








-
The central challenge of modern biology is no longer data generation, but data interpretation. The 3.2 billion-base-pair human genome, once the initial blueprint has given way to a cascade of dynamic data streams that depict life in motion. Technologies like single-cell RNA-seq (scRNA-seq) generate millions of individual cellular profiles, a scale being tackled by AI-driven curation efforts like scBaseCount[1]. Simultaneously, spatial omics methods weave these narratives into the tissue's fabric, creating multi-terabyte mosaics of histology and gene expression. The result is not merely a data deluge, but a crisis of interpretation. The intricate patterns of health and disease are encoded in these datasets, yet they exist at a scale and complexity far beyond the reach of traditional, manual analysis. Human expertise, once the gold standard, is now fundamentally mismatched to this new reality, creating a chasm between data generation and biological discovery (Fig. 1a). This sets the stage for a new class of computational interpreter—one capable of navigating this complexity to automate and accelerate the extraction of knowledge.
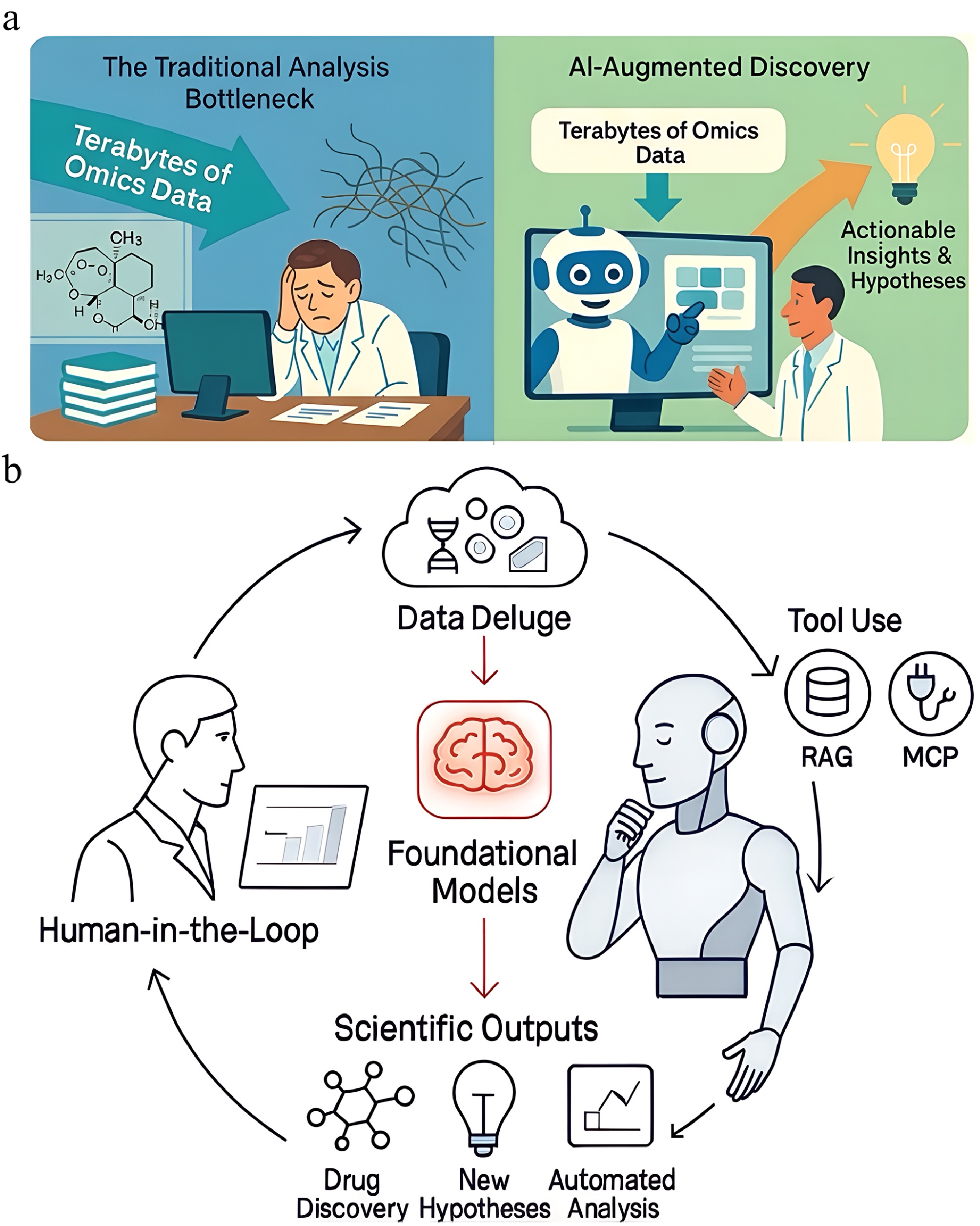
Figure 1.
The dawn of the digital biologist: a new paradigm for scientific discovery. (a) The shift from the traditional analytical bottleneck to AI-augmented discovery. On the left, a human scientist is overwhelmed by the complexity and scale of modern omics data, leading to a bottleneck that slows the pace of discovery. On the right, an AI agent acts as a collaborative 'digital biologist', empowering the human scientist by processing the same terabyte-scale data to generate actionable insights and testable hypotheses, thereby accelerating the research cycle. (b) A schematic of the iterative and collaborative workflow powered by AI. The cycle begins with the 'data deluge' from modern experiments, which is interpreted by 'foundation models' that serve as the core reasoning engine. The AI agent, operating within this framework, leverages specialized tools, such as those enabled by technologies like Retrieval-Augmented Generation (RAG) for accessing up-to-date knowledge and the Model Context Protocol (MCP) for interfacing with software to produce diverse 'scientific outputs'. These specific technologies (RAG, MCP) represent key opportunities for the field and will be detailed later in this review. These outputs can include automated data analysis, the generation of new hypotheses, and candidates for drug discovery. The 'human-in-the-loop' guides the process, evaluates the results, and refines the objectives, creating a powerful, cyclical partnership between human intuition and artificial intelligence.
The application of artificial intelligence (AI) in biology is not a new phenomenon[2]. The field has witnessed a steady progression from classical machine learning algorithms used for classification and regression, to early deep learning models like convolutional neural networks (CNNs) for image analysis and recurrent neural networks (RNNs) for sequence data. However, the recent advent of the transformer architecture and the subsequent development of Large Language Models (LLMs) represent a qualitative, not merely quantitative, leap forward. Unlike their predecessors, the LLMs possess two transformative characteristics: they are generative and exhibit remarkable in-context learning. Pre-trained on vast, unlabeled corpora, such as the entirety of sequenced genomes or the scientific literature, to learn the fundamental statistical patterns, or 'grammar', of their respective domains. This pre-training phase creates powerful 'foundation models' that encode a general understanding of data modality. This foundational knowledge can then be adapted to a wide array of specific downstream tasks, such as predicting DNA function or classifying cell types, often with minimal task-specific labeled data. This ability of transfer learning is a significant advantage in biology, where labeled data is often scarce or expensive to acquire.
While LLMs can serve as advanced analytical tools, the frontier is now shifting towards the development of AI agents, which exhibit autonomy, planning, and tool use in the pursuit of scientific goals[3]. The AI agent in this context is more than a static model; it actively decides which actions to take, and can chain together multiple steps to accomplish a high-level objective. Crucially, these agents can interface with external tools and data sources, enabling them to retrieve relevant literature, call a statistical software package, or even control laboratory robotics. This paradigm extends beyond a 'smart algorithm', to an AI that functions as a research assistant or even a junior scientist. The key characteristics that distinguish agents from passive tools are: (1) Autonomy: agents can operate without step-by-step human instructions, making decisions based on intermediate results. (2) Planning and memory: agents maintain a working memory of the project and plan multi-step workflows to achieve a goal. (3) Tool integration: agents can use other software and data sources in a coordinated manner. In essence, an AI agent imitates the research process of a human scientist rather than solving a single, narrow problem. Early demonstrations of this concept, discussed later, include systems that can take a research idea and produce a complete analysis and report. This paradigm opens the door to fully automated experiments, where an AI might design and even execute laboratory work when paired with automation. This new workflow, often with a human scientist providing critical oversight (a 'human-in-the-loop'), forms a powerful iterative cycle of discovery (Fig. 1b). Although this journey is just beginning, it signals a fundamental shift from using AI within predefined workflows to having AI orchestrate the workflows themselves.
