









-
Colorectal cancer (CRC) ranks as the third most prevalent malignancy globally and the second major contributor to mortality from cancer. In 2020, it was estimated to have affected around 1.9 million individuals, resulting in approximately 935,000 fatalities[1] . Nowadays, a concerning trend of increasing incidence is observed that the number of new CRC cases worldwide is expected to surge to 3.2 million in 2040 due to economic progression and the adoption of Western lifestyles. This surge in CRC incidence challenges global public health[2,3]. Although colonoscopy could help to detect early-stage CRC patients, 25%−50% of them will still progress to metastatic tumors, even when the lesion in situ is resected in time[4]. Metastatic CRC most often targets the liver as its primary organ[5].
The liver's susceptibility to CRC metastasis is multifactorial. Its rich blood supply, coupled with unique anatomical features, increases the likelihood of circulating cancer cells colonizing. The liver's dual blood supply from the hepatic portal vein and the hepatic artery, along with the properties of its sinusoidal endothelial cells—discontinuous, highly permeable, and sluggish blood flow—facilitate the metastatic process[6]. Second, perivascular hepatic stromal cells (fibroblasts around the portal veins and hepatic stellate cells around the hepatic veins) provide unique support for metastatic cancer cell proliferation, and angiogenesis[7]. However, many researchers have illustrated that circulatory patterns cannot fully explain the mechanisms driving cancer metastasis to specific organs[8]. As a critical immune organ and the metabolism center, the liver harbors a distinct immune microenvironment populated by various immune cells. The liver's routine exposure to dietary antigens and intestinal bacterial components necessitates its tolerance to antigenic stimuli[9]. This suppressive immune microenvironment, together with the cancer cells' influence, contributes significantly to the development of colorectal cancer liver metastases (CRLM).
In this review, we will elaborate on the function of the liver immune microenvironment in CRLM, which may help explore therapeutic approaches targeting the immune microenvironment and provide ideas for future research.
-
The progression of CRLM encompasses a complex and multifaceted series of events, broadly categorized into five distinct phases, each with its unique characteristics.
Pre-metastatic phase
-
The journey of CRLM begins with the pre-metastatic phase, which contains tumor cells entering the circulation and primary tumor-derived factors facilitating a premetastatic ecological niche. Bacteria residing within tumors like Escherichia coli undermine the intestinal vascular barrier, enabling tumor cell entry into circulation[10]. Tumor cells can also detach from each other (by downregulating E-cadherin) and then enter the circulation through new blood vessels and lymphatic vessels attributed to degrading the basement membrane[11]. Meanwhile, specific gene expression profiles of the primary tumor induce CRC cells to secrete soluble factors and exosomes, thereby promoting remote activation of hepatocytes and pre-metastatic niches of circulating cancer cells. This process involves multiple immune cells, signaling pathways, and molecules, such as Granulocyte-macrophage colony-stimulating factor (GM-CSF), macrophage migration inhibitory factor (MIF), TGF-β and TNF-α signaling, S100 family proteins S100P and S100A8 (Fig. 1), and other chemokines and cytokines (like CCL20, CCL5, CXCL2, CXCL5, and CXCL12)[12−16]. The exosome can also downgrade Kupffer Cells' (KCs) matrix metalloproteinases (MMPs) expression to enhance vascular permeability, contributing to the formation of the pre-metastatic niche[17]. Figure 1 describes in detail the process of this stage, i.e. how the pre-transfer niche is formed.
