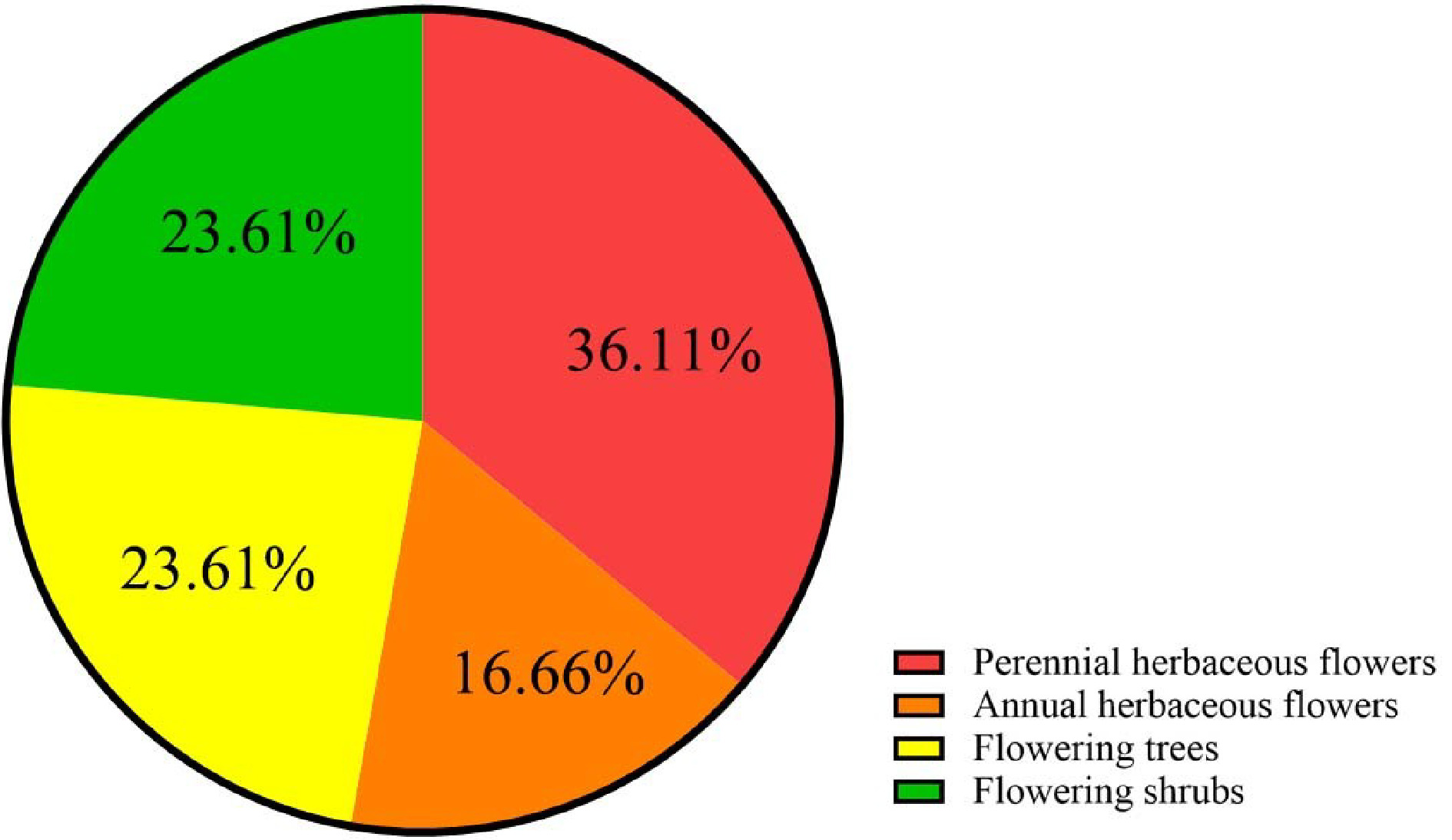
Figure 1.
The reported 72 ornamental plants species fall into 4 categories.
Table 2. Representative species of different family of ornamental plants.
Representative species Rosaceae Prunus mume, Prunus yedoensis, Rosa chinensis Fabaceae Ammopiptanthus nanus, Mimosa pudica Asteraceae Chrysanthemum lavandulifolium, Chrysanthemum nankingense, Helianthus annuus Orchidaceae Dendrobium catenatum, Phalaenopsis aphrodite 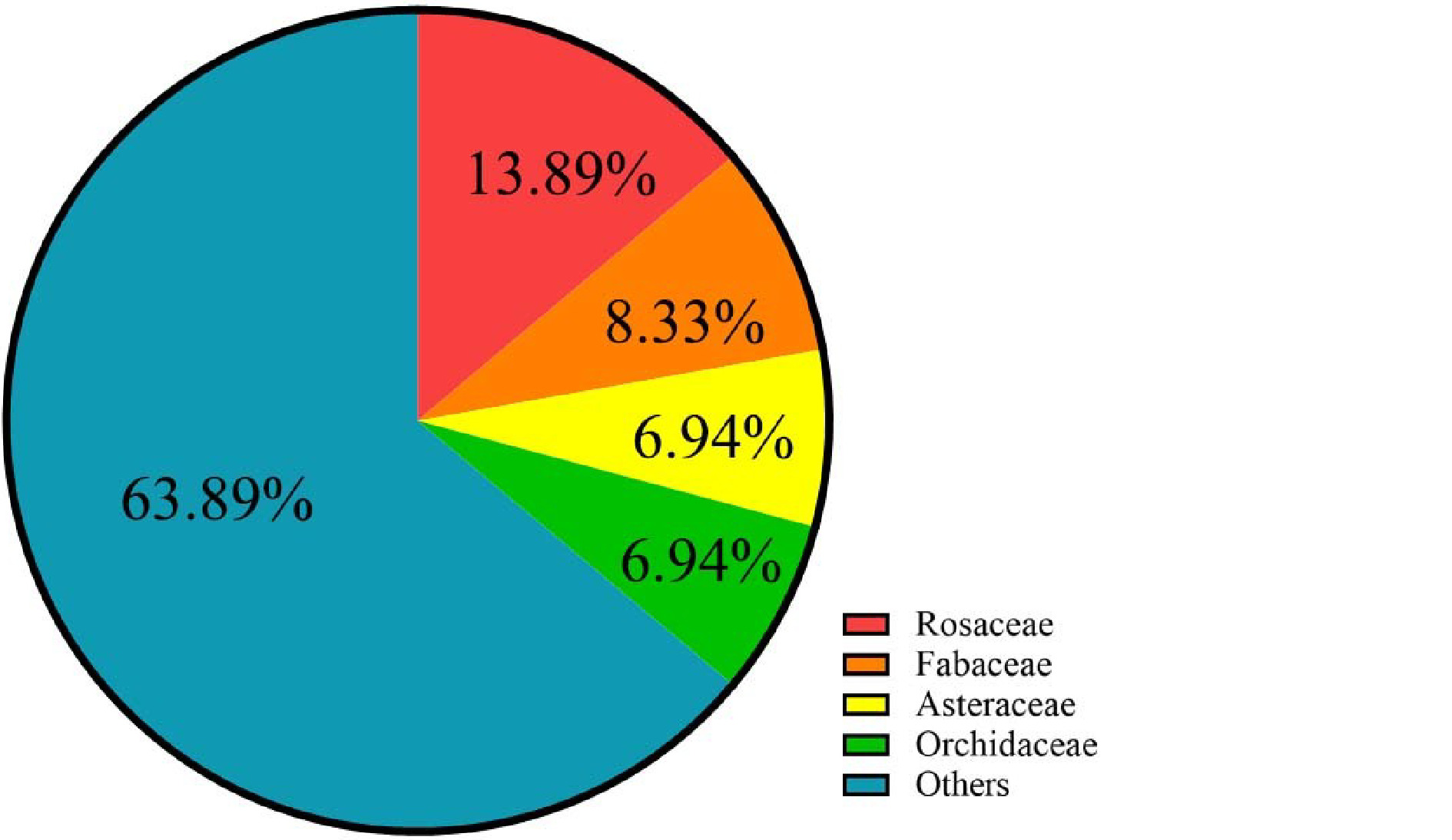
Figure 2.