In this review, we comprehensively survey this rapidly evolving landscape. We begin by examining the foundational language models that serve as the engines of this new paradigm, from those that decipher the grammar of DNA/RNA/protein, to those that interpret the complex language of single-cell, and spatial omics. We then chart the rise of autonomous AI, from single-purpose tools to collaborative multi-agent systems that tackle complex research problems. We categorize these agents by their function: automating data analysis, designing novel experiments, accelerating drug discovery, and pursuing the ambitious quest for a generalist 'AI Scientist.' Finally, we addressed critical challenges that must be overcome, including spanning reliability, security, and ethics. We then explore the immense opportunities that lie ahead, highlighting key technologies like Retrieval-Augmented Generation (RAG) for ensuring factual grounding, the Model Context Protocol (MCP) for seamless tool integration, and the push towards causal reasoning and fully autonomous 'self-driving labs.' Our goal is to provide a structured overview of the state-of-the-art, offering a glimpse into a future where a 'digital biologist' becomes an indispensable partner in scientific discovery (Fig. 2). To complement this review, we have also created a publicly available and actively maintained resource list that includes the models, agents, and datasets discussed, available on our project website at:
http://awesomebio.webioinfo.top .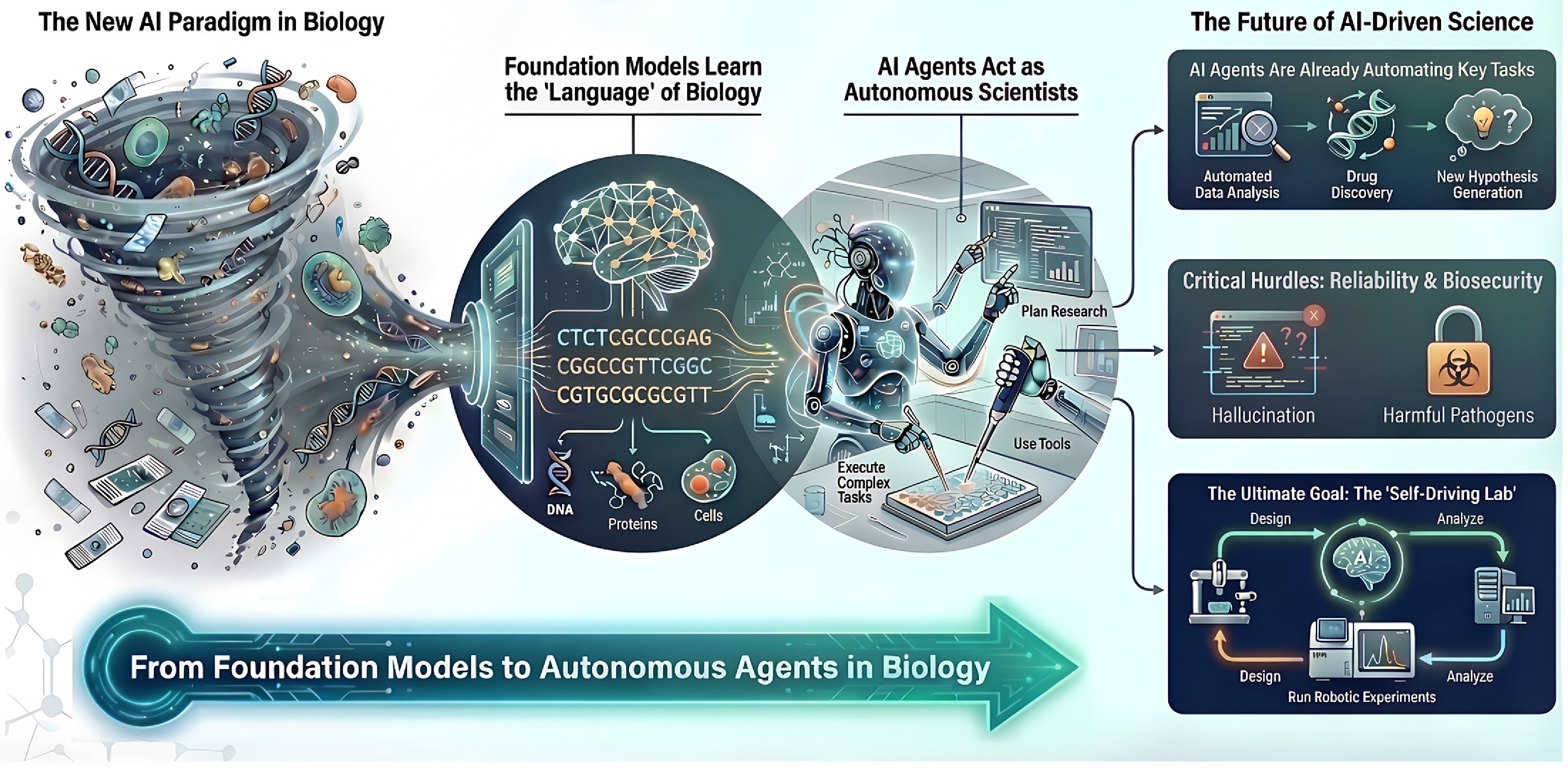
Figure 2.
Strategic roadmap for the era of AI-driven biology. This diagram outlines the structure of this review and the evolutionary trajectory of the field. The journey begins with foundation models (left), which ingest the 'data deluge' to learn the languages of sequences and cells. It progresses to autonomous agents (center), which leverage these models to act as active scientists capable of planning, tool use, and hypothesis generation. Finally, it points towards the future of discovery (right), addressing critical hurdles such as reliability and biosecurity, and envisioning the 'self-driving lab', where AI and robotics close the loop on scientific experimentation.
-
Foundation models are large-scale models trained on massive data using unsupervised or self-supervised learning to capture general and transferable knowledge. Most modern foundation models are based on the transformer architecture[4], whose attention mechanism models relationships between tokens, and enables highly scalable pre-training on large corpora. The power of modern biological AI stems from foundation models pre-trained on the fundamental 'languages' of biology. These models learn deep, context-aware representations of biological entities—from DNA sequences to entire cells—that can be leveraged for a vast array of downstream tasks (Fig. 3). A critical step that defines a model's capabilities is the choice of how to represent biological data as 'tokens'. The evolution from simple, human-defined tokens to more data-driven, semantically rich representations is a recurring theme and a key driver of progress. Below, we explore foundation models across two main categories: DNA/RNA/protein sequence models, and single-cell and spatial omics models.
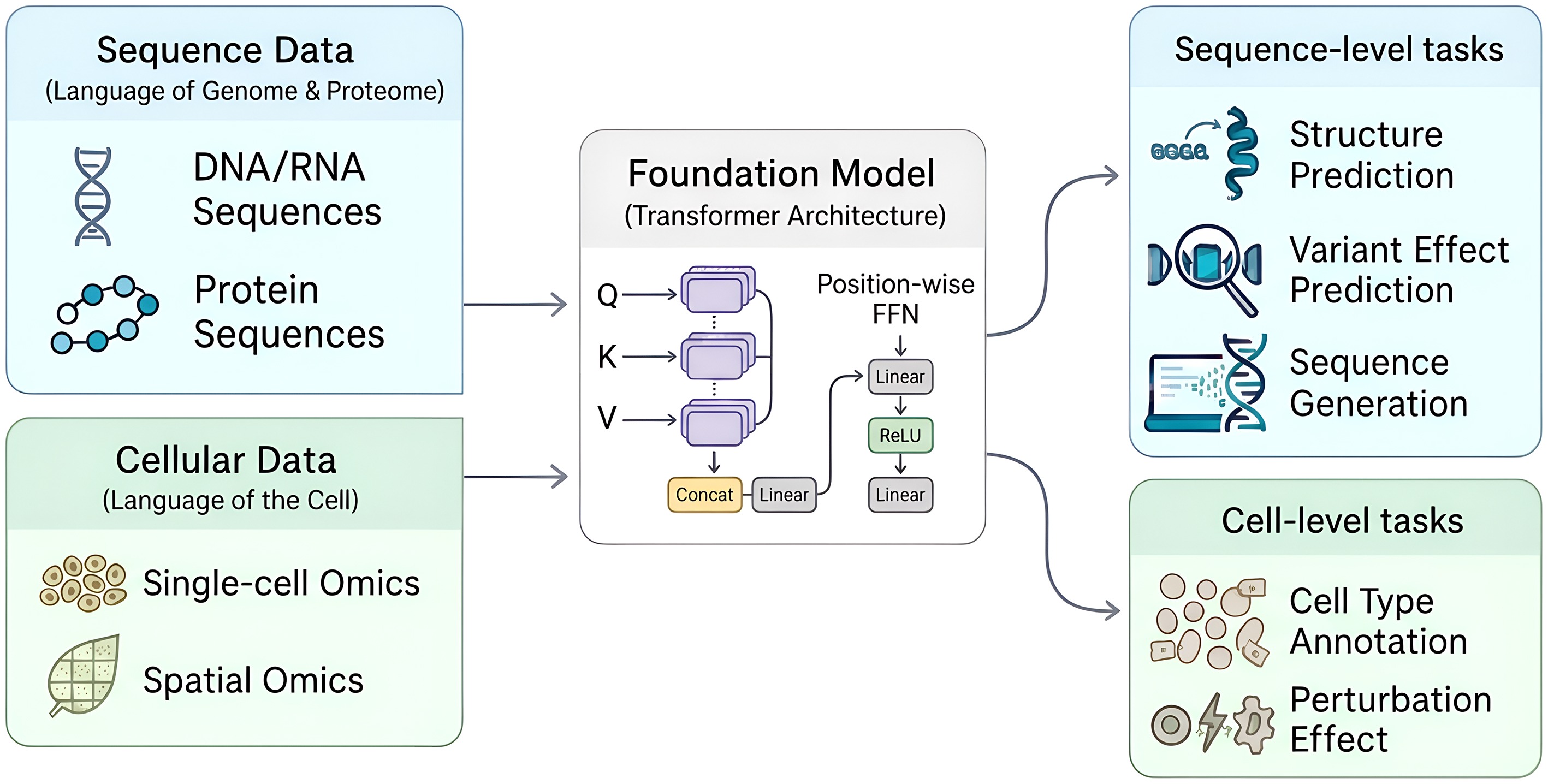
Figure 3.
Application of foundation models in biology. Foundation models are pre-trained on diverse biological data, including sequence data (DNA, RNA, proteins), and cellular data (single-cell, spatial omics). Using transformer architecture, these models learn fundamental representations of biological systems. The resulting models can then be applied to a wide range of downstream tasks (sometimes task-specifically fine-tuned), such as structure prediction and sequence generation at the sequence level, or cell type annotation and perturbation effect predictions at the cell level.
Learning the language of the genome (DNA and RNA), and proteome
-
Early efforts to apply transformers to DNA demonstrated that nucleotide sequences have a latent 'language' of regulatory signals and patterns. DNA Bidirectional Encoder Representations from Transformers (DNABERT)[5] was a landmark—a BERT-based model trained on the human genome using a k-mer tokenization. Notably, it showed particular strength in low-data regimes, outperforming specialized tools that were trained from scratch on small, task-specific datasets. The successor, DNABERT-2, introduced crucial innovations for efficiency and performance[6]; it replaced the rigid k-mer tokenization with Byte Pair Encoding (BPE), a data-driven algorithm that builds a vocabulary of variable-length tokens by iteratively merging the most frequent adjacent characters. A further adaptation, DNABERT-S, tailored this architecture specifically for the task of differentiating species from their genomic sequences[7]. Other projects include the Nucleotide Transformer (NT) project[8], which trained transformers up to 2.5 billion parameters on 850 genomes from across species, using a 6-mer tokenization. And the latest iteration, Nucleotide Transformer v3 (NTv3), advances this paradigm by employing a U-Net–like architecture to enable single-base tokenization across 1 Mb contexts[9].