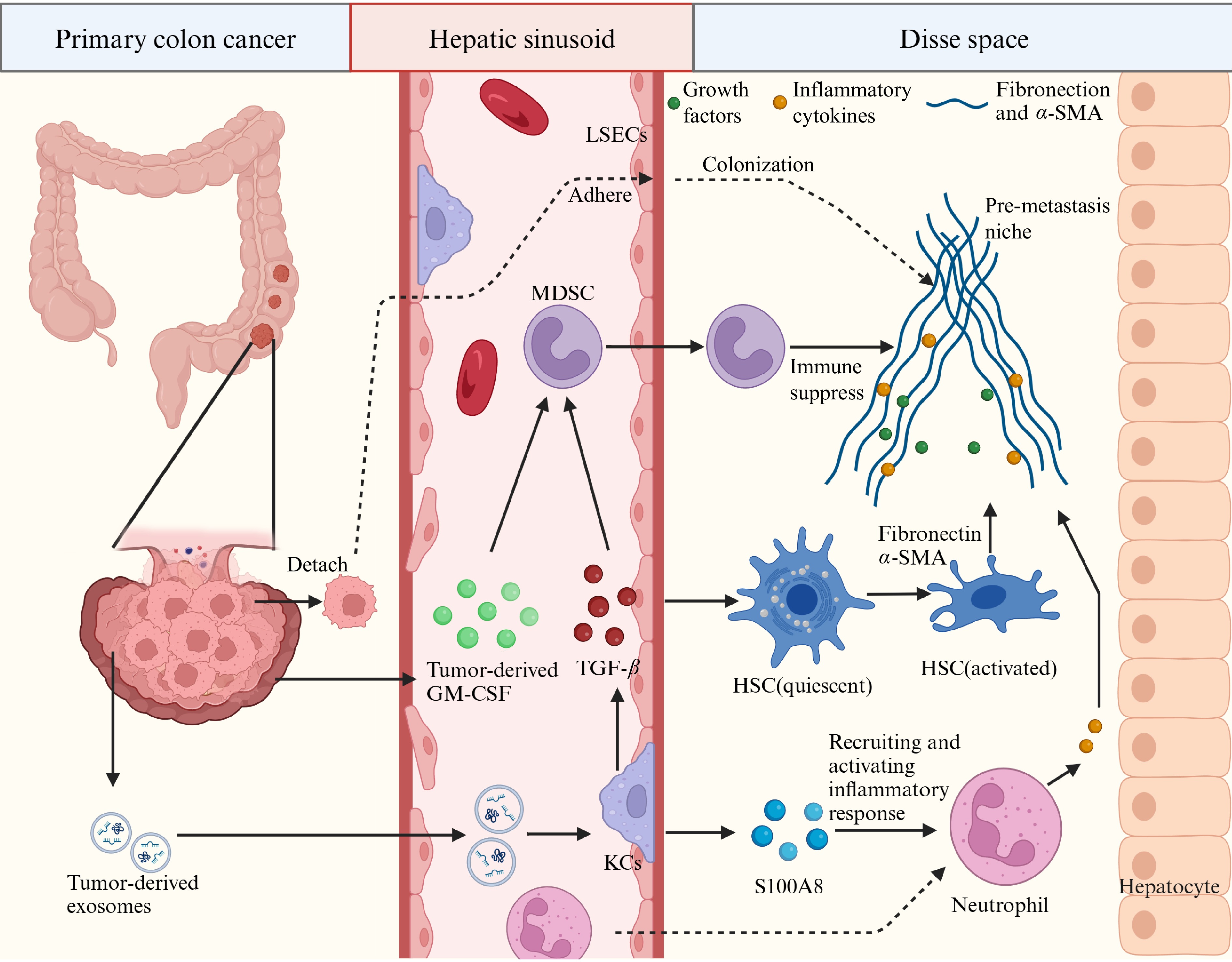
Figure 1.
The pre-metastatic niche. Shown is a diagrammatic representation of the cells and molecules involved in the formation of premetastatic niches in the liver. Disse space, also known as perisinusoidal space, is the liver blood sinusoidal endothelial cells and hepatocytes between a narrow gap.