The genome-sequenced ornamental species belong to 33 families.
Sequencing strategy of ornamental plants
-
In 1977, Maxam & Gilbert[13] reported a sequencing method by chemical degradation. In the same year, Sanger et al.[14] developed a method of DNA sequencing based on the selective incorporation of chain-terminating dideoxynucleotides by DNA polymerase during in vitro DNA replication. The application of these technologies implies the birth of a new generation of sequencing. The earliest plants sequenced, such as Arabidopsis thaliana[15], Oryza sativa[16], Zea mays[17], Prunus persica[18] adopt the method of first-generation sequencing. However, high cost, time-consuming and low-throughput limit its application, so second-generation sequencing with high throughput and low cost arises at a significant moment[19]. So far, second-generation sequencing represented by Illumina is still a popular method of sequencing. Among the sequenced ornamental plants, 36 species have adopted the second-generation sequencing strategy (Supplemental Table S1, Table S2). However, their limited ability to resolve large complex genomes derived from complex whole-genome duplications leads to many problems. First of all, short read-lengths make it hard to distinguish a large number of repeated sequences, it often leads to incomplete assembly, which brings great difficulties to the subsequent gene annotation and other bioinformatic analysis[20]. Secondly, second-generation sequencing methods depend on PCR amplification during library preparation on the flow cells[19], PCR is a bias-prone process that leads to GC bais. Under the requirement of high throughput, low cost and long read lengths, third-generation sequencing technology is gradually developing (Supplemental Table S4).
HeliScope is the first commercial single molecule sequencing platform, as the method was relatively slow, expensive, and produced short reads, it did not prove viable[19]. Single-Molecule Real-Time Sequencing (SMRT) and Oxford Nanopore Technologies (ONT) are two commercially successful third-generation sequencing techniques, both of which can produce genome assemblies of high quality without PCR amplification. Single-Molecule Real-Time Sequencing (SMRT) was developed by Pacific Biosciences, and takes advantage of the natural process of DNA replication, using zero-mode waveguide (ZMW) technology and gamma-labeled phosphonucleotides for direct observation of DNA synthesis on single DNA molecules in real time[21]. Single-Molecule Real-Time Sequencing offers longer read lengths than the second-generation sequencing technologies, making it well-suited for genome, transcriptome, and epigenetics research[21]. In 2014, Oxford Nanopore Technologies (ONT) released the MinION as a Nanopore sequencing device, it is much smaller than the current NGS platforms. MinION directly observe DNA or RNA bases by means of pores that are embedded in a membrane separating two compartments[22]. Although the MinION is easy to carry and can produce ultra-long reading length[23], because of the lower read accuracy and high cost, the application of Oxford nano-hole sequencing in plants is still in its infancy[19,24]. Although third-generation sequencing has the advantage of longer read length, its error rate is significantly higher than that of second-generation sequencing, which can be combined to make up for their respective shortcomings, which is especially beneficial to assemble the genome from scratch. This method is called hybrid sequencing[25]. Hybrid sequencing is widely used in plant sequencing. Among the sequenced ornamental plants, four species use Illumina+Nanopore strategy for genome sequencing, 31 species use Illumina+PacBio strategy and one species adopt BGISEQ-500+PacBio strategy (Supplemental Table S1, Table S2).
Considering species with complexity and high repeat sequence, it is very difficult to assemble a complete genome only by sequence reading[20], many researchers use Hi-C data to assist genome assembly. High-throughput chromosome conformation capture (Hi-C) is a technology that can study the three-dimensional architecture of whole genomes. By coupling proximity-based ligation with large parallel sequencing, Hi-C allows unbiased identification of chromatin interactions across an entire genome[26]. The many advantages of Hi-C technology have attracted the wide attention of researchers, the assembled genomes of Rhododendron williamsianum[27], Rhododendron simsii[28], Rosa chinensis[9], Osmanthus fragrans[29], Chimonanthus praecox[30], Chimonanthus salicifolius[31] are assisted by Hi-C technology. At present, two sequenced white lupine genomes provide us with a good example. The genomes using Hi-C data not only have higher assembly integrity, but also have better chromosome collinearity[32,33].
Genome database of ornamental plants
-
The Arabidopsis Information Resource (TAIR,
http://www.arabidopsis.org ) was launched in 2001, and is the first angiosperm genome database[34]. With the development of the ornamental plant genome sequencing project, various ornamental plant genome databases have been established. The ornamental plant genome database can be further classified as four different types: family database[18,35]; single species database[18,33,35]; clade-oriented database[36,37] and comprehensive database (Supplemental Table S1, Table S2).The single species database of individual species not only provides the updated genome sequence, but also provides a series of bioinformatics tools for online analysis. For example, the Nelumbo Genome Database (NGD,
http://nelumbo.biocloud.net )[38] contains the most updated lotus genome assembly and information on both gene expression in different tissues and coexpression networks. In addition, genetic variants and phenotypes of 88 key lotus cultivars were integrated. GBrowse provided by NGD can easily view genes, DNA sequences, amino acids, SNPs and Indels. Moreover, the website also provides tools such as blast, blat, and Primer for sequence analysis and search. The database is helpful to the study of population genetics and molecular breeding of lotus, as well as a comprehensive understanding of the genetic variation characteristics of the Nelumbo nucifera genome. The history of cultivation and domestication of lotus can be traced back to the data in order to cultivate new varieties with high yield and high quality and to improve the level and speed of breeding techniques. Unfortunately, among the sequenced ornamental plants, only a few plants such as Helianthus annuus[38], Ipomoea nil[39] and Dianthus caryophyllus[40] have an established database, and the database of many species is far less perfect than that of the Nelumbo genome database.Compared with the single species database, the database integrated according to families/clade-oriented pays more attention to the integration and unification of different plant data. The Genome Database for Rosaceae (GDR,
https://www.rosaceae.org )[41] first established in 2003, after decades of development, has significantly expanded data and functions centering on Rosaceae plant interaction. GDR contains detailed data on published trait loci in Rosaceae, based on these trait terms, the GDR team developed the Rosaceae Trait Ontology that is tightly linked to the Plant Trait Ontology (TO). Except for this, QTL in GDR also includes aliases, curator-assigned QTL labels, published symbols, trait names, taxa, trait descriptions, screening methods, map positions, associated markers, statistical values, datasets, contact information and references. At the same time, the website supports viewing and statistics of these data according to genus units. The Breeding Information Management System (BIMS) is a Tripal module developed by the GDR team, it provides breeding teams with tools to store, manage, archive and analyze their private or public breeding data, and other breeders can also use BIMS to query and download public breeding data. At present, GDR includes genome assembly and annotation data of five ornamental plants, including Cerasus × yedoensis, Malus baccata, Rosa multiflora, Rosa chinensis, Prunus persica, as well as 24 crops, including apricot, apple and strawberry.A family database or clade-oriented database has the disadvantage of incomplete data. At present, GDR lacks the genome data of Prunus mume, Dryas drummondii and Prunus yedoensis. OrchidBase lacks the genome data of Dendrobium officinale and Phalaenopsis Aphrodite. The pomegranate genome lacks two cultivars, 'Tunisia' and 'Dabenzi', in the Hardwood Genomics Project. Many comprehensive plant databases have been established, and the more representative ones include Phytozome[42], PGDD[43], PLAZA[44], and Ensemblplants[45], containing most of the published plant genome information.
The genome data of ornamental plants are growing rapidly in quantity and complexity, and its main function has evolved from data storage to online analysis[46]. The excellent genome database will provide great convenience for the study of structural genomics, comparative genomics and functional genomics of ornamental plants.