A major bottleneck for early transformers was their computational complexity, which scales quadratically with input sequence length, making it prohibitive to model the long-range dependencies essential for gene regulation. Several models introduced architectural breakthroughs to overcome this. HyenaDNA replaced the costly self-attention mechanism with a sub-quadratic design inspired by signal processing, enabling it to process sequences of up to 1 million tokens at single-nucleotide resolution on a single GPU[10]. Similarly, the GENA-LM family of models handles long sequences using sparse attention mechanisms and recurrent memory mechanisms[11]. Following these architectural advances, the field has rapidly moved towards scaling both model size and data volume. Evo2 is a 40-billion parameter model trained on 9.3 trillion DNA base pairs with an unprecedented 1-million-token context window[12,13]. Expanding the scope to the vast diversity of environmental DNA, GenomeOcean, a 4-billion-parameter model trained on 219 TB of global metagenomic data, investigates the 'Genomic Manifold Hypothesis.' By demonstrating a strong linear correspondence with Evo2's embedding space, it suggests that these models are converging on a universal, low-dimensional evolutionary manifold governed by biochemical constraints[14]. Concurrently, Googles DeepMind's AlphaGenome processes 1-megabase DNA sequences to predict thousands of properties related to gene regulation, serving as a 'one-stop shop' for variant effect prediction[15]. In parallel, researchers have developed models for RNA sequences, such as RiNALMo, a 650-million-parameter model trained on 36 million noncoding RNA sequences that captures the unique 'grammar' of RNA secondary structures and motifs[16].
Parallel to the advances in genomics, an equally transformative revolution has occurred in learning the language of proteins. Foundation models like Evolutionary Scale AI's ESM series have demonstrated that transformers trained on massive databases of protein sequences can learn deep representations that capture evolutionary, structural, and functional information. The latest iteration, ESM3, is a multimodal generative language model that unifies sequence and structure modeling. Instead of predicting coordinates directly, it generates discrete structure tokens, which are then decoded into high-resolution all-atom protein structures.[17]. The field has also seen an explosion in scale and capability, exemplified by Baidu-backed BioMap's rapid advancement from the 100-billion-parameter xTrimoPGLM. These powerful foundation models are capable of both understanding and designing novel protein structures and functions[18]. Furthermore, the definition of biological 'language' is rapidly expanding beyond 1D sequences to include the continuous semantics of 3D geometry and molecular interactions. While AlphaFold demonstrated the initial potential of deep learning in Critical Assessment of Structure Prediction (CASP) 13[19], the paradigm shifted dramatically with AlphaFold2[20,21], which first achieved near-experimental accuracy for single-chain protein structure prediction in CASP14 benchmarks; AlphaFold3 subsequently extended this framework to a broad range of biomolecular complexes and interactions[22]. By integrating learned representations with a diffusion-based architecture, it can predict the joint structure of complexes involving proteins, DNA, RNA, ligands, and ions with markedly higher accuracy than previous methods across most complex types, providing a comprehensive view of molecular machinery in action[22]. Similarly, generative foundation models are tackling protein dynamics; for instance, Microsoft's BioEmu efficiently emulates equilibrium protein conformations, achieving thermodynamic accuracy (~1 kcal/mol) at a fraction of the computational cost, though its scope is currently limited to soluble proteins under fixed conditions[23].
The frontier of sequence modeling is now pushing beyond single data types towards true integration, a trend foreshadowed by models like ProCALM, which blur the lines between modalities by training on both biological sequences and free-text descriptions to generate novel proteins from natural-language prompts[24,25]. Other architectures like IsoFormer advance this by employing a multi-modal transfer learning framework. Instead of training from scratch, it aggregates embeddings from specialized pre-trained encoders, specifically Enformer for DNA, Nucleotide Transformer for RNA, and ESM-2 for proteins, to connect these three core biological modalities. This approach allows it to tackle complex problems like predicting tissue-specific transcript isoform expression[26]. This trend has accelerated with the integration of natural language to make sophisticated analysis more accessible. ChatNT, for instance, is a conversational agent that understands both biological sequences and english prompts, allowing it to solve complex bioinformatics tasks interactively[27]. Supporting this drive are open-source efforts like Geneverse[28] and industry-led frameworks like NVIDIA's BioNeMo[29], which provide blueprints for ambitious integrative tasks such as enabling integrated workflows, where natural language specifications of molecular function can be combined with protein language models, structure-conditioned generators, and optimization modules to design candidate protein sequences.
Deciphering the language of biological systems: from single cells to clinical phenotypes
-
Beyond the linear sequence of the genome, the collective state of a biological system forms a complex, multi-layered 'language' defined by gene expression, spatial organization, and phenotypic outcomes. A new class of foundation models aims to learn this language from large single-cell omics datasets. One pioneering work was Geneformer, a transformer pre-trained on 30 million (and later 100 million) single-cell gene expression profiles[30,31]. It introduced a rank-based encoding of gene expression and was trained with a masked learning objective, allowing it to learn fundamental gene network dynamics and enabling powerful zero-shot performance on downstream tasks. Another key model, scGPT, employs a generative pre-training approach and has proven effective for cell type annotation, data integration, and predicting perturbation responses[32]. Its successful continual pre-training on spatial transcriptomic profiles (called scGPT-spatial) demonstrates the adaptability of these architectures to new data modalities[33]. Other notable models include CellFM[34], scFoundation[35], stFormer[36], and Nicheformer[37]. A particularly novel approach to tokenization is presented by Cell2Sentence[38,39], which converts a cell's expression profile into a ranked list of gene names, creating a literal 'cell sentence'. This clever transformation allows standard Natural Language Processing (NLP) models (like GPT-2) and popular libraries (like Hugging Face) to be directly fine-tuned on single-cell data for tasks such as conditional cell generation and complex cell type prediction, elegantly bridging the gap between the NLP and biology domains. Complementing these single-cell approaches, GeneRAIN leverages 410,000 bulk RNA-seq samples and introduces a novel 'binning-by-gene' normalization strategy to mitigate expression bias. By ensuring equitable learning probabilities across the transcriptome, it generates multifaceted gene embeddings that encode diverse biological contexts, such as protein domains and disease associations, thereby enabling effective knowledge transfer to characterize understudied elements like long noncoding RNAs[40].
Beyond novel tokenization strategies, another key avenue for enriching the 'language' of the cell is to integrate it with curated biological knowledge. Rather than relying on the model to learn gene functions from expression patterns alone, models like Scouter[41] ingest textual information by utilizing high-dimensional gene embeddings derived from LLMs. This integration of biological and sequence-based priors is further advanced by specialized generative frameworks such as scDiffusion[42], which leverages foundation model-based autoencoders within a latent diffusion framework to generate high-fidelity, conditional gene expression data, and reconstruct continuous developmental trajectories via a gradient interpolation strategy. To address the complexities of multi-modal data, scDiffusion-X introduces a Dual-Cross-Attention module that adaptively learns hidden relationships between molecular layers, facilitating high-fidelity modality translation and the discovery of cell-type-specific regulatory networks[43]. Additionally, the Bag-of-Motifs framework provides a minimalist yet highly predictive sequence code for cell identity by representing distal regulatory elements as unordered counts of transcription factor motifs[44]. By conditioning the model on these fixed semantic embeddings alongside control cell expression data, it captures complex gene–gene interactions to accurately predict transcriptional responses to perturbations.
Single-cell foundation models are also expanding to incorporate spatial context, which is crucial as a cell's state is influenced by its neighbors and microenvironment. Models like OmiCLIP[45] bridge histology images and gene expression by learning to align visual and molecular features. This allows the model to predict the gene expression profile from an H&E image and to perform cross-modal retrieval, unlocking capabilities like identifying regions in a tumor image that likely express a drug target. Expanding visual-linguistic pathology, the TITAN multimodal foundation model aligns hundreds of thousands of whole-slide images with reports using visual self-supervised learning[46]. This yields general-purpose representations that bridge pixel features and clinical semantics, enabling zero-shot or few-shot retrieval for rare diseases and cancer prognosis, while substantially reducing the need for task-specific labels. SPATIA[47] is a multi-scale generative model that directly fuses morphological tokens from cell images with transcriptomic tokens, learning spatially aware representations from the single-cell to the whole-tissue level.
A particularly exciting direction is modeling perturbations and dynamics, aiming to provide a causal, rather than purely correlational, understanding of biology. For generative models, AI-driven virtual cells (AIVC) have gained increasing attention in recent years. Envisioned as multi-scale, multi-modal foundation models, AIVCs integrate diverse biological data to represent and simulate the behavior of molecules, cells, and tissues across varying states[48,49]. Beyond prediction, the field is advancing towards a 'Predict, Explain, Discover' paradigm, where AIVCs not only forecast functional responses to perturbations but also provide mechanistic explanations and generate testable hypotheses for lab-in-the-loop validation[50]. This evolution is supported by foundational 'data pillars'—including a priori knowledge, static architecture, and dynamic states—that nourish the growth of these silicon-based entities. Emerging methodologies are addressing the 'black box' limitation of earlier models; for instance, VCWorld introduces a 'white-box' simulator by combining LLMs with structured biological knowledge graphs to offer interpretable, step-by-step reasoning[51]. Simultaneously, new human-interpretable grammars are democratizing the field by allowing researchers to encode complex multicellular systems biology models using natural language rules[52]. Within this rapidly expanding landscape, models like UniCure[53] and Arc Institute's STATE[54] continue to push the boundaries of perturbation biology. STATE was trained on the unprecedented Tahoe-100M, a large atlas of 100 million perturbed single-cell profiles[55]. It uses a State Transition (ST) module to predict how a cell's state will shift under a given perturbation, significantly outperforming prior methods in predicting experimental outcomes. Broadening the scope of prediction, Prophet learns a unified representation of the experimental space to predict diverse phenotypes, from gene expression to cell morphology, encompassing various genetic and chemical perturbations[56]. Moving towards personalized medicine, ODFormer acts as a 'virtual organoid' for pancreatic cancer; by integrating transcriptomic and mutational profiles, it accurately predicts patient-specific drug responses and identifies potential biomarkers without physical testing[57]. The creation of such powerful predictive models is only possible due to the availability of large, harmonized perturbation datasets like scPerturb[58], Cell Painting Gallery[59], RxRx3[60], and the genome-wide scale of newer resources like X-Atlas/Orion[61]. The development of rigorous evaluation frameworks like PerturBench[62] and Cell-Eval[54] is also critical for benchmarking and comparing these predictive models.