Microvascular phase
-
In this phase, the cancer cells get to the liver and become embedded in the hepatic blood sinusoid, where they might evade immune surveillance. Deformation and mechanical stress injury of cancer cells in hepatic blood sinusoids can lead to cancer cell death. Cancer cells are also killed by phagocytosis of KCs and antitumor cytotoxicity of NK cells[18−20]. During CRC colonization, the liver's sinusoidal endothelial cells (LSECs) adopted a tumor-derived phenotype as adhesion molecules (ICAM-1, VCAM-1) expression and activation of mannose receptor-mediated endocytosis, which may help CRC cells avoid immune killing[21,22]. Tumor-activated hepatic stellate cells (HSCs) produce soluble inflammatory factors (like IL-6, TGF-β, and TNF) that facilitate more HSCs differentiation and myofibroblast recruitment into hepatic micrometastasis, which will contribute to the next phase[18]. The metastatic niche is key for promoting cancer metastasis since it provides a suitable microenvironment, including organotropism, vascular permeability, inflammation, and immunosuppression, in the liver for disseminating tumor cells.
The extravascular, pre-angiogenic phase
-
During this phase, cancer cells that escape immune surveillance post-microvascular invasion commence their growth in the metastatic niche. Within the pre-metastatic niches, cancer cells are surrounded by HSCs and KCs. The niches' microenvironment is characterized by hypoxic portal blood, enriched with metabolic substrates, pro-inflammatory cytokines, and reactive oxygen species. Despite challenges in endothelial adhesion and extravasation, metastatic cancer cells have developed strategies for survival within the vasculature[23]. Metastatic cells also enhance their spread by proliferating within the capillaries, disrupting the liver's sinusoidal structure. A pattern of replacement growth characteristic of metastatic lesions follows this disruption. Moreover, HSCs, when activated by tumors, release soluble factors like MCP-1 and TGF-β. These factors stimulate inflammatory responses and attract myofibroblasts to hepatic micrometastases. The micrometastases then secrete fibronectin and collagen, crucial for forming new blood vessels[24,25].
Angiogenic phase
-
With the activation of HSCs and the release of proangiogenic factors, the metastatic niche undergoes vascularization, providing the necessary nutrients and oxygen for tumor growth. At the tumor-liver interface, activated LSECs, together with vascular endothelial growth factor (VEGF) from activated HSCs, foster the development of intra-metastatic blood vessels[26]. Proangiogenic factors released by tumor-activated myofibroblasts and cancer cells drive angiogenesis, countered by endogenous liver-derived anti-angiogenic factors like endostatin that suppress this process[27]. Endothelial cell migration is selectively directed towards avascular micrometastases with a dense myofibroblastic infiltrate, ensuring that these early metastatic sites receive the support they need to develop further[18]. The immunosuppressive microenvironment is formed by various immunosuppressive cells like immunosuppressive regulatory T (Treg) cells, myeloid-derived suppressor cells (MDSCs), and macrophages, which are recruited and activated through multiple mechanisms in this phase. The balance between pro- and anti-angiogenic factors determines the success of this phase, with therapeutic opportunities targeting the formation of new blood vessels within the tumor.
The growth phase
-
The final phase is characterized by the establishment of clinically apparent metastases, where the tumor's expansion begins to significantly impact liver function[28]. Shielded by the liver's natural immune tolerance and the suppressive immune environment, CRC cells establish an adequate blood supply. This support facilitates their swift proliferation, culminating in the development of a metastatic tumor that becomes clinically apparent[29]. The liver's tissue architecture, physiological roles, and the clinical manifestations of patients can vary significantly based on the location of liver metastatic lesions. Therapies targeting this phase aim to disrupt the tumor's blood supply and immune evasion mechanisms, to slow or halt tumor growth.
As we deepen our understanding of the liver's immune landscape and the phases of CRLM, new avenues for immunotherapy are emerging. These include strategies that target the early pre-metastatic niche to prevent the initial seeding of metastatic cells, as well as approaches that disrupt the later angiogenic and growth phases. Therapies targeting the angiogenic phase and immune checkpoints extensively investigated, we can potentially develop more effective treatments that not only limit tumor growth but also reactivate the immune system to combat CRLM by targeting specific molecular pathways and cellular interactions[30]. Moreover, the stromal cells of the liver, such as LSECs and HSCs, play a vital role in the colonization and growth of CRLM, so targeting them may directly inhibit the development of metastatic liver cancer.