-
In recent years, a large number of plant genome sequencing and resequencing projects have promoted the development of comparative genomics. By comparing the genome sequences of different related species, we can identify the contraction and expansion of gene families[47], estimate the divergence time of species[48], identify gene fusion and gene cluster[10,49] and Analysis system evolution[8]. The study of comparative genomics of ornamental plants will help to speed up the basic research of their biology and improve the efficiency of the development and utilization of wild related species resources.
Gene replication event
-
A variety of evidence shows that gene replication can promote the rapid recombination of plant genome, lose a large number of genes and increase structural variation, which is extremely important for plant evolution[50]. The study of gene replication in ornamental plants has important guiding significance for their genetics and breeding.
Gene duplication includes tandem, tetraploid, segmental and transpositional[51]. Tandem repeats are highly prevalent at centromeres[52], to create a cluster of homologous genes with similar sequence and function, which are arranged in series on the chromosome in a head-to-tail manner[53]. There is evidence that tandem duplication leads to widespread TS and CYP gene clusters in the chrysanthemum genome, which greatly promotes the diversity of terpenoids in chrysanthemum[10]. Segmental duplication is the most important structural variation on chromosomes[54]. Repetitive region caused by segmental duplication is usually the repetition of all genes in a large region, rather than the duplication of single or several genes. For individual gene families, fragment replication also plays a major role in species-specific expansion[55−57]. DNA binding with one figure (Dof) proteins play important roles in various biological processes and defense regulatory networks in plants. Dof genes of many plants came primarily from segmental and duplication events[55,56]. A total of 24 full-length RchDof genes were identified in Rosa chinensis, seven pairs of RchDof duplicated genes (12 RchDof genes) were generated by segmental duplication[57]. Transposable elements (TEs) is an important part of the plant genome. They can replicate independently and insert replicative fragments into many different loci on other DNA sequences in the same cell[58]. The long terminal repeat (LTR) retrotransposon in Phalaenopsis equestris were identified and six high-copy retrotransposons were found[59]. Among them, Gypsy1, Gypsy2, Gypsy3, and Orchid-rt1 have amplified within Phalaenopsis orchids concomitant with the expanded genome sizes, and Orchid-rt1 and Gypsy1 may experience various events of mutation and homologous recombination. Polyploid or whole genome duplication is a common phenomenon in plants, which promotes the evolution and diversity of organisms[60]. WGD events played an important role in regulating the evolution of chrysanthemum. It was found that many ornamental and medicinal traits of chrysanthemum were related to gene replication[10]. In addition, gene replication is closely related to flower fragrance[30], flower shape[61], flowering[62], flower development[10,63], resistance[64] and circadian rhythm[65].
Gene duplication usually results in three different situations: becoming a pseudogene; acquiring a novel function; or subfunctionalization[66]. However, random mutations may not be helpful to human social production. Some studies have suggested that the polyploidy formed by whole genome replication leads to a greater phenotypic breadth in which natural and artificial selection can work, thus making successful domestication possible[67]. Through the domestication of ornamental plants, the variation traits that meet people's needs have been continuously accumulated and strengthened, and the wild varieties have finally formed high-quality modern cultivated species.
Evolution and domestication
-
Domestication and evolution have profoundly shaped the genomic structure and genetic diversity of crops today. The in-depth study and exploration of the genetic mechanism of crop domestication can help us to understand and identify the wild ancestral species of ornamental plants[68]; determine the time and place of the domestication[69]; clarifiying whether it comes from a single domestication event or multiple domestication events[69,70]; understanding the differences in morphological, physiological and biochemical characteristics between ornamental plants and their ancestral species, as well as the genetic basis for such differences[71]; evaluating the changes and causes of the genetic structure of crop populations compared with their ancestral species[72].
Rosaceae contains nearly 3,000 species, many of which have been domesticated by humans thousands of years ago. The whole genome sequence of Prunus mume was assembled and nine ancestral chromosomes of Rosaceae were reconstructed[7]. It is speculated that they evolved from the same ancestor[7]. Based on the resequencing of Prunus mume, the phylogenetic tree of Prunus mume was constructed by using the core genes of 13 species of Prunus and three related species of Rosaceae, and the evolutionary history of Prunus was reconstructed. The results show that the divergence times between Prunus mume and other Prunus species as ~3.8 Mya and that between wild Prunus mume and cultivated Prunus mume is ~2.2 Mya. It is concluded that divergent selection may have played a role in the differentiation of these two subspecies long before the domestication of Prunus mume[48]. Four thousand to five thousand years ago, peach experienced a primitive domestication bottleneck in China, which showed a significant decline in the diversity of wild Prunus davidiana and the domesticated Asian cultivar[73]. The cultivation and domestication path of peach was analyzed based on the whole genome of peach and 14 other species of Prunus resequenced. It is considered that the effect of the second bottleneck is clearly reflected in the reduction of nucleotide diversity observed during the transfer from eastern to western cultivar. Compared with eastern and wild relatives, these bottlenecks seem to lead to a serious loss of diversity of western varieties, as well as obvious deficiencies of rare varieties and relatively slow LD decay[18]. The de novo chromosome-level reference genome of Cerasus serrulata was assembled[74]. Through 656 single-copy orthologs, the authors constructed a phylogenetic tree containing 10 species. It was found that Cerasus serrulata diverged from Prunus yedoensis about 17.34 million years ago, while Cerasus serrulata and Cerasus avium diverged from 21.44 million years ago. These results confirm that Cerasus serrulata are more closely related to Prunus yedoensis.
Lotus is an important aquatic plant, which has many values such as medicine, edible and ornamental. Nineteen individuals of lotus, including three cultivated temperate lotus (rhizome lotus, seed lotus and flower lotus), one wild temperate lotus (wild lotus), one tropical lotus (Thai lotus) and one outer group (Nelumbo lutea). Through the analysis of genetic diversity, it was confirmed that wild Thai lotus showed greater differentiation and higher genomic diversity than cultivated lotus[75]. A recent re-sequencing study of 296 lotus germplasm resources further confirmed this conclusion. The analysis of population structure and genomic diversity of three types of cultivated lotus: rhizome, flower, and seed lotuses showed that the seed and rhizome lotus groups had not originated from a single source, but had a more complex multi-source[76].
-
With economic development, people have put forward more requirements for the color, fragrance and shape of ornamental plants. In recent decades, breeders are committed to identifying important ornamental gene loci and regulation patterns. Thanks to the high-quality reference genome, once the ornamental major genes are located, the corresponding molecular markers can be developed, and the mutant plants can be obtained by genetic transformation (Supplemental Table S5). These genes related to important traits are reviewed below.
Phenological traits
-
Plant phenology, including flowering phenology, is an important parameter for crop growth monitoring, yield prediction and growth simulation[77]. Understanding the phenology of ornamental plants, especially flowering phenology, can help producers determine the time of sowing, hybridization and harvest. Therefore, many studies have been devoted to analysis of the molecular mechanisms of bud dormancy, flowering, flower development and flowering regulation.
Dormancy is an adaptive process that enables plants to withstand difficult environmental conditions in winter temperate climate[78]. The study of the molecular mechanism of dormancy is of great significance for perennial temperate ornamental plants. However, compared with other crops, the molecular mechanism of ornamental plants affecting dormancy is still in the primary stage, and the dormancy studies of many important ornamental plants remain in the stage of physiological experiments. At present, only Paeonia suffruticosa[79], Prunus mume[80], Prunus persica[81] and a few others have isolated dormancy-related genes. The verification of gene function is not enough due to the lack of genetic transformation systems. The CBF gene of Prunus persica was isolated and overexpressed in Malus × domestica. Compared with the untransformed apple lines, the transgenic plants with the PpCBF1 gene showed growth cessation and leaf senescence under short-day treatment. This is the first example of short-day light-induced dormancy caused by structural overexpression of the CBF gene[82]. MADS-box gene (PmDAM6) was found expressed in the lateral buds of Prunus mume[83]. Overexpression of PmDAM6 in transgenic poplars showed that the plants exhibited growth cessation and terminal bud set when the stem tips of the control plants continued to grow, indicating that PmDAM6 has the function of growth inhibition[84].