While generalist models strive for universality, specialized architectures are proving superior in specific domains. Nephrobase Cell+[63], a kidney-specific foundation model trained on nearly 40 million profiles, outperforms generalist counterparts like scGPT in tasks such as batch correction and cross-species alignment, suggesting that organ-focused pre-training creates higher-fidelity representations for specialized biology.
As seen across the domains of genomics, proteomics, and cellular analysis, the clear trajectory is towards integration. A cell, after all, is not defined by its transcriptome or genome alone; its proteomic state, spatial environment, and regulatory state are all critical. Reflecting this trend, scTranslator acts as a generative 'translator' that infers single-cell proteomic profiles directly from transcriptomic data, effectively synthesizing multi-omics views from RNA-seq alone[64]. While many models still operate within a single domain, the most advanced architectures like AlphaFold3 and OmiCLIP are already bridging modalities. The ultimate goal remains a holistic, unified model that can encode an entire cell or tissue state, combining sequence, expression, structure, spatial context, and even clinical data, and reasoning together. This multi-modal integration is the critical steppingstone toward the generalist AI agents discussed next, which must handle diverse data types to solve complex research problems.
Benchmarking analysis: navigating the landscape of biological foundation models
-
As the ecosystem of biological foundation models expands, rigorous benchmarking has become essential to delineate their practical utility against established baselines. Recent comprehensive evaluations highlight a nuanced landscape where increased model complexity does not always guarantee superior performance. In the realm of zero-shot learning—where models are applied to new tasks without further training—evaluations of prominent single-cell models like scGPT and Geneformer reveal significant limitations. Studies indicate that, for fundamental tasks such as cell type clustering and batch integration, these complex transformers often underperform compared to simpler, established methods like scVI, Harmony, or even identifying highly variable genes (HVG)[65]. Furthermore, the assumption that performance scales linearly with pre-training data volume is being challenged; recent investigations into dataset size and diversity suggest that model performance on downstream tasks frequently plateaus using only a small fraction (e.g., 1%) of the available training corpora, implying that current architectures may not yet be fully leveraging the vast scale of single-cell data[66]. For specific high-stakes applications like Drug Response Prediction, the choice of model depends heavily on the evaluation scenario. The scDrugMap benchmark demonstrates that while scFoundation achieves state-of-the-art performance in 'pooled-data' settings (where test data resembles training distributions), models like UCE and scGPT demonstrate superior generalizability in 'cross-data' scenarios, effectively handling unseen datasets[67]. Crucially, this study emphasizes that fine-tuning consistently, yields better results than using frozen embeddings, highlighting the necessity of task-specific adaptation. Meanwhile, the integration of generalist LLMs into this domain presents a mixed picture. The CELLVERSE benchmark reveals that while massive generalist models (e.g., GPT-4, DeepSeek) surprisingly outperform smaller, specialized bio-LLMs (like C2S-Pythia) on language-centric biological tasks, they still struggle with complex reasoning; notably, in drug response prediction tasks, these generalist models often fail to outperform random guessing, underscoring the gap between linguistic fluency and deep biological insight[68]. These findings collectively suggest that researchers should prioritize rigorous, task-specific benchmarking over blind adoption of the largest available models.
-
While the foundation models described above represent a monumental leap in understanding the languages of biology, they are inherently passive reasoners. They can interpret a DNA sequence or predict a protein structure when prompted, but they cannot independently decide to then take that structure and screen it against a drug library. To unlock this potential, these models must be integrated into a larger, active framework. This is the role of the AI agent: an architecture that wraps a powerful LLM 'brain' in a system that provides it with memory, planning abilities, and access to external tools (Fig. 4a). However, not all agents operate with the same degree of independence. As outlined by Gao et al.[3], these systems can be classified into distinct levels of autonomy, ranging from Level 0, where ML models function merely as tools, to Level 1 'assistants' that execute specific, narrow tasks. More advanced Level 2 agents act as 'collaborators' capable of refining hypotheses, while aspirational Level 3 agents function as 'autonomous scientists' capable of de novo discovery and skeptical reasoning. This hierarchy highlights the shift from using LLMs as an analytical tool to deploying them as a research partner. In this section, we survey concrete examples of such agents, organized by their primary functions (Fig. 4b). It's worth noting that many agents could fit in multiple categories (for instance, a drug discovery agent might also generate new hypotheses), thus we will discuss each in its most salient context.
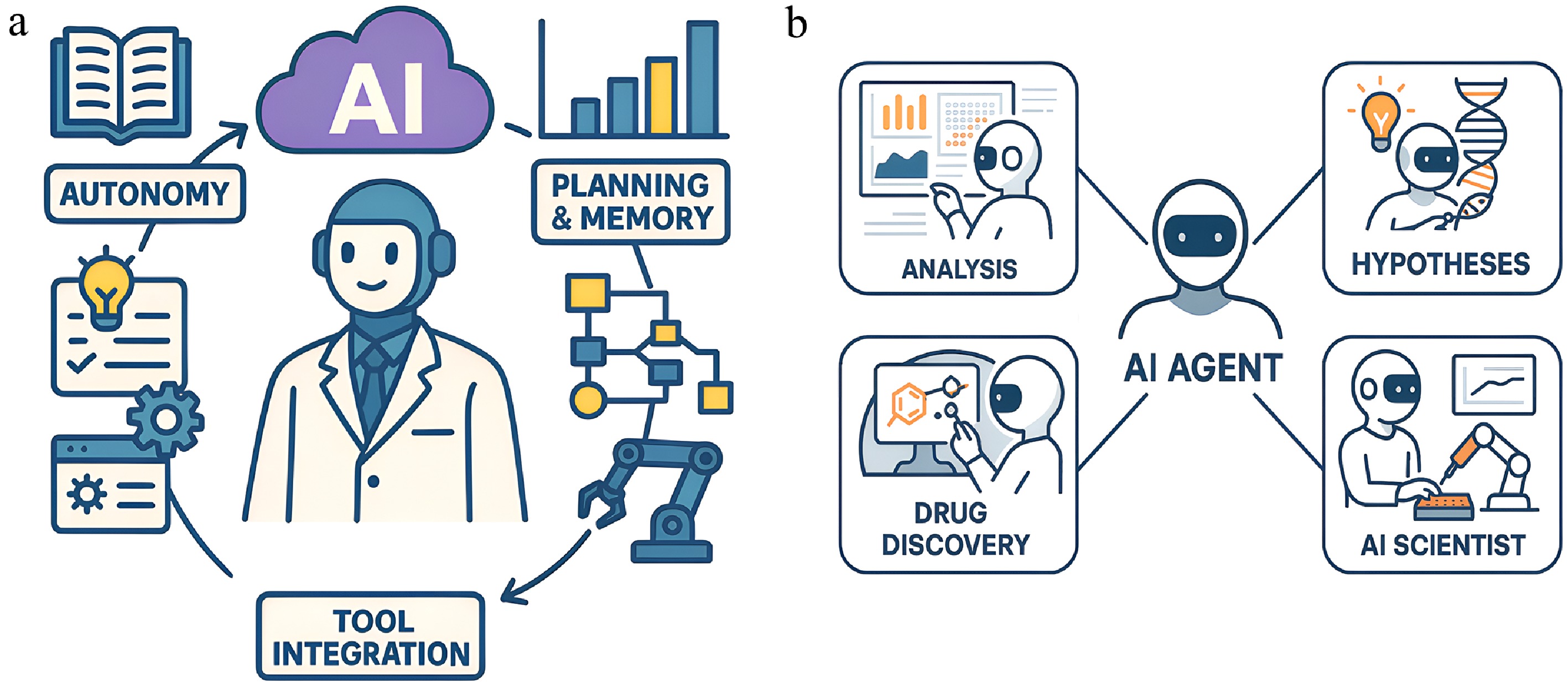
Figure 4.
Anatomy and applications of the AI agent in biology. (a) Core characteristics of an AI agent. An AI agent is defined by a set of core capabilities that enable it to act as an autonomous research partner. These include: (1) autonomy, the ability to operate without step-by-step human instruction by leveraging knowledge; (2) planning and memory, the capacity to devise multi-step strategies and maintain a working memory of a project; and (3) tool integration, the ability to interact with, and control external software, databases, or laboratory robotics. (b) Key application areas. By leveraging these core characteristics, AI agents are being deployed across a wide range of biological applications. These include automating complex data analysis, accelerating drug discovery by navigating vast chemical spaces, generating novel scientific hypotheses, and pursuing the ultimate goal of a generalist AI scientist that can design and orchestrate entire experimental workflows.