-
The liver possesses a unique immune environment teeming with immunocompetent cells. This complex and dynamic microenvironment is a network of both immune and non-immune stromal cells that work together to maintain liver homeostasis and respond to various stimuli. It comprises Kupffer cells (resident liver macrophages), LSECs, HSCs, natural killer (NK) cells, dendritic cells (DCs), diverse T cell populations, and bone marrow-derived immune cells that migrate to the liver. Additionally, the liver hosts suppressive cells like regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs)[31]. Therefore, the hepatic immune system plays a dual role in CRLM: it can eliminate cancer cells while potentially fostering their colonization and growth[32,33].
Non-immune stromal cells
Kupffer cells
-
KCs, the liver's resident macrophages, form the initial defense of the hepatic immune. KCs exhibit a dual role in tumor metastasis, initially inhibiting and later promoting cancer spread[34,35]. During the early stages of CRLM, KCs phagocytose cancer cells and release cytokines such as tumor necrosis factor-alpha (TNF-α), interleukin-1 alpha (IL-1α), and IL-1β (Fig. 2). These cytokines activate neutrophils, NK cells, and other immune cells, contributing to the suppression of liver metastasis from colorectal cancer[36,37].
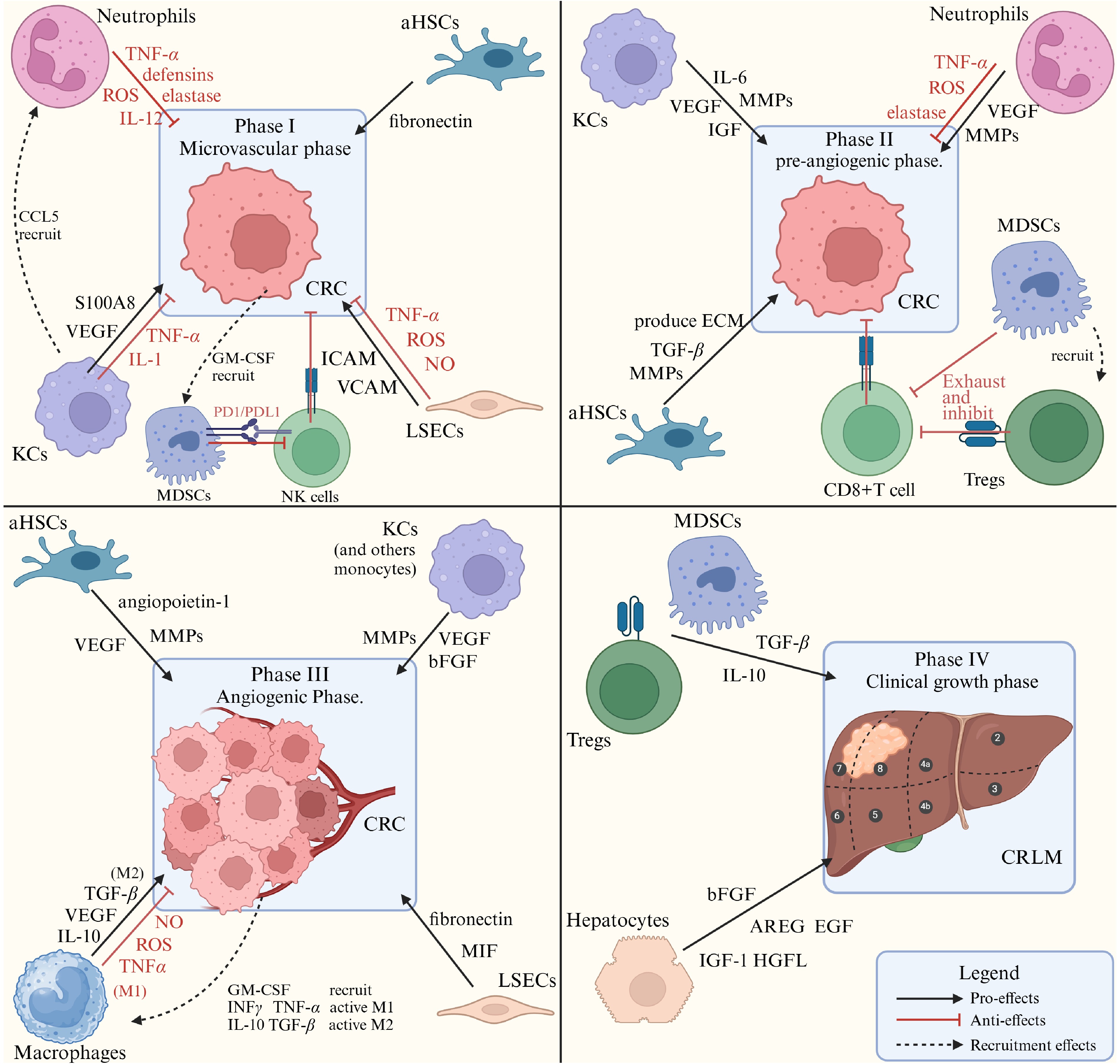
Figure 2.