After the release of dormancy, flowering is a key event in the life cycle of seed plants, and there are six main ways to regulate flowering, including vernalization and autonomous pathways, photoperiod pathway and the circadian clock, gibberellin pathway, ambient temperature pathway, age pathway and meristem responses[85].The mechanisms of the six pathways have been well studied, many genes associated with them have been located in ornamental plants. Among them, a large number of genes have been verified. FLC, a MADS-box protein, is a key inhibitor in vernalization and autonomous pathways[86]. An AGL6-like gene CpAGL6, was cloned from Chimonanthus praecox. The transgenic experiment showed that the ectopic expression of CpAGL6 inhibited the vegetative growth of Arabidopsis thaliana and led to early flowering, which was mainly related to the inhibition of floral inhibitor FLC and floral promoters AP1 and FT[87]. In 1998, the LHY gene was confirmed to be related to the photoperiod pathway and the circadian clock of Arabidopsis thaliana[88]. LHY, TOC1 and CCA1 constitute the core negative feedback loop of the plant circadian clock[89]. A LHY/CCA1-like gene was identified in chrysanthemum and named CsLHY, CsLHY was fused with a gene encoding a short transcriptional repressor domain (SRDX) and constitutively expressed in chrysanthemum. The results showed that although the flowering transition was insensitive to photoperiod in CsLHY-SRDX transgenic plants, the further development of capitulum was blocked and no flowering was observed[90]. Gibberellin (GA) is an important plant hormone, which can promote flowering in annual and biennial plants, and may inhibit flowering in woody perennials[91]. Della protein is a repressor directly acting on the downstream of GA receptor, regulating plant growth and development induced by GA[91].Two Della homologous genes PmDELLA1 and PmDELLA2 in Prunus mume are overexpressed in Arabidopsis thaliana, resulting in plant dwarfing and flowering delay, indicating that these two Della proteins are negative regulators of GA signal in Prunus mume[92]. SVP gene plays an important role in the control of flowering time by ambient temperature[93]. A SVP homologous gene LaSVP, in Lavandula angustifolia was confirmed. The expression of LaSVP in Arabidopsis thaliana delayed flowering and affected the floral dosage-dependent in a dosage-dependent manner. MicroRNA156-mediated spl transcription factors can ensure flowering time under non-inductive conditions, which is an important age pathway[94]. It is found that CmNF-YB8 gene can regulate the early transformation of chrysanthemum from juvenile to adult, as well as early flowering, regardless of day-length conditions. In this process, members of the cmo-miR156 and SPL families, the core components of the age pathway, are involved[95]. During floral induction, the apical meristem changes from vegetative meristem to inflorescence meristem, which is related to the increased expression of SOC1 encoding MADS-box transcription factors[85]. Six homologous SOC1 genes were isolated from four tree peony cultivar, including PrSOC1, PdSOC1, PsSOC1, PsSOC1-1, PsSOC1-2 and PsSOC1-3. The ectopic expression of PsSOC1 in tobacco showed that the high expression of PsSOC1 in transgenic tobacco not only promoted plant growth, but also advanced flowering time[96]. It should be pointed out that in the process of flowering induction, several genes play a role in several tissues more than once, and these genes converge into a complex regulatory network of flower development.
The longevity of cut flowers is an important trait that determines the sale of ornamental plants. Consumers appreciate long-lasting flowers, and the sales industry hopes to reduce the deterioration of flower quality in the distribution chain[97]. Senescence, as the last stage of flower development, determines the lifespan of ornamental plants. It has been found that the occurrence of aging is regulated by a number of phytohormones, including ethylene, abscisic acid (ABA), jasmonic acid (JA) and polyamine (PA), and cytokinin (CTK), which can inhibit aging[98]. The pathway of ethylene synthesis is relatively simple, in which ACC serves as the unique precursor, ACC synthase (ACS) and ACC oxidase (ACO) are key enzymes[99]. Many studies on the molecular mechanism of ethylene-mediated flower senescence focus on these three key enzymes. At present, several studies have confirmed that ACC synthase and ACC oxidase are closely related to the senescence of ornamental plants[100,101], furthermore, the regulation mechanism of ethylene on flower senescence has been well verified in many plants, such as Petunia hybrida[102], Ipomoea nil[103], Paeonia suffruticosa[104], and Dianthus caryophyllus[105,106]. However, many ornamental plants are not regulated by ethylene, ethylene has little effect on petal senescence in lilies, tulips, chrysanthemums and gladiolus[107]. ABA is considered to be an important regulator of petal senescence in ethylene-insensitive ornamental plants[108], and the effect of abscisic acid on senescence has been proved in many ethylene-insensitive ornamental plants such as gladiolus[109] and lily[110]. It should be noted that it is not a single phytohormones that regulates senescence in ornamental plants. Multiple hormones antagonize each other and jointly regulate the senescence process of plants[111]. At present, there have been many studies on the molecular mechanism of the antagonistic role of plant hormones such as gibberellin[112,113] and salicylic acid[114] in the process of petal senescence.
Flower shape
-
Flowers are unique reproductive organs of angiosperms. For the wild species, the evolutionary direction of plants has always been to promote the reproductive success of plants by changing the shape, color, smell and reward to attracting pollinators[115]. However, the wild species do not fully meet people's needs, and the customer's demand for purposeful and unique horticultural plants urges scientists to constantly improve the shape of flowers. The morphology of ornamental plants mainly includes floral organ structure, flower branch growth state, inflorescence type and plant morphology. Improving floral size, double flowered, number of flower branches and inflorescence type is an important direction of morphological improvement.
In angiosperms, the basic developmental system of floral organs is explained by the ABCDE model[116]. In this model, the genes related to flower development are divided into five categories, namely A, B, C, D and E, in which A- and E-class determine sepal development, A-, B- and E-class determine petal development, B-, C- and E-class determine stamen development, C- and E-class determine carpels development and D- and E-class determine ovule development. Plants such as Antirrhinum majus, Petunia hybrida and Arabidopsis thaliana have made important contributions to the proposal and perfection of flower development models[117−119], its own genes related to flower development have also been well studied. A large number of genes related to flower development are located in model plants which provides great convenience for the subsequent study of genes related to other ornamental plants. Taking Antirrhinum majus as an example, after the B-class genes DEF and GLO were first located in Antirrhinum majus, its homologous genes were also cloned in Phalaenopsis Aphrodite[120], Eustoma grandiflorum[121], Torenia fournieri[122] and other plants. At present, A-[123], B-[124], C-[125,126], D-[126], and E-[87,127] class genes in ornamental plants has been verified by transgenic plants. In chrysanthemum, 14 C-class genes were cloned, of which seven belong to the CAG1 gene and seven belong to the CAG2 gene[128]. The chimeric repressors silencing technique was used to knock out C-class in chrysanthemum to form a multiple-petal phenotype. The expression of CAG1s and CAG2s chimeric repressors led to the morphological changes of pistil and stamens forming petaloid organs in disk florets[128]. In orchid, four SEP-like genes were cloned and divided into PeSEP1/3 and PeSEP2/4. Transcriptome data showed that all PeSEP genes were expressed in all flower organs. When PeSEP3 was silenced by VIGS, the sepal became leaf-like organs, and the characteristics of the epidermis and the content of anthocyanin and chlorophyll changed, while the silencing of PeSEP2 had little effect on flower phenotype[129].