Agents for automated data analysis
-
The first breed of AI research agents focused on automating entire data analysis pipelines, effectively acting as an autonomous bioinformatician. The vision is to democratize bioinformatics, allowing non-experts to perform sophisticated analyses with minimal effort. A prime example is CellAgent[69], an LLM-driven multi-agent system for scRNA-seq analysis. It consists of specialized agents, including Planner, Executor, and Evaluator, who collaboratively design strategies, execute steps using tools like Scanpy, and check the results, adjusting the plan as needed. Similarly, SpatialAgent autonomously navigates the workflow of spatial biology, from experimental design to hypothesis generation[70]. CellVoyager is an agent that autonomously explores scRNA-seq datasets, uniquely conditioned on a human user's prior analyses to build upon existing work and explore complementary hypotheses[71]. To further enhance interactivity, CellWhisperer introduces a multimodal approach, aligning transcriptomes with text to enable natural-language interrogation of gene expression directly within the CELLxGENE browser[72]. Addressing the 'black-box' nature of automated annotation, new agents prioritize transparency alongside accuracy. GPTBioInsightor integrates differential expression genes with biological context and pathway functions via an LLM reasoning pipeline and rigorous scoring to predict cell types or states with quantitative confidence and transparent explanations[73]. Similarly, CASSIA integrates reasoning and quality assessments to calibrate confidence, effectively mitigating hallucinations and improving accuracy across rare cell populations[74]. For proteomics, DrBioRight 2.0 offers a conversational interface for exploring large-scale cancer proteomics data[75]. AutoBA is designed for fully automated analysis of various omics data types, requiring only a data path and objective to self-design and execute a complete workflow[76]. Demonstrating that powerful agents need not rely on the largest proprietary models, BioAgents is a multi-agent system built on Microsoft's small Phi-3 language model with RAG, designed to help users design and debug complex bioinformatics workflows while running locally, thereby enhancing accessibility.[77]. Reinforcing this trend towards efficiency, the Nano Bio-Agents (NBA) framework further validates that small language models (< 10 B parameters) can achieve high accuracy in genomics question answering, by orchestrating tools like NCBI and AlphaGenome, with significantly lower computational overhead[78].
Agents for hypotheses and experimental design
-
Moving beyond the automation of established analytical pipelines, this next class of agents is designed to generate fundamentally new and testable scientific hypotheses. This class of 'active' agents represents a critical step up in autonomy, moving beyond analyzing existing data to proposing novel experiments and generating new hypotheses. BioDiscoveryAgent is designed to navigate the vast hypothesis space of genetic perturbation experiments, using an LLM-based agent, together with tools and past experimental results, to propose small, targeted sets of genes to perturb to achieve a desired phenotype[79]. In domains where data is scarce, such as in combinatorial therapy, Coated-LLM employs a 'scientific collaboration' metaphor. By utilizing specialized agents acting as Researcher, Reviewer, and Moderator, it systematically debates and evaluates hypotheses to ultimately predict efficacious drug combinations for complex diseases like Alzheimers[80]. Focusing on a specific technique, CRISPR-GPT is an agent that automates the detailed design of gene-editing experiments. Given a high-level goal, it decomposes the task and uses external tools to suggest target exons, design guide RNAs, predict off-target effects, and even design validation primers[81]. In protein science, ProtAgents is a multi-agent platform for de novo protein design that employs a collaborative system of planner, assistant, and critic agents to tackle complex design problems[82].
Agents in drug discovery and therapeutics
-
This area highlights the application of agentic AI to the high-stakes, multi-step process of developing new medicines. DrugAgent is a multi-agent framework that automates the complex programming tasks in AI-aided drug discovery, allowing researchers to apply sophisticated machine learning techniques without being expert coders[83]. LIDDIA (Language-based Intelligent Drug Discovery Agent) is designed to navigate the in silico drug discovery process, balancing the exploration of novel chemical space with the exploitation of known scaffolds to generate new molecules[84]. Complementing these efforts with mechanistic reasoning, BioScientist Agent unifies a massive biomedical knowledge graph with reinforcement learning. This architecture allows it to traverse complex biological relationships, identifying drug repurposing candidates while automatically generating causal reports that elucidate the putative mechanisms of action[85]. Such agents often draw upon large, curated dataset collections like the Therapeutics Data Commons (TDC), which aggregates dozens of datasets for therapeutic science[86]. For therapeutic reasoning, TxAgent represents a new level of sophistication. It is an AI agent designed to make personalized treatment recommendations by integrating real-time knowledge from a curated, 'ToolUniverse' of 211 specialized tools, including sources like the FDA (Food and Drug Administration) drug database and Open Targets. Rather than relying on static training data, TxAgent performs multi-step reasoning, calling on these external tools to analyze drug interactions, contraindications, and patient-specific factors, providing transparent, evidence-backed decision traces[87]. Similarly, the NVIDIA Biomedical AI-Q Research Agent is a blueprint for creating on-premise agents that combine literature research with virtual screening capabilities, allowing a researcher to investigate a disease, identify a target, and autonomously screen for novel therapies.
The quest for a generalist AI scientist
-
The ultimate vision of this field is the creation of a general-purpose AI research agent—a 'digital biologist', that can work across multiple biomedical domains. Agent Laboratory is an autonomous framework that takes a human-provided research idea and then carries out the literature review, experimentation, and report writing[88]. The AI Scientist project from Sakana AI has pushed the boundaries of autonomy even further; in a landmark experiment, a paper fully generated by its AI Scientist-v2 system received scores above the acceptance threshold at an International Conference on Learning Representations (ICLR) workshop[89]. Other similar studies include AI Co-scientist[90], and The Virtual Lab[91]. In the realm of biomedicine, Biomni represents a project to build an AI with encyclopedic knowledge of biology that can autonomously execute tasks across 25 different domains using 150 specialized tools[92]. Most recently, they introduced Biomni-E1—a unified, curated 'action universe' that aggregates all the sources used in Biomni into a single programmatic environment, giving AI agents on-demand access to expert resources across their defined 25 biomedical subfields, and enabling tasks that span variant prioritisation, drug repurposing, and multi-omics analysis. OriGene is another sophisticated architecture: a self-evolving multi-agent system that functions as a 'virtual disease biologist' for therapeutic target discovery. Critically, it features a framework that allows it to continuously integrate human and experimental feedback to iteratively refine its own reasoning templates and analytical protocols. OriGene's capabilities were validated by its successful nomination of novel cancer targets that showed significant anti-tumor activity in patient-derived models, representing a major step towards truly intelligent and adaptive scientific agents[93]. Finally, extending the AI scientist's capabilities into the physical realm, LabOS represents an end-to-end co-scientist that unites digital reasoning with wet-lab execution[94]. Unlike agents confined to in silico tasks, LabOS integrates its self-evolving multi-agent core with a physical lab module, utilizing robotic automation and multimodal perception to actively orchestrate experiments—from hypothesis generation to physical validation—thereby closing the loop between the digital 'brain' and the laboratory reality.
-
A fundamental weakness of current LLMs is their propensity to 'hallucinate'—generating plausible but factually incorrect information. In a scientific context where accuracy is paramount, this is a critical failure mode[95]. An incorrect gene function or a flawed experimental protocol generated by an AI could waste months of research or, in a clinical setting, lead to dangerous outcomes[96]. Beyond mere correctness, a lack of model interpretability poses another barrier; for an AI's output to be trusted, scientists must be able to understand the 'reasoning' behind its conclusions, a feature often lacking in complex 'black box' models[97]. Ensuring the scientific accuracy of AI outputs and developing methods for models to provide calibrated confidence scores are urgent research priorities[98]. To address this, Explainable AI (XAI) and attribution methods are emerging as essential tools. Feature attribution techniques, such as SHAP and Integrated Gradients allow researchers to quantify the contribution of a specific biological input, like nucleotide sequences or chemical substructures to a model's prediction, distinguishing valid biological reasoning from spurious correlations[99]. Furthermore, there is a recurring tension between the power of massive, generalist pre-training and the superior performance of specialized models on niche tasks. Some benchmarks have shown that simpler statistical models can outperform large foundation models on specific, well-defined problems, suggesting a 'No Free Lunch' theorem for biological LLMs[65,100]. This indicates that while large-scale pre-training provides a powerful starting point, deep domain-specific knowledge remains essential, and reinforces the idea that scale isn't everything.
Evaluation
-
Evaluating the performance of an AI scientist is a complex challenge. Unlike tasks with static benchmarks, the output of a research agent can be a multifaceted hypothesis, a dataset analysis, or a draft paper. Rigorous evaluation is difficult, as success might be a novel finding that is only recognizable in hindsight. One solution is to use simulated environments where the ground truth is known. For example, hiding a known pathway in simulated data and testing if the agent can discover it. Frameworks like scMultiSim, which can generate realistic single-cell and spatial data guided by known gene regulatory networks, are essential for creating these controlled environments for validation[101]. Another approach is direct competition, where AI agents and human teams enter the same scientific challenges[102]. Developing standardized benchmarks for 'AI as a scientist' is an active and crucial area for tracking progress.
Data privacy and bias
-
Biomedical AI models are trained on vast datasets that often contain sensitive patient information and reflect historical inequities in healthcare, creating two major ethical risks. The first is the potential for violating patient privacy, which necessitates strict adherence to regulations and robust cybersecurity. The second is the risk of perpetuating or amplifying existing biases. A model trained on data that overrepresents certain demographic groups may perform poorly for underrepresented populations, leading to unequal health outcomes. Algorithmic bias can also reinforce scientific dogma; for instance, an agent might prioritize well-studied drug targets over novel ones, thereby narrowing the scientific horizon. Careful data curation and explicit debiasing strategies are needed to mitigate these risks.
Safety and security
-
The generative power of biological AI comes with a significant dual-use risk. The GeneBreaker framework has demonstrated that DNA foundation models, including the powerful Evo2, are vulnerable to 'jailbreak' attacks[103]. Through carefully crafted prompts, these models can be coerced into bypassing their safety filters and generating DNA sequences with high homology to known human pathogens, such as the SARS-CoV-2 spike protein. Although end-to-end physical synthesis from such jailbreaks has not yet been demonstrated, this in silico ability to generate potentially harmful biological material poses a profound biosecurity threat by significantly lowering the technical barrier for designing hazardous agents, highlighting the urgent need for the development of more robust safety alignment techniques, monitoring, and tracing mechanisms for these powerful generative models. As agents become capable of not just designing, but also orchestrating wet-lab experiments, whether via robotics or by guiding human operators, the risk graduates from generating harmful information to initiating harmful synthesis.