Different kinds of cells in the liver immune microenvironment and their roles in CRLM phases. The secreting factors that promote and inhibit tumor colonization and growth, from varieties of liver cells are listed. MMPs: matrix metalloproteinases; VEGF: vascular endothelial growth factor; MIF: macrophage migration inhibitory factor; AREG: amphiregulin, an EGF-like protein; bFGF: basic fibroblast growth factor; HGFL: hepatocyte growth factor-like protein.
KCs also promote tumor metastasis and growth. They activate and expand Tregs through antigen presentation and promote immune tolerance by upregulating PD-L1. KCs also contribute to chronic inflammation by secreting IL-6, which facilitates pro-tumor microenvironments[38]. Studies have shown that exosomal ANGPTL1, derived from KCs, typically suppresses the JAK2-STAT3 signaling pathway, reducing MMP9 production in these cells. However, in the context of CRC, exosomes from tumor cells have been observed to contain significantly lower levels of ANGPTL1. This decrease in ANGPTL1 leads to an upregulation of MMP9, which, alongside VEGF from KCs, increases vascular permeability and facilitates tumor cell extravasation, thereby promoting tumor growth and metastasis[36,39,40].
Moreover, in the context of CRLM, KCs exhibit considerable plasticity, which allows them to shape the liver metastasis microenvironment after differentiating into M2-type macrophages. The secretion of IL-6 by metastatic CRC cells can promote the polarization of KCs towards the M2 phenotype. This polarization is crucial for the progression of liver metastasis, as M2 macrophages play a distinct role in the tumor microenvironment, which will be detailed further below[41].
The liver's sinusoidal endothelial cells (LSECs)
-
LSECs play an important role in physiological and immunological processes. They act as filters, removing harmful substances from the bloodstream and engaging in immune responses by endocytosing pathogens and presenting antigens[42,43]. Additionally, LSECs function as a selective barrier, facilitating the uptake of molecules like proteins and drugs, while effectively blocking the passage of whole cells[8,44].
LSECs also play a bimodal role in CRLM. While CRC retaining in hepatic sinusoid, mechanical stress injury, and deformation of cancer cells can lead to cancer cell death. LSECs can activate mannose receptor-mediated endocytosis, which kills cancer cells and initiates the antigen presentation pathway. LSECs also secrete inflammatory factors (such as IFNγ, NO, ROS, and TNFα, shown in Fig. 2), to recruit immune cells, upregulate Fas, and promote CRC apoptosis through the Fas/FasL pathway[45,46].
The pro-inflammatory response to cancer can however lead to elevated levels of vascular adhesion factors, including E-selectin, integrins, cadherins, and members of the cell adhesion molecule (CAM) family, such as VCAM-1 and ICAM-1. These molecules enhance the adhesion of cancer cells to LSECs, a process critical for the initial steps of metastatic colonization[47−49]. Additionally, LSECs secrete a unique adhesion molecule termed LSECtin, which potentially dampens the cytotoxic capabilities of T cells and suppresses the immune system's ability to target and eliminate CRC[50]. LSECs also play a vital role in epithelial-to-mesenchymal transition (EMT) and cancer angiogenesis, which are strongly associated with CRC invasion, metastasis, and growth[51].