The floral size and the number of flowers may be the most easily observed traits, which directly affect the visual effect of the whole plant. At the same time, the size of flowers and the number of flowers also have an important effect on plant pollination[130]. However, there are few studies on the gene mapping of these traits in ornamental plants, only in Petunia hybrida[131,132], Helianthus annuus[133,134] and Aquilegia[135].
The perianth, including the calyx and corolla, are the most prominent parts of a flower. The corolla of most plants has a beautiful shape and bright colors, which is the main ornamental part. A small number of plants, such as Strelitzia reginae, Begonia fuchsioides belong to calyx ornamental plants. The study of the number of petals (including double flower and other traits) and the number of calyx is of great importance to improve the ornamental quality of flowers. Through GWAS, QTL and other methods, scientists have mapped a number of genes related to the number of petals in Dianthus caryphyllus[125], Rosa chinensis[8,136,137], Prunus mume[48], Prunus persica[138] and other plants, and genetic transformation verification is also under way. An APETALA2 homologous gene RcAP2 from rose was overexpressed in Arabidopsis thaliana, increasing the number of petals. In addition, after silencing the RcAP2 gene in 'Old Blush', the petal number decreased significantly[136].
The corolla of ornamental plants has a variety of shapes. Except for a few asymmetric flowers, most of the corolla are classified as radially symmetrical, Cruciform, Caryophyllaceous, Rosaceous, Campanulate, Tubular, Infundibuliform, Hypocrateriform, Urceolate, Rotate, and bilaterally symmetrical Butterfly-like or Papilionaceous, Ligulate, Bilipped or Bilabiate corolla. The molecular basis of flora symmetry, including related processes such as development, life cycle, and metabolism, is regulated by a specific transcription factor (TF)[139]. In core dicotyledonous plants, CYC2 is the main regulator of flower symmetry, which can independently control the bilaterally symmetrical traits of flowers[140−142]. Some studies have shown that the CYC2 gene is at least involved in the evolutionary transformation of the symmetry of Brassicales, Malpighiales, Dipsacales, Asterales and Lamiales[143]. In addition, some genes such as RAD and DIV belonging to the MYB family also control symmetry[144]. In Antirrhinum majus, the antagonistic action that RAD has over DIV by competing for the DRIF proteins preventing the formation of the DRIF-DIV complex and, consequently, in the establishment of an asymmetric pattern of gene activity in the Antirrhinum flower meristem[145]. Six CmCYC2 genes in chrysanthemum were identified[146]. It was found that the ectopic expression of the CmCYC2 gene in Arabidopsistcp1 mutant changed the symmetry and flowering time of Arabidopsis thaliana[146].
Organ fusion is another way of flower shape change, which can lead to greater morphological diversity of closely related species[147]. Fusion occurs in two ways: when the organ primordium cannot be separated from the floral meristem, or at the later stage of the individual primordium or even the full formation of the organ, the 'fusion program' was initiated to enable the establishment of a permanent connection between them[148]. The genesis of fusion organs is thought to be mediated by NAC transcription factors from the NAM/CUC3 subfamily and microRNA from the miR164 family, which mediates the formation of boundaries during meristem development[149−151]. This conclusion has been further confirmed in model plants such as Antirrhinum majus[152], Petunia hybrida[153], Arabidopsis thaliana[154] and so on.
Flower color
-
Flower color is the most important feature of plants to attract pollinators[155]. At the same time, the bright color of flowers is also an important factor to attract consumers. Although natural ornamental plants have plentiful colors, the colors of some important ornamental plants are limited. For example, roses and chrysanthemums lack blue varieties[156], mei and morning glory lack bright yellow[157], Cymbidium hybrida lack orange and brick-red[158]. Therefore, flower color improvement has always been an important goal for breeders. The main substances that determine flower color are the following three kinds: flavonoids, carotenoids and betaine[159].
The most important substance of flavonoids is anthocyanins, which determine the pink, red, violet and blue of flowers[160]. It is the main contributor to color designation. It has been found that the key structural genes that catalyze the early and later stages of anthocyanin biosynthesis include phenylalanine ammonia lyase (PAL), cinnamate-4-hydroxylase (C4H), 4-coumarate: CoA ligase (4CL)chalcone synthase (CHS), chalcone isomerase (CHI), flavanone-3-hydroxylase (F3H), flavonoid 3'-hydroxylase (F3'H) and flavonoid 3',5'-hydroxylase (F3'5'H), flavonol synthase (FLS), UDP-glucose: flavonoid glucosyltransferase (UFGT) and methyl transferase (MT)[159,161−163]. Many of these genes have been identified in ornamental plants. In chrysanthemum, seven structural genes CHS, F3H, F30H, DFR, ANS, 3GT and 3MT regulated by transcription factors CmMYB5-1, CmMYB6, CmbHLH24 and CmMYB7-1 were identified as key genes for anthocyanin biosynthesis[164,165]. In cultivated chrysanthemum, due to the lack of delphinidin-based anthocyanins, no violet/blue chrysanthemum was found. F3′5′H gene was introduced into eight chrysanthemum lines by Agrobacterium tumefaciens transformation, and it was found that the petal color of all cultivars turned blue due to the accumulation of delphinidin[166].
Carotenoids are isoprenoid molecules related to plant colors such as yellow, and orange to red[167] and accumulate in nonferrous bodies in the form of fat-soluble products. Structural genes in the carotenoid biosynthesis pathway have been well described in a variety of plant model systems[168−170]. Phytoene synthase (PSY), phytoene desaturase (PDS), ζ-carotene isomerase (Z-ISO), carotene isomerase (CRTISO), geranylgeranyl pyrophosphate synthase (GGPPS) are important enzymes involved in the synthesis of carotenoids[171]. The crtW gene encodes β-carotene ketolase (4,4'-b-oxygenase), which can guide the synthesis of pink to red carotenoid pigments. This gene is commonly found in bacteria, fungi and unicellular algae[172]. The crtW gene was isolated from marine bacteria and modified the carotenoid biosynthesis pathway of lotus. In transgenic plants, the color of petals changed from light yellow to dark yellow or orange[173]. In addition, carotenoid biosynthesis genes in many ornamental plants such as Narcissus pseudonarcissus[174], Primula vulgaris[175] and Dianthus caryophyllus[176] have also been identified. In addition, there is evidence that the biosynthesis pathway of flavonoids and carotenoids may be regulated by MYB transcription factors[175,177,178].
Betaine is a nitrogenous compound whose colors range from red-violet betacyanins to yellow betaxanthins[179]. Similar to flavonoids, betaine accumulates in the vacuoles. At present, only in Caryophyllales (except for Caryophyllaceae and Molluginaceae) are stained with betaine[180,181]. Compared with other major types of plant pigments, the study on betaine biosynthesis lags seriously[180], the core biosynthetic pathway of these pigments has not been fully elucidated until recently[182,183]. At present, the research on the regulation and synthesis of betaine genes is mainly focused on crops such as Beta vulgaris[184,185] and Hylocereusundatus[186,187]. Only Mirabilis jalapa[188,189] has been researched in ornamental plants. The key enzyme gene in betaine biosynthesis was isolated from Mirabilis jalapa: MjDOD, and transformed it into E. coli by constructing a vector containing MjDOD gene, which induced its expression, and successfully obtained betaine[189].
Flower fragrance
-
Flower fragrance is an important trait of ornamental plants. The fragrance of plants can make people feel good, and the flower volatile compounds related to flower aroma are also widely used in perfumes, medicine and condiments among other things. At the same time, flower fragrance can also attract pollinators and promote flower fertilization[190]. More than 1,700 flower volatile organic compounds have been identified in 1,000 species of angiosperms[191], which come from a few synthetic metabolic pathways, including terpenoids, phenylpropanoids/benzenoids, and fatty acid derived biosynthesis pathways.