Besides, it is crucial to differentiate the urgency of these threats. Cybersecurity risks are immediate and pervasive: an AI agent writing analysis code can already inadvertently or maliciously damage infrastructure or leak proprietary data. In contrast, autonomous biological synthesis remains an emerging, though high-stakes, future threat. To mitigate these distinct risks, a layered defense strategy is required: developers must implement strict sandboxing for AI-generated code to prevent digital damage, while the community must enforce rigorous screening of gene synthesis orders and hardware-level access controls for robotic platforms to prevent physical misuse.
A recent study from OpenAI also identified a 'toxic persona' feature that most strongly controls emergent misalignment, which could potentially be exploited to make AI systems malicious[104]. On the cybersecurity side, an AI agent that writes code for analysis presents another attack surface. If an adversary poisons a dataset or library, the agent could inadvertently execute malicious code, damage compute infrastructure, or leak data. Another subtle security issue is intellectual property: if an agent is trained on proprietary databases or papers, does it 'know' things that are under patent or confidential? This necessitates 'AI IP firewalls' to ensure, for example, that a pharmaceutical company's AI doesn't inadvertently incorporate a competitor's private data that it may have seen in public leaks during its training.
Cost and accessibility
-
The development of state-of-the-art foundation models is an immensely resource-intensive endeavor. Training a model like GPT-3 is estimated to cost millions of dollars, and the computational demands continue to grow exponentially[105]. This high cost concentrates the power to build and train these models in the hands of a few large, well-funded technology companies and research institutions. This creates a significant barrier to entry for most academic labs, startups, and researchers in lower-resource settings, risking the creation of a new digital divide that could stifle innovation and competition. To quantify these practical barriers, we summarize the computational requirements and accessibility of representative models in Table 1. As illustrated, the landscape is nuanced. To mitigate resource constraints, developers of massive models like Evo2 and xTrimoPGLM often release scaled-down versions (e.g., Evo2 offers 1B, 7B, and 40B variants), enabling broader adoption on modest hardware. However, accessibility remains bifurcated by licensing and deployment models: while some are fully open-source, others, like AlphaGenome, are restricted to API access, and AlphaFold3 is currently limited to non-commercial academic use via application. Consequently, while 'lightweight' options exist, access to the most powerful frontiers sometimes remains gated. Energy use is another aspect: widespread use of huge models has an environmental footprint, and the scientific community will need to consider the trade-off between compute usage and the benefits of discoveries made faster. Some have suggested that AI should tackle the sustainability of its own workflows, perhaps by having agents optimize their experiments to use less compute by intelligently pruning the search space.
Table 1. Computational landscape of major bio-foundation models.
Model name Type Parameters Training hardware Accessibility DNABERT-2 Genome 117 M 8 RTX 2080Ti https://huggingface.co/zhihan1996/DNABERT-2-117M GenomeOcean Genome 100 M, 500 M, 4 B 64 NVIDIA A100 https://huggingface.co/DOEJGI/GenomeOcean-4B HyenaDNA Genome 1 K, 16 K, 32 K, 160 K,
450 K, 1 M8 NVIDIA A100 https://huggingface.co/collections/LongSafari/hyenadna-models Nucleotide Transformer Genome 100 M, 500 M, 2.5 B 128 NVIDIA A100 https://huggingface.co/collections/InstaDeepAI/nucleotide-transformer Nucleotide Transformer v3 Genome 8 M, 100 M, 650 M / https://huggingface.co/spaces/InstaDeepAI/ntv3 Evo2 Genome 1 B, 7 B, 40 B > 2,000x NVIDIA H100 https://huggingface.co/collections/arcinstitute/evo AlphaGenome Genome 450 M 512 GOOGLE TPU +
64 NVIDIA H100API only RiNALMo Genome 650 M 7 NVIDIA A100 https://zenodo.org/records/15043668 ESM3 Proteome 1.4 B, 7 B, 98 B / https://huggingface.co/EvolutionaryScale/esm3-sm-open-v1 xTrimoPGLM Proteome 1 B, 3 B, 7 B, 100 B 768 NVIDIA A100 https://huggingface.co/biomap-research/proteinglm-100b-int4 AlphaFold2 Proteome 93 M 128 GOOGLE TPU https://github.com/google-deepmind/alphafold AlphaFold3 Proteome / / Apply for academic usage BioEmu Proteome 31 M / https://huggingface.co/microsoft/bioemu ProGen2 Proteome 151 M, 764 M, 2.7 B, 6.4 B / https://github.com/salesforce/progen/tree/main/progen2 -
While large foundation models possess a remarkable breadth of knowledge, their true power is unlocked through specialization and grounding. The opportunity lies in moving beyond general-purpose models to create highly accurate, domain-specific experts. Fine-tuning remains a cornerstone strategy, allowing pre-trained models like Evo2 or CellFM to be adapted to specific biological contexts, such as a particular cancer type, a novel sequencing technology, or a unique experimental dataset. This process sharpens their predictive accuracy and tailors their generative capabilities to the precise needs of a research question. Domain-specific adapters or low-rank fine-tuning (LoRA) layers can be swapped in or out to keep pace with the field while controlling cost[106].
To combat the critical issue of hallucination and ensure that AI-generated insights are scientifically valid, RAG is becoming an important tool[107]. The opportunity here is to build systems where AI agents are dynamically grounded in the latest, most reliable information (Fig. 5, left panel). Instead of relying solely on their internal, static knowledge, agents can query and incorporate data from external, curated sources in real-time. This could include the entire PubMed corpus, proprietary clinical trial data, up-to-the-minute genomic databases, or even a lab's own internal experimental results. RAG helps shift the role of the LLM from primarily a 'knower' to more of a 'reasoner' that synthesizes trusted information, providing more transparent, citable, and up-to-date outputs. As mentioned previously, BioAgents demonstrated that even a smaller open-source LLM (Phi-3) can effectively assist with complex bioinformatics pipeline design by employing RAG to inject domain knowledge on the fly. Other recent biological RAG systems, such as BioRAG, outperforms both vanilla GPT-4 and search-engine-only pipelines on life-science question answering, underscoring how crucial specialized retrieval and embeddings are for the bench biologist[108]. Furthermore, self-BioRAG is a framework that specializes in generating explanations, retrieving domain-specific documents, and self-reflecting on generated responses for biomedical text[109].
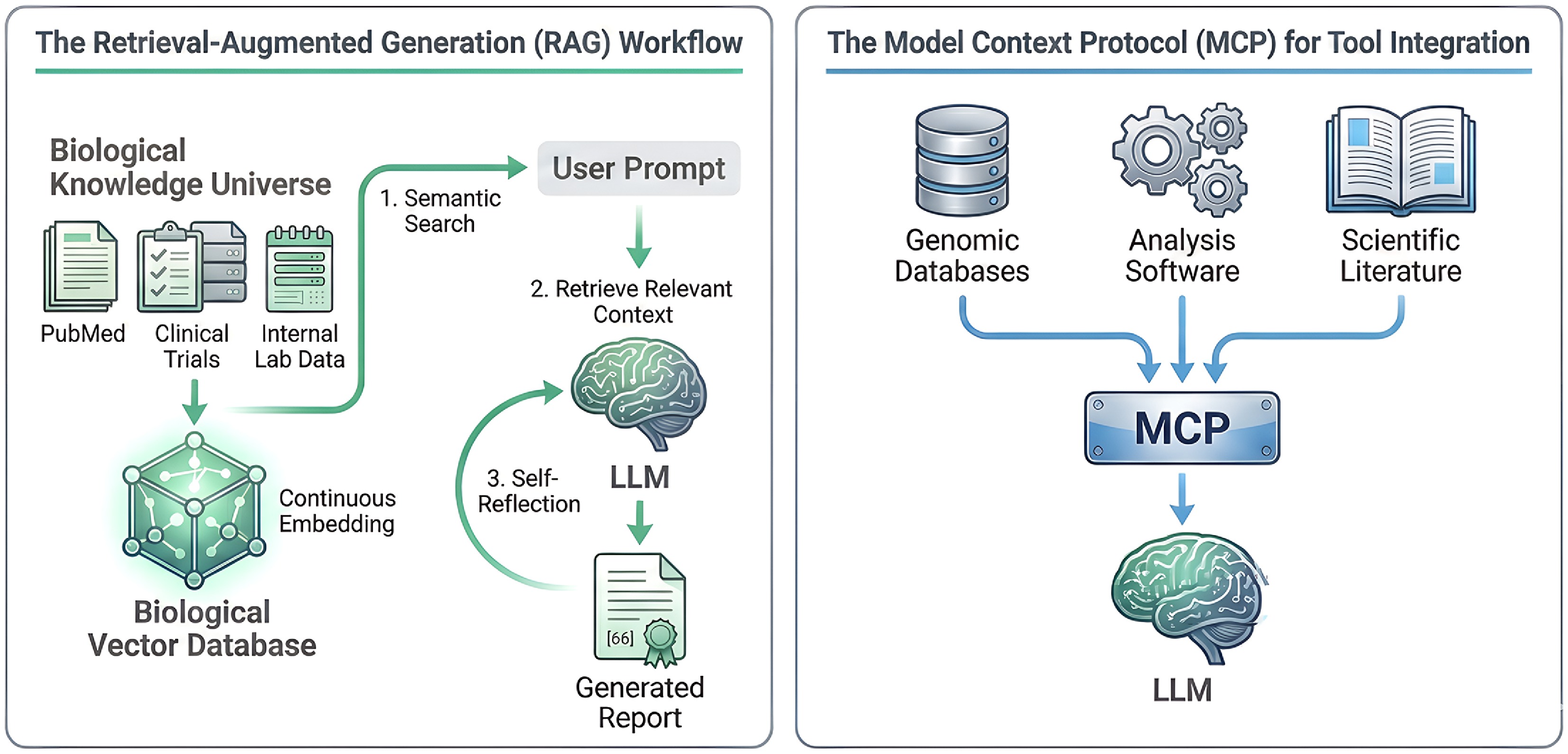
Figure 5.