Hepatic stellate cells (HSC)
-
Hepatic stellate cells, as resident unactivated hepatic mesenchymal stromal cells, can promote liver fibrosis and cancer when activated by exosomes (EV miR-181a-5p), TGF-β, IL-6, and many other mechanisms[52−54]. HSCs contribute to the immune cells' recruitment and formation of the pre-metastatic niche by secreting various cytokines and chemokines that create a conducive environment for incoming cancer cells[55,56].
During CRLM, HSCs undergo myofibroblast transdifferentiation, acquiring cancer-associated fibroblast (CAF) characteristics. In this state, they produce fibronectin and alter the extracellular matrix (ECM), providing structural support through α-SMA and acting as a reservoir for growth factors that promote angiogenesis[57,58]. Additionally, activated hepatic stellate cells (aHSCs) secrete TGF-β, which inhibits the activation of CD4+ Th1 cells and dampens cytotoxic T-cell responses, fostering an immunosuppressive environment that protects cancer cells from immune detection[55,59]. Moreover, aHSCs' secretion of MMP-8 and MMP-9 is crucial for cancer progression in CRLM. These matrix metalloproteinases (MMPs) facilitate cancer cell invasion and activate growth factors, thereby stimulating tumor growth and angiogenesis[60,61]. Research indicates aHSCs can promote the CRLM through the CCL20/CCR6 axis. Moreover, CRC cells secrete miR-181a-5p, which increases CCL20 expression in aHSC, suggesting a positive feedback mechanism[54].
Recent studies have demonstrated that QGAP1 can inhibit the activation of HSCs by suppressing TGF-β receptor II signaling. QGAP1 downregulation was observed in myofibroblasts associated with human CRLM, suggesting a role in HSC activation[62]. These pieces of evidence suggest that HSCs play an important role in the CRLM process and hold promise as new therapeutic targets. However, there is currently a scarcity of direct evidence supporting the effectiveness of targeted therapies for HSCs, largely attributed to the absence of clinical drugs that can specifically target HSCs without causing toxicity.
Hepatocyte
-
Hepatocytes, the primary cellular constituents of the liver, play a multifaceted role in the metastatic spread of CRC to the liver. Firstly, hepatocytes mediate the adhesion of CRC cells by integrins, which facilitate the formation of liver metastasis (LM)[63]. They also contribute to the tumorigenic process by producing growth factors such as Insulin-like growth factors (IGF-I) and hepatocyte growth factor-like protein (HGFL), which stimulate the proliferation and survival of cancer cells[64,65]. Moreover, hepatocytes have been shown to alter the transcriptome of tumors, facilitating LM through the activation of IL-6–STAT3 signaling and the overexpression of serum amyloid A (SAA), which in turn reshapes the hepatic tumor microenvironment (TME) to create a pre-metastatic niche conducive to the establishment of metastatic foci[66,67]. Moreover, hepatocytes contribute to the invasiveness of cancer cells by undergoing EMT, offering stromal support. This process also involves the promotion of angiogenesis, which is crucial for tumor growth. Hepatocytes secrete basic fibroblast growth factor (bFGF), thereby stimulating the formation of new blood vessels and reinforcing their integral role in the intricate dynamics between liver tissue and CRLM[68].
Immune cells
Neutrophils
-
Neutrophils in CRLM exhibit a dualistic role, divided into N1 and N2 subtypes. Through the CXCR2 receptor, neutrophils are drawn to the tumor microenvironment by soluble factors released by KCs, LSECs, and tumor cells[18]. Specifically, IFN and IL-1β drive the polarization toward the N1 phenotype, while TGF-β, IL-6, and IL-8 steer toward the N2 phenotype[69]. Activated N1 neutrophils, known as tumor inhibitors, can eliminate cancer cells by generating inflammatory mediators such as ROS, NO, TNF-α, IL-12, and defensins, as well as by recruiting CD8+ cytotoxic T cells and macrophages[70].