Terpenoids, especially monoterpenes such as linalool, limonene, myrcene, and trans-b-ocimene, but also some sesquiterpenes such as farnesene, nerolidol, and caryophyllene, are common components of floral fragrance[192]. In orchids, the PbGDPS gene was identified in Phalaenopsis bellina, which can produce both precursor of monoterpenes (GDP) and the precursor of sesquiterpenes (FDP)[193,194]. Chan et al. identified the transcripts related to fragance in Vanda Mimi Palmer and found that the clones of 32 transcripts were related to sesquiterpene synthase, germacrene D synthase and tyrosine decarboxylase[195]. In peony, four wild tree peony species were profiled as integrative volatile, including Paeonia ostii, Paeonia rockii, Paeonia delavayi and Paeonia lutea. A total of 67 floral volatiles were identified and the terpenoids in Paeonia ostii and Paeonia rockii were the most abundant. Transcriptome sequencing showed that there were 17,967 DEGs, of which 116 were related to the accumulation of terpenoids. Among them, 1-deoxy-D-xylulose 5-phosphate synthase, geranyl pyrophosphate synthase, farnesyl pyrophosphate synthase and terpene synthase may be the main regulatory enzymes of Paeonia ostii and Paeonia rockii terpene biosynthesis[196].
Phenylpropanoids/benzenoids are the second largest group of flower fragrance components[191]. The biosynthesis of these compounds is regulated not only by the time and space of flower development, but also by the rhythmic and the biosynthesis of precursors[197]. Phenylpropanoids/benzenoids are the main sources of fragrance in plants such as Prunus mume[198], Petunia hybrida[199], Syringa vulgaris[200]
and Magnolia champaca[201]. At present, many coding enzyme genes involved in this pathway have been cloned and verified. Taking Petunia hybrida as an example, Ph BSMT1, Ph BSMT2, Ph BSMT3, Ph IGS1, Ph EGS1, Ph PAAS, Ph BPBT, Ph CFAT, Ph BSMT1 and Ph BSMT2 are known to catalyze the formation of benzoic acid and salicylic acid, which are the components of many plant fragrances[197,202−207]. At the same time, the transcription factors ODO1[199], EOBI and EOBII[208] of phenylpropanoid-related compounds have been proven by genetic transformation experiments. In Prunus mume, benzyl alcohol and benzyl acetate were found to be the main sources of the aroma of Prunus mume. No benzyl alcohol or benzaldehyde reductase (BAR) activity was detected in the fragrant variety 'Fenghou'. It is inferred that the lack of benzyl alcohol synthesis of the 'Fenghou' variety is due to the low activity of PmBAR1-Fen and the low expression of PmBAR3[209]. The proportion of fatty acid derivatives in plant volatiles is lower than that of terpenoids, phenylpropanoids / benzenoids. It is found that aliphatic compounds play an important role in the aroma sources of Dianthus caryophyllus[210], Centaurea aeolica[211], Plumeria rubra[212] and Antirrhinum majus[213]. Fatty acid derivatives in ornamental plants are mainly synthesized by C18 fatty acids, including linolenic and linoleic acid. The biosynthesis of fatty acid derivatives begins with stereospecific oxygenation catalyzed by the lipoxygenase (LOX) pathway[214]. At present, the studies on fatty acid derivatives are mainly focused on genes involved in the lipoxygenase pathway[215], but there are few studies on expression in flower organs.
Stress resistance
-
Stress generally refers to biotic or abiotic stresses. Abiotic stresses including diseases, pests and weeds, and abiotic stresses include cold, high temperature, drought, waterlogging, and salinization. The earths' climate is diverse and complex, and the vegetation in different regions face different stresses, which pose a severe challenge to the production of ornamental plants.
At present, the main strategies for crop disease control are still highly dependent on chemical pesticides. However, all kinds of pesticides usually cause direct or indirect harm to human beings and the natural environment, and it is a strong desire of consumers to move towards an environmentally friendly and healthy production system. In this context, the use of gene editing technology to obtain disease-resistant varieties accurately and efficiently has become the primary breeding strategy. A variety of ornamental plants have acquired the ability to resist diseases and insect pests through transgenic technology. In chrysanthemum, insect-resistant transgenic chrysanthemum plants were obtained by transferring the Bacillus thuringiensiscol1Ab gene[216]. Chrysanthemum containing the rice chitinase gene was transformed by Agrobacterium tumefaciens. It was found that their resistance to Botrytis cinerea was enhanced[217]. Transformants of chrysanthemums with resistance to cucumber mosaic virus can produce better flowers when attacked by the virus[218]. In lily, in addition to the study of anti-fungal[219] and anti-viral[220], there are also studies on nematode resistance[221,222]. Genetic transformation of disease resistance was also studied in Gladiolus[223−225], Orchidaceae[226−228], Petunia hybrida[229], Rosa chinensis[230] and other ornamental plants.
Different climatic conditions put forward different requirements for the survival of ornamental plants, such as cold tolerance in northern areas and drought tolerance in desert areas. In order to ensure ornamental plants have a broader living space, it is imperative to improve plant resistance. In many plants/organisms, some genes have been found which encode and synthesize these stress protective compounds, which are mainly divided into three categories: 1) genes that encode the synthesis of osmolytes such as mannitol, glycine betaine, proline, heat shock proteins; 2) genes responsible for ion and water uptake and transport like aquaporins and ion transporters; and 3) genes regulating transcriptional controls and signal transduction mechanisms, for example MAPK and DREBI[231]. A heat shock protein synthesis gene RcHSP17.8 from Rosa chinensis was obtained and transformed into Arabidopsis thaliana by Agrobacterium tumefaciens transformation. It was found that these transgenic plants were more tolerant to to high temperature, salt, osmotic and drought stress[232]. The salt tolerance of three chrysanthemum varieties with wild chrysanthemum were compared through physiological experiments. The results showed that the salt tolerance of Chrysanthemum lavandulifolium and 'Jinba' were better than that of the other two varieties, while 'Xueshan' was salt sensitive. Based on the differential expression analysis of genes, it was found that the genes related to signal transduction, ion transport, proline biosynthesis, reactive oxygen species scavenging systems and flavonoid biosynthesis pathway were related to salt tolerance of chrysanthemum. Activation of mitogen-activated protein kinases (MAPKs) is an important pathway in eukaryotic signaling events, which plays a key role in plant defense response and growth and development[232]. Studies have shown that CmMKK4-CmMPK13 and CmMKK2-CmMPK4 may be involved in the regulation of salt tolerance in chrysanthemum, and the relationship between CmMKK9 and CmMPK6 and temperature stress in chrysanthemum[233].
Plant architecture
-
Plant architecture generally refers to the morphology of the aboveground parts of higher plants, which are mainly determined by factors affecting branch branches, plant height and inflorescence morphology. At present, in the field of ornamental plants, the research on plant architecture mainly has two purposes: to make the plant shape more beautiful, to meet people's aesthetic needs (Weeping Prunus mume, turtuosa Prunus mume, Styphnolobium japonicum), and to make plants more adapted to the development of the horticulture industry, and to reduce management costs (dwarfing plants and compact branches). The weeping trait is very popular due to their beautiful shape and common appreciation of flowers and trees. Prunus persica, Prunus mume, Cerasus subhirtella and other Rosaceae plants as well as Ulmus pumila, Betula pendula have the phenomenon of hanging branches. The weeping tree phenotype in Prunus persica is located in the LG2 linkage group and is controlled by the Ppa013325 gene. It may be a potential plant gene regulator of gravity perception or response[234]. In the study of Prunus mume, the major QTL loci related to weeping traits have been mapped within a 0.29 Mb region on chromosome 7, and the only specific expression of Pm024213 in buds and stems was detected in PmWEEP interval. Functional annotation and membrane structure prediction showed that the gene was a transmembrane protein located in the chloroplast and containing thioredoxin domain[235]. In recent years, miniaturized and compact potted flowers and bonsai are becoming more and more popular, as the stalks of many ornamental plants are easily damaged, the dwarfing technology of ornamental plants shows more and more vitality. Dw/dw[236] and Tssd/tssd[237] have been identified to be related to the dwarfing of peach. In addition, SHI, rol and other genes have been well transformed in ornamental plants such as Euphorbia pulcherrima[238], Angelonia salicariifolia[239], and Mecardonia procumbens[240].