Grounding AI agents with knowledge and tools. (Left) the Retrieval-Augmented Generation (RAG) workflow. To ensure factual accuracy, a diverse 'Biological Knowledge Universe' (e.g., PubMed, clinical trials, internal lab data) is converted into a 'Biological Vector Database'. When a user provides a prompt, a semantic search retrieves the most relevant context from this database. This information is then fed to the LLM, which uses it to generate a grounded and verifiable report, often incorporating a 'Self-Reflection' step to check its own output. (Right) the Model Context Protocol (MCP) for tool integration. MCP acts as a universal, standardized interface that allows an LLM to seamlessly interact with a wide array of external resources. This includes querying 'Genomic Databases', calling 'Analysis Software', or accessing the latest 'Scientific Literature'. By creating a common language for tool use, MCP simplifies the process of building context-aware agents that can leverage the best available resources for a given task.
A complementary development is the rise of the MCP as a flexible interface layer connecting models to tools and data sources[110]. Introduced in late 2024, MCP is an open standard that allows AI systems to securely access external services (like databases, computational tools, or lab instruments) through a unified protocol (Fig. 5, right panel). One can think of MCP as a 'USB-C port' for AI models—a standardized connector that replaces ad hoc, fragmented integrations with a consistent interface. By using MCP, a biology-focused LLM agent can, in principle, query a genomic database, call a protein folding API, or send commands to a microscope, all through a unified protocol[111]. An MCP could define how to package a patient's data—genomics, transcriptomics, imaging, and clinical history—into a single, coherent 'context' that an agent like TxAgent can ingest. This promises to simplify the tool augmentation of LLMs, enabling context-aware agents that automatically bring the right resources into their context window. Standardizing this process will be crucial for interoperability, allowing different AI agents and tools to seamlessly communicate and build upon each other's analyses, ensuring that the AI has the full picture before making a recommendation or generating a hypothesis[112]. Adopting MCP-style interfaces in bio-LLM services would let users chain fine-tuned models, RAG modules, and wet-lab schedulers without bespoke code, fostering an open ecosystem analogous to the early web. Today, multiple MCP servers and directories already expose tools that can be accessed through HTTP-based streaming APIs, making it easier to plug external capabilities into AI workflows.
Taken together, improved fine-tuning methods, RAG pipelines, and MCP-based tool integrations delineate a toolkit for adapting general models into biomedical specialists. A future 'digital biologist' agent might combine all three: a foundation model fine-tuned on biomedical texts and omics data, augmented by the retrieval of patient or literature data, and interfacing with lab equipment via MCP to perform experiments, thereby tightly coupling knowledge, context, and action in service of scientific discovery.
Multi-agent systems
-
The complexity of biological research rarely allows for a single expert to suffice; it requires a team. The future of AI in biology mirrors this reality, moving from monolithic, single-AI systems to collaborative multi-agent systems. As seen in frameworks like CellAgent, ProtAgents, and OriGene, the opportunity lies in creating digital research groups composed of specialized agents, each with a distinct role: 1) a Planner Agent that outlines the high-level research strategy; 2) a Literature Agent that performs comprehensive background research using RAG; 3) an Executor Agent that writes and runs code to perform data analysis using tools like Scanpy or Bioconductor; 4) a Critique Agent that evaluates the results, checks for errors, and suggests revisions; and 5) a Visualization Agent that generates publication-quality figures and charts. Notably, MCP may provide very broad possibilities for these agents to invoke a wide variety of tools. Likewise, environments like Biomni-E1 enables AI agents to access a wide range of specialized tools and knowledge. This modular approach cannot only be more robust and scalable, but also more transparent. By breaking down a complex task like 'discover a new drug target for liver cancer' into a series of sub-tasks handled by specialized agents, the overall reasoning process becomes easier to audit, debug, and validate by human experts. This collaborative architecture allows AI to tackle far more ambitious, multi-step research programs than any single agent could alone.
Human-AI collaboration
-
The future is unlikely to be one where AI fully replaces human scientists. Rather, it points towards a 'centaur' model of collaboration, where the unique strengths of humans and AI are combined. AI systems excel at processing vast amounts of data, executing complex analyses, and exploring enormous hypothesis spaces. Humans, in contrast, provide high-level strategic direction, domain intuition, creativity, and critical judgment—the scientific 'taste' that current AI lacks. Humans also provide the essential high-level direction, defining what constitutes a meaningful scientific question or a valuable discovery, thereby guiding the AI's vast analytical power. This partnership implies a distinct evolution for different roles: wet-lab biologists will increasingly focus on foundational research design and identifying the core scientific questions to be solved, validating AI-driven protocols, while bioinformaticians will transition from writing routine analysis pipelines to managing the agentic systems and curating the high-quality data that fuels them. The most powerful systems will be interactive, incorporating a human-in-the-loop for continuous feedback, guidance, and refinement, creating a synergistic partnership that outperforms either human or AI alone[113,114]. Technological strides are now physically bridging this partnership. Connecting the digital reasoning agents to the physical world, LabOS introduces an AI-XR framework where agents equipped with smart glasses can 'see' the experimental context[94]. By uniting computational reasoning with physical perception, it enables AI to actively assist in real-time execution of processes ranging from stem-cell engineering to cancer therapy, transforming the lab into a shared, intelligent workspace where human and machine discovery evolve together.
Causality: the next frontier
-
A major limitation of many current machine learning models is that they are masters of correlation, but novices at causation. They can identify that gene A's expression is associated with a disease, but they cannot inherently determine if gene A causes the disease. The next great frontier for AI in biology is to bridge this gap. Most current bio-LLMs operate on correlations. Embedding causal reasoning would let them propose interventions (CRISPR edits, small-molecule perturbations) with mechanistic justifications rather than heuristic confidence scores. Recent surveys on causal discovery for LLMs discuss various approaches to integrate causal reasoning, including interpretations of self-attention as Pearl-style structural causal models, and the application of invariant risk minimization in debiasing frameworks[115]. Perturbation models like STATE are a critical step in this direction. By learning to predict the cellular outcomes of specific genetic or chemical perturbations, these models are implicitly learning the causal wiring of the cell. In other fields, such as materials science, closed-loop autonomous platforms have demonstrated the ability to accelerate experimental cycles through real-time experiment-theory integration, suggesting potential pathways for biology.[116]. An AI armed with a robust causal model could move beyond descriptive analysis to perform in silico experiments. A scientist could ask, 'What is the likely outcome if I knock out this gene in this specific cell type?', or 'Which compound in this library is most likely to reverse this disease signature?'. Answering these questions is the essence of mechanistic understanding and therapeutic development. Achieving true causal reasoning will be a profound leap, enabling AI agents to generate novel, testable, and mechanistically-grounded hypotheses that drive discovery forward.
Closing the loop: self-driving labs
-
The ultimate culmination of these opportunities is the creation of 'self-driving labs'—fully autonomous, closed-loop research systems. This visionary concept integrates all the previously discussed elements into a continuous cycle of scientific discovery, powered by AI and robotics. The workflow would be as follows: (1) Hypothesis: an AI agent, using causal reasoning and knowledge from literature, generates a novel hypothesis (e.g., 'inhibiting protein X will reverse drug resistance in this cancer cell line'); (2) Design: a specialized agent designs a series of experiments to test this hypothesis, generating the precise protocols for robotic execution; (3) Execution: the protocols are sent to automated lab robotics, which perform the experiments (e.g., cell culturing, drug application, high-throughput imaging, or sequencing); (4) Analysis: the raw data from the experiment is fed back to a data analysis agent, which processes the results; (5) Iteration: the results are interpreted by the hypothesis agent, which then updates its internal model of the biological system. It either validates, refutes, or refines its original hypothesis and immediately begins the cycle anew. Such a closed-loop system would represent a fundamental paradigm shift, compressing research cycles that currently take months or years into a matter of days or weeks. By allowing AI to autonomously explore the vast landscape of experimental possibilities 24/7, self-driving labs hold the promise of dramatically accelerating the pace of discovery, and ushering in an era of unprecedented progress in medicine and biology[117].
-
The convergence of massive biological datasets with the powerful representational and reasoning capabilities of LLMs and AI agents represents a genuine paradigm shift in the life sciences. This review has charted the rapid and remarkable journey from foundation models that learn the fundamental languages of the genome and the cell, to specialized assistive tools that democratize complex analysis, and finally to the dawn of autonomous agents that are beginning to function as active participants in the scientific process. This evolution is giving rise to a new entity: the 'digital biologist', an AI collaborator that can augment and amplify human intellect. The trajectory of AI in biomedicine, as outlined in this review, is one of accelerating abstraction and autonomy (Fig. 6). The journey begins with foundation models learning the basic representations of biological data, progresses to the application of these models in discrete, assistive tools, and culminates in the emergence of autonomous agents that can plan, act, and even create. Several key trends are clear: (1) a move towards integrating multi-modal data for a more holistic view; (2) the rise of a dominant architectural pattern where a generalist LLM acts as a tool-using orchestrator, and (3) a conceptual evolution from static, one-shot systems towards dynamic, adaptive agents that can learn from feedback and collaborate with human scientists.