Conversely, N2 neutrophils, or tumor-promotion neutrophils, contribute to pre-metastatic niche formation when interacting with S100A proteins[71]. Influenced by tumor-derived cytokines like IL-8, N2 neutrophils increase the expression of integrins CD11b/CD18, enhancing their adhesion to tumor cells via ICAM-1 and facilitating CRC trans-endothelial migration[72]. Furthermore, N2 neutrophils release MMP-8, MMP-9, cathepsin G, and neutrophil elastase, which promote cancer cell extravasation and metastasis[73,74]. They also secrete molecules such as arginase, and myeloperoxidase (MPO), which suppress T cell and NK cell function, thereby promoting tumor growth[75].
Tumor-associated macrophages (TAMs)
-
Tumor-associated macrophages (TAMs) are macrophages that infiltrate malignant tissues, conventionally categorized into two subtypes: M1 and M2 macrophages[76]. Dormant macrophages were activated by GM-CSF and stimulated by LPS, IFN-γ, and TNF-α to form M1-type macrophages[77]. M1 macrophages can produce NO, TNFα, and IL-12 to kill cancer cells directly and recruit T cells and NK cells. M1 can also regulate T cell activation through MHC aids[78−80].
On the other hand, cytokines such as TGF-β, IL-4, IL-10, IL-13, and macrophage colony-stimulating factor (M-CSF) are key in the initial activation of M2-like macrophages[81]. Within the tumor microenvironment, M2 macrophages stimulate the generation of Tregs by releasing prostaglandin E2 (PGE2), IL-10, and indoleamine 2,3-dioxygenase (IDO), and attract Tregs through chemokines CCL17, CCL18, and CCL22. Moreover, IL-10 from Tregs induces monocytes to express PD-L1, which collectively helps in suppressing effector T cells[82,83]. M2 macrophages also aid in cancer invasion and metastasis by producing MMPs for matrix remodeling and by secreting growth factors like VEGF, EGF, and bFGF, which promote tumor growth[84].
Recent studies have shown that exosomes from CRC cells, rich in miRNAs, can reprogram macrophages metabolically, pushing them toward an M2 phenotype[85]. Additionally, the secretion of Wnt ligands by cancer cells enhances the polarization of TAMs towards the M2-type via the Wnt/β-catenin signaling pathway, thereby promoting CRLM[86]. As the immune editing of the TME and the shift in macrophage subtypes emerge as potential anticancer strategies, a detailed understanding of macrophage populations in CRLM is increasingly important. A previous single-cell analysis study identified MRC1+, CCL18+ and SPP1+ macrophages as the predominant M2 subsets within liver metastatic tissue[15,87].
Myeloid-derived suppressor cells (MDSCs) and Treg cells
-
CRCs and associated stromal cells release multiple chemokines and cytokines like IL-10 and TGF-β. These factors facilitate the recruitment and proliferation of MDSCs within the tumor microenvironment. MDSCs express galectin-9, which binds to the T cell surface antigen TIM-3, triggering T cell apoptosis. MDSCs also suppress cytotoxic T-cell function by recruiting Treg cells and mediate immune evasion by upgrading PD-L1[88,89]. The accumulation of MDSCs is a hallmark immunological feature of CRC, playing a role in pre-metastatic niche formation[90]. MDSCs are classified into polymorphonuclear MDSC (PMN-MDSC) and monocytic MDSC (M-MDSC) phenotypes; however, their specific roles and defining markers within the hepatic immune microenvironment require further exploration[91].