-
Ornamental plants are of great commercial value to the horticultural industry, and people have been committed to their ornamental genetic improvement for a long time, but the traditional breeding methods often have a long cycle and low breeding efficiency. Although researchers have obtained a large number of candidate genes by means of GWAS and genetic linkage analysis, only some of them have been verified by transgenic experiments. With the development of biotechnology, gene editing technology can accurately edit the DNA sequence and shorten the breeding cycle, which plays a great role in the breeding of ornamental plants. This part provides reference for modern ornamental plant breeding through an overview of general biotechnology.
Gene mapping
-
Quantitative traits are continuous phenotypic variations, which cannot be strictly classified. Quantitative traits controlled by polygenes and are easily affected by the environment. Many ornamental traits of plants, including flowering time[241], double flower[8] and flower color[48], belong to quantitative traits. At present, several methods such as quantitative trait loci mapping (QTL), genome wide association study (GWAS), bulked segregant analysis (BSA), and other methods have been developed for the study of these quantitative traits.
Quantitative trait locus analysis (QTL) is a widely used tool to analyze the genetic basis of complex traits based on the construction of genetic maps. The basic idea of QTL is to use the linkage information between markers and QTL, to detect the linkage degree between molecular markers and QTL by maximum likelihood method or regression analysis, so as to locate QTL location and estimate QTL effect[242]. Paterson et al. reported the first work on plant QTL[243]. Since then, QTL mapping has gradually become an important tool for functional gene mapping. After more than 30 years of development, the QTL location method has developed from single marker analysis to multi-interval mapping, and from static QTL location to dynamic QTL location. In the pas decade, combined with the continuous development of high-throughput sequencing technology, marker resources have become more abundant, and the construction of high-density genetic maps of some ornamental plants[244−247] has become possible. On this basis, more and more major QTL of important ornamental traits of ornamental plants have been cloned[127,248], and considerable progress has been made in the study of QTL of ornamental plants. The F1 hybrid population 'Liuban' × 'Fentaichuizhi' of Prunus mume was selected as experimental material, and a large-scale molecular marker was developed and constructed with a high-density genetic linkage map of Prunus mume using the SLAF-seq technique, and then QTL mapping analysis was carried out on 15 important ornamental traits, such as growth, plant architecture and flower related quantitative. A total of 66 QTLs loci were detected, and 58 possible candidate genes were screened using the genome annotation information of Prunus mume[244].
The genome wide association study (GWAS) is based on linkage disequilibrium. By analyzing the segregation characteristics of high-density molecular markers of a large number of individuals, researchers can select molecular markers associated with phenotypic variation of complex traits, and then analyze the genetic effects of these molecular markers on phenotypes[249]. This method directly uses the natural population, which can save a lot of time in constructing the population. Besides that, the method has large recombination variation and high mapping accuracy, and can identify many multi-allele/gene loci that have not been found in QTL mapping[249]. Although GWAS was originally mainly used for the analysis of complex genetic diseases in humans, over the past decade it has been gradually extended to plants, including some ornamental plants (Supplemental Table S6). In Prunus mume, researchers have identified significant quantitative trait loci through GWAS technology. The candidate regions associated with traits including petal color, stigma color, calyx color, bud color, staminal filament color, wood color, petal number, pistil character, bud aperture, and branching phenotype were identified. It is the first time the genetic structure of floral size, color, and structure, in terms of the number of loci, their genomic distribution and the magnitude and pattern of their effects in a woody plants has been clarified[48]. At present, researchers have mapped more than a hundred loci (Supplemental Table S6) in seven species of ornamental plants by using GWAS technology, which shows that the application of GWAS has greatly promoted the research of alleles of important traits in ornamental plants.
Bulked segregant analysis (BSA) is a mapping method for rapidly functional gene mining using extreme phenotypic individuals. The main idea is to sequence two groups of individuals with opposite extreme phenotypes in the segregated population, and to compare whether there is a significant difference in allele frequencies at polymorphic sites between the two groups[250]. Compared with traditional genetic analysis methods, BSA only needs to identify individuals with different extreme phenotypes in the target population, regardless of the accuracy of other individuals, which greatly reduces the scale and cost by simplifying the program[251]. Because BSA does not need a large population, it is favored in the study of plant type traits of woody plants, such as the weeping trait of Malus halliana[252], Prunus persica[234] and Prunus mume[235], the dwarf trait of Prunus persica[234] and gene mining and marker development. Twentyt individuals each of weeping and upright F1 generation of 'Liuban' × 'Fantaichuihzi' were selected. Using the strategy of RNA-seq based on BSA, five QTL related to the weeping trait were detected on chromosome 7 and combined with WGCNA to identify a core candidate gene PmUGT72B3. The Pm024074 (PmUGT72B3) allele encodes a protein containing conserved coniferyl-alcohol glucosyltransferase, which is a key protein regulating lignin and IAA. Now, BSA analysis has become a powerful tool for mapping functional genes in addition to QTL and GWAS.
VIGS
-
Virus-induced gene silencing (VIGS) is a technique based on RNA interference, which uses the instant knock-down of the target gene expression of the modified plant virus genome[253]. Compared with traditional gene function research methods, such as transgene, gene knock-down and antisense inhibition, VIGS has the advantage of a short test cycle, being independent of transgenic operationand has low cost and high throughput[254]. The photoperiod flowering responses of plants can be divided into three types: long-day (LD); short-day (SD); and day-neutral (DN). Under the condition of SD and LD, the expression of RCCO and RcCOL4 in rose was reduced by VIGS, which successfully delayed the flowering time[255]. The expression of PeERF1 was down regulated in orchids using CymMV-based virus-induced gene silencing (VIGS), and it was found that the nanoridges of the silenced plants reduced. It is proved that PeERF1, as a SHN ortholog transporter, participates in the morphological formation of lip epidermis at the end of flowering by regulating the development of nanoridges structure of Phalaenopsis[256].
Gene editing
-
The development of gene editing technology is the demand of modern plant breeding, which rapidly modifies the plant genome in an accurate and predictable way[257]. Many gene editing techniques have been developed, including Zinc-finger nucleases (ZFNs)[258], transcription activator-like effector nucleases (TALENs)[259] and clustered regulatory interspaced short palindromic repeat (CRISPR)[260]. Some studies have shown that CRISPR/Cas9 is obviously superior to TALENs and ZFNs in terms of design, construction difficulty, time, cost and modification efficiency. CRISPR has gradually become the first choice for plant gene editing[261−263].
Dihydrofavonol-4-reductase-B (DFR-B) is an important enzyme in the anthocyanin biosynthesis pathway. In Ipomoea nil, DFR is present as a small, tandemly arrayed three-gene family (DFR-A, DFR-B and DFR-C), in which DFR-B is the major gene that controls flower color. The DFR-B gene was removed from Ipomoea nil with CRISPR/Cas9. Among the 32 transgenic plants, 24 (75%) had low anthocyanin content whose performance is white[264].This is the first report to use CRISPR/Cas9 technology on flower color research. In lily, CRISPR/Cas9 was used to knock out the LpPDS gene of two kinds of lily and obtained completely albino, pale yellow and albino–green chimeric mutants[265]. CRISPR/Cas9 will significantly promote the development of improved ornamental horticultural crops and bring innovative solutions to sustainable and competitive production of novel ornamental plants.