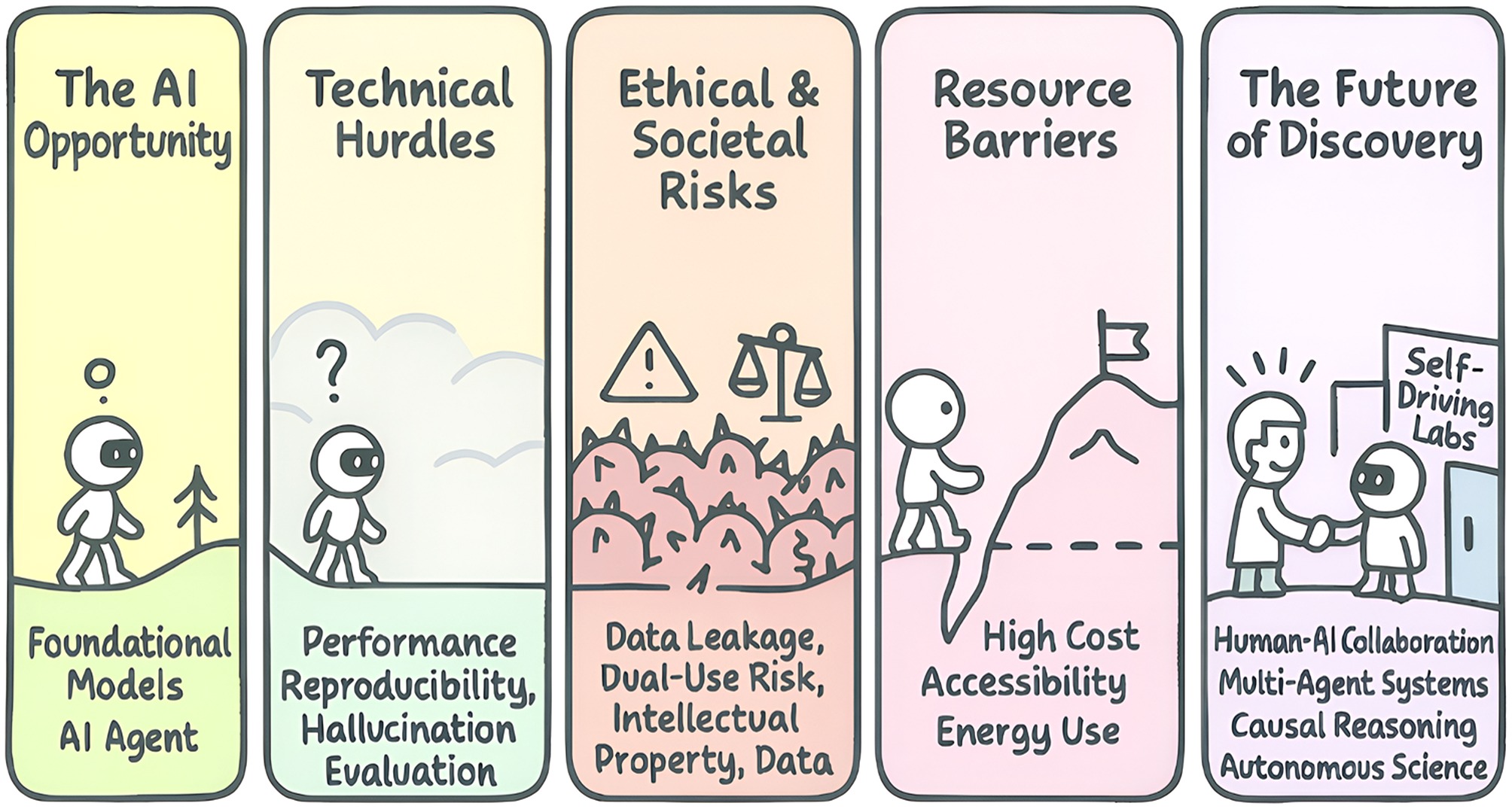
Figure 6.
The Path to trustworthy AI in biology: navigating challenges to unlock opportunities. This roadmap illustrates the progression from the core AI opportunity (foundation models and agents) to the future of discovery. This path requires overcoming technical hurdles (e.g., performance, hallucination), navigating ethical and societal risks (e.g., data privacy, dual-use), and breaking down resource barriers (e.g., cost, accessibility). Successfully doing so will enable a new era of science defined by human-AI collaboration, causal reasoning, and autonomous labs.
Besides, we must critically distinguish between high-potential directions and overhyped expectations. While scaling parameters improves general fluency, the blind pursuit of scale appears increasingly overhyped, as recent evidence suggests that specialized, smaller models often outperform larger generalist models in biological tasks. Conversely, the most promising avenue lies in the 'modular expert' approach, integrating reasoning layers via RAG and MCP with specialized tools. This distinction directly frames the bottlenecks in evolving from Level 1 'assistants' to Level 3 'autonomous scientists'. The transition to Level 2 is currently gated by the challenge of robust tool integration, whereas the ultimate leap to Level 3 is stalled not by data volume but by the causality gap. Without the ability to distinguish correlation from causation and verify findings in closed-loop systems, agents cannot safely graduate from suggesting hypotheses to autonomously executing them.
However, realizing the full potential of this paradigm requires confronting a set of formidable and interconnected challenges. The path forward is laden with significant issues of reliability, rigorous evaluation, data privacy, algorithmic bias, and the profound biosecurity risks of generative biology. These must be navigated with foresight, responsibility, and a commitment to ethical principles. Furthermore, the high cost of developing these technologies also risks creating a new digital divide that could limit the scope of scientific inquiry. If these challenges are met, the potential is immense. The future of science will likely be a hybrid one, characterized by a deep, synergistic collaboration between human researchers and AI agents. By connecting these intelligent agents to robotic automation in self-driving labs and by pushing them beyond correlation towards a true understanding of causality, we stand on the cusp of a new era. This AI-augmented science promises to dramatically accelerate the pace of discovery, deepen our understanding of the intricate machinery of life, and ultimately usher in a new age of truly personalized and predictive medicine. In closing, one cannot help but feel that we are witnessing the emergence of a new era of science, in which discoveries are accelerated by orders of magnitude. The convergence of big data and big models means that an ambitious question (like 'How do we regenerate organs?', 'How does the microbiome affect mental health?', or 'How can we finally conquer cancer?') might be tackled not by decades of slow incremental work, but by a concerted human–AI team rapidly iterating through hypotheses and experiments. If the 20th century was defined by the molecular biology revolution, the 21st may be defined by this AI-driven research revolution. The 'digital biologist', armed with LLMs and AI agents, will not replace the creative intuition of human scientists, but will empower it, helping us see patterns we couldn't, generating options we wouldn't, and carrying out work we didn't have time for. Together, human and AI agents can push the frontiers of knowledge faster and further, ushering in new understandings of life and new solutions for health. The dawn has broken; it will be up to us to ensure that this new light is used wisely and for the benefit of all.
-
During the preparation of this work, the authors used Gemini 3.0 Pro (January 2026) for language refinement and stylistic editing, and Gemini Nano Banana (January 2026) for scientific figure enhancement and visualization. The authors reviewed and edited all content produced with the assistance of these tools, verified its accuracy, and take full responsibility for the integrity and originality of the final manuscript. This work represents the authors' own intellectual contribution, and no AI tool is credited as an author.
-
The authors confirm their contributions to the paper as follows: design and writing of the manuscript and shape the scope of the review: Huang S, Peng Y; preparation of this manuscript: Huang S, Peng Y, Lang M; Provided critical feedback and revised the manuscript: Lang M. critically revised the manuscript for important intellectual content: Chen Z, Yang C, Huang X, Mohtashaminia Z. All authors reviewed and approved the final version of the manuscript.
-
The comprehensive resource list of foundation models, AI agents, and datasets discussed in this review is publicly available and actively maintained to support the research community. The project repository is hosted on GitHub at https://github.com/Webioinfo01/Awesome-AI-Meets-Biology. Additionally, a user-friendly web interface for accessing these resources is available at http://awesomebio.webioinfo.top.
-
We thank all the members of the Webioinfo team.
-
The authors declare that they have no conflict of interest.
- Copyright: © 2026 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Huang S, Lang M, Chen Z, Yang C, Huang X, et al. 2026. From foundation models to autonomous agents in biology. Genomics Communications 3: e006 doi: 10.48130/gcomm-0026-0005 

From foundation models to autonomous agents in biology
- Received: 13 December 2025
- Revised: 27 January 2026
- Accepted: 25 February 2026
- Published online: 28 March 2026
Abstract: Advances in sequencing and multi-omics have unleashed exponential biological data growth, from genomes and transcriptomes to single-cell and spatial profiles. Traditional pipelines, reliant on manual curation, strain under this deluge. This bottleneck hampers discovery from terabyte-scale datasets. Large Language Models (LLMs) and AI agents are emerging as a powerful paradigm to address these challenges. Breakthroughs in foundation models pre-trained on biological 'languages' offer in-context learning and generative capabilities far beyond prior bioinformatics tools. By coupling LLM reasoning with multi-agent systems, Retrieval-Augmented Generation (RAG), and the Model Context Protocol (MCP), autonomous AI research agents can plan experiments, execute analyses, and even generate hypotheses with minimal human guidance. While these technologies promise to augment human intellect, their deployment presents critical challenges in reliability, biosecurity, and accessibility. Navigating these obstacles is key to ushering in an era of accelerated discovery and personalized medicine. By moving from static models to active agents, we are witnessing the rise of the 'digital biologist'—an AI collaborator poised to reshape biomedical research. We trace this paradigm's rapid evolution, from foundation models learning the language of biological sequences and single-cell data, to autonomous agents capable of automating analysis, designing experiments, and driving drug discovery. By synthesizing these developments, we offer a strategic roadmap for researchers to navigate the opportunities and challenges of this AI-driven era. Finally, to support the community, a public, actively maintained resource list of models, agents, and datasets is available on our project website: http://awesomebio.webioinfo.top.
-
Key words:
- Large Language Models /
- AI Agents /
- Foundation Models /
- Computational Biology /
- Autonomous Science