-
It is predicted that nearly 600 species of flowering plants will be sequenced by July 2021 (
www.plabipd.de/plant_genomes_pa.ep ). Many research groups have proposed to sequence more plants[266−268]. With the development of sequencing technology and the reduction of sequencing cost, plant genome data will show explosive exponential growth in the future. As such, comparative genomics and functional genomics, which are based on whole genome data, will develop more rapidly and infiltrate into all aspects of plant genetic research. Although the development of plant genomics technology has promoted the research of ornamental plants, there are many problems in genome sequencing, data utilization and achievement transformation.First, the sequencing and assembly of genomes is still a difficult problem. Many ornamental plants have experienced a long history of evolution and domestication, especially plant with homologous polyploids or extremely large genome sizes[269]. For genome assembly based on short reading (75_700 bp) data, repeat content, and high heterozygosity sequences are usually not well solved, resulting in the formation of chimeric sequences and fragmentation contigs[270]. Even if third-generation sequencing technology with longer read-lengths is used, the integrity of genome assembly of some ornamental plants is only about 80%[62]. Secondly, it is difficult to map genes related to key traits from a large amount of genome data in the process of molecular breeding of ornamental plants, which is largely due to the lack of suitable phenotypic data. The gene mapping of many crops only needs to obtain simple phenotypic data such as plant height, fruit weight and leaf area, while the ever-changing morphology of ornamental plants and the twisted branches of turtuosa Prunus mume and complex flower shapes of chrysanthemum not only make the measurement work extremely difficult, but also the effective estimation of the recorded character data is low, resulting in weak correspondence between the trait data and the allelic variation of candidate genes in the germplasm set. Thirdly, many ornamental plants (especially woody plants) lack effective transformation systems, so the new genes cannot be transformed into plants for cultivar creation. Finally, whether transgenic ornamental plants meet safety and health needs is also one of the problems facing ornamental plant breeders, although at least 50 species of ornamental plants have been transformed[271], few GM ornamental cultivar have passed field trials and obtained regulatory approval. In the biotechnology/GM crop database approved by (ISAAA), an international agricultural biotechnology application service, only three ornamental plants are listed: petunia, rose, and carnation[271]. This may be due to the potential toxicity and allergies to humans, potential environmental risks (such as opportunities for gene flow, adverse effects on non-target organisms, resistance evolution of weeds and insects). Genetically modified crops that are widely used to carry foreign genes face obstacles to promotion[272]. Although these problems have existed in the many years of development of ornamental plant genomics, researchers continue to use new methods and new ideas to overcome these difficulties, such as gene editing technology, with the rapid development of this technology.
In the aspect of sequencing, DNA sequencing technology in the past produced either highly accurate short reads or less-accurate long reads. The long high-fidelity (HiFi) readings recently launched by PacBio broke this balance. The accuracy of HiFi readings generated consensus sequence from multiple passes of a single template molecule as high as 99.8%, and can produce readings with an average length of 13.5 kb[273]. After the progress of third-generation sequencing technology, the innovation of a genome assembly algorithm followed. HiFiasm[274], a new de novo assembler that strives to preserve the contiguity of all haplotypes. And HiCanu[275] is a modification of the Canu assembler which can accurate assemble segmental duplications, satellites, and allelic variants from high-fidelity long reads. The advantages of these new techniques have been shown in challenging sequencing tasks such as hexaploid California redwood[274] and autotetraploid cultivated alfalfa[276]. On this basis, the gaps, collapsed regions and unassigned sequences in the plant genome will be less, which will provide a guarantee for the comprehensiveness and accuracy of phenotypic analysis in the future. In the mapping of complex traits, researchers are no longer satisfied with the location of simple one-dimensional traits such as plant height and fruit diameter, but the combination of image analysis and QTL mapping to study complex phenotypic traits has gradually become a research hotspot. Many scholars have studied the gene mapping of plant morphology by using the morphometric model based on elliptic Fourier, and have made many gratifying achievements in the location of fruit shape[277] and leaf shape[278,279]. And a mature geometric morphometry platform has been established[280]. Unfortunately, there are few studies on the location of characters of ornamental plants, and the transformation of three-dimensional flower structure into available one-dimensional data is still one of the difficulties that need to be overcome.
During the 1990's, with the revolution of genomics and the rise of systems biology, biosynthesis emerged[281]. Synthetic biology is the science of how to design and synthesize organisms. Specifically, synthetic biology is an advanced form of gene editing. It applies the principles of modularity and engineering design to synthesize cells by changing their DNA[282]. The study of synthetic biology was first carried out on bacterial cells such as Mycoplasma mycoides[283], and now it has been gradually extended to eukaryotes, including plants. We believe that with the analysis of more gene regulatory networks and metabolic networks of ornamental traits and the progress of biological gene synthesis, ornamental plants designed according to the needs of consumers will emerge in the near future. China has a long history of ornamental plant cultivation and is rich in ornamental plant resources, which is known as the 'mother of gardens'. It is estimated that there are at least 31,000 species of vascular plants in China, of which more than 6,000 species have ornamental value. Most of the ornamental plants, such as rhododendron, rose, camellia and mei, originated and are cultivated in China[284].
The rapid development of genomics has greatly promoted the research of horticultural plants[285]. In recent years, with the continuous reduction of the cost of genome sequencing technology, more and more ornamental plants have their own genome data, but it is still a small portion of ornamental plant germplasm resources. It has become an arduous task for modern ornamental plant researchers to sequence ornamental plants in an orderly and reasonable manner, to carry out in-depth mining of important genetic resources, and to protect and utilize them. Therefore, breeders need to make full use of genomic theories and methods (including molecular markers, whole genome selection, genome editing and synthetic biology) to alleviate the bottleneck of existing variety innovation and utilization, analyze the genetic regulation mechanism of important ornamental characteristics, establish an efficient biological breeding technology system, and carry out variety creation.
This study was supported by the National Natural Science Foundation of China (No. 31870689), Forestry and Grassland Science and Technology Innovation Youth Top Talent Project of China (No.2020132608), and the National Key Research and Development Program of China (2018YFD1000401).
-
The authors declare that they have no conflict of interest.
- Supplemental Table S1 List of the representative public accessible genomes of ornamental plant and their database construction status.
- Table S2
- Supplemental Table S2 List of the other public accessible genomes of ornamental plant and their database construction status.
- Supplemental Table S3 The genome-sequenced ornamental species belong to 33 families.
- Supplemental Table S4 Comparison of different sequencing technologies.
- Supplemental Table S5 Overview of important genetically modified ornamental plants.
- Supplemental Table S4
- Supplemental Table S6 List of current GWAS analysis in ornamental plant.
- Supplemental Fig. S1 Annual increase in the genome-sequenced method.
- Copyright: © 2022 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Li M, Wen Z, Meng J, Cheng T, Zhang Q, et al. 2022. The genomics of ornamental plants: current status and opportunities. Ornamental Plant Research 2:6 doi: 10.48130/OPR-2022-0006 

The genomics of ornamental plants: current status and opportunities
- Received: 23 November 2021
- Accepted: 22 February 2022
- Published online: 23 March 2022
Abstract: With the rapid development of sequencing technologies, followed by the reduction of sequencing cost, numerous ornamental plants have been sequenced, resulting in their genomic studies shifting from gene cloning and marker development to whole genome profiling. A profound understanding of genome structure and function at the whole genome level can not only help to modify ornamental traits, such as fragrance, color and flower shape, through genetic engineering, but also infer the genetic relationship and evolutionary history of ornamental plants via comparative genomics analysis. In this paper, we review the current situation of sequencing strategies and the application of genomics to study the origin and evolution of ornamental plants. We highlight challenges of ornamental plant genomic research. The use of cutting-edge technologies, such as genomics, gene editing and molecular design polymerization breeding, can facilitate our understanding of genetic regulation mechanisms and the germplasm innovation of important traits in ornamental plants. The results can be expected to significantly increase the breeding efficiency of ornamental plants.
-
Key words:
- Ornamental plants /
- Genome sequences /
- Genetic modification /
- Genomics /
- Ornamental traits
{kind=link}