T lymphocytes, natural killer (NK) cells, and dendritic cells
-
It's widely known that NK cells, abundant in the liver, are capable of directly eliminating tumor cells through the expression of TNF-related apoptosis-inducing ligand, IFN-γ, and FasL[92]. Similarly, cytotoxic T cells can destroy tumor cells by releasing perforin and TNFα, and by attracting macrophages[93]. The diminished activity of T cells and NK cells within the TME is a significant concern, prompting research aimed at rejuvenating their function to curb tumor growth and spread. One such approach involves inhibiting transmembrane 4 L six family member 5 (TM4SF5), which can decrease STAT3 signaling and enhance NK cell surveillance, resulting in reduced fibrotic and cancerous phenotypes and extended survival[94].
Dendritic cells (DCs) play a significant role in activating naïve T cells and initiating adaptive immune responses as specialized antigen-presenting cells. A subset of regulatory DCs within tumors induces Treg differentiation, fostering immune tolerance through CTLA-4-dependent IL-10 and IDO production[95]. However, DCs' unique role in CRC liver metastasis requires further investigation.
Currently, the roles of various cells within the liver immune microenvironment have become increasingly clear (shown in Fig. 2), yet the classification of these cells remains a subject of investigation. The simplistic N1/N2 and M1/ M2 classifications may not suffice for the selection of cells to study their specific functions, as this binary division might not fully capture their biological diversity. Exploring additional phenotypes and biomarkers, as well as promoting cellular phenotype transitions to enhance tumor immunity, may be the main direction for current TME research.
-
CRC has a high propensity for liver metastasis, a phenomenon influenced not only by the liver's immune microenvironment but also by the cancer cells themselves. Exosomes, in particular, play a significant role in this process. These lipid bilayer vesicles are instrumental in mediating communication between CRC cells and the liver environment, promoting the formation of pro-metastatic niches and a suppressive immune milieu[96].
CRC-derived exosomes contain various molecules such as protein (FasL, TGFβ, angiopoietin-like protein 1 (ANGPTL1), AKT, tumor necrosis factor-α (TNF-α), β-catenin), RNA (such as miR-21, miR-25-3p, miR-375-3p, miR-934, and miR-181a-5p)[97,98]. Upon liver arrival, exosomal contents can trigger the development of pre-metastatic niches and enhance cell activities such as propagation, inflammation, invasion, and EMT, which in turn promote the spread of CRC[15,86].
Interactions between CRC-derived extracellular vehicles (EVs) and resident liver cells, such as Kupffer cells and HSCs, trigger signaling pathways that enhance the secretion of immunosuppressive cytokines. This results in the recruitment and expansion of MDSCs and the polarization of TAMs towards an M2 phenotype, which are both instrumental in creating a conducive environment for tumor growth. Furthermore, the presence of CRLM induces the transformation of HSCs into cancer-associated fibroblasts (CAFs), which secrete factors that enhance extracellular matrix remodeling and chemotactic gradients, thereby facilitating the migration and invasion of metastatic cancer cells[54,99,100]. TGF-β and FasL from exosomes inhibit T cell activity and promote apoptosis, further reinforcing the immunosuppressive environment[101].
CRC's immune evasion
-
Primary CRC acquires immune-evasive character mainly by epithelial-to-mesenchymal transition (EMT), which is the process of transformation of epithelial cells to mesenchymal cells. Over 90% of human CRC cells exhibit EMT, making EMT a promising target for preventing primary cancers from becoming invasive or for halting metastasis and recurrence after tumor resection. Figure 3 shows the cause of EMT activation and the main signaling pathways (including TGF-β, Wnt, Notch, and others). Activation of EMT in tumor cells induces the dissolution of inter-cell adhesion, disruption of apical-basal polarity, and reorganization of the cytoskeleton, thereby promoting cell migration, invasion, survival in the circulation, and eventual growth of distant metastases[102]. Recent research indicates that EMT is linked to the creation of immunosuppressive tumor microenvironments and heightened levels of immune checkpoint inhibitors[103]. Cancer cells undergoing EMT display a decrease in major histocompatibility complex class I (MHC-I) and an upregulation of PD-L1, while also increasing the secretion of pro-inflammatory mediators that draw in immunosuppressive cells like Tregs and MDSCs, and guide TAMs towards the M2 phenotype[104,105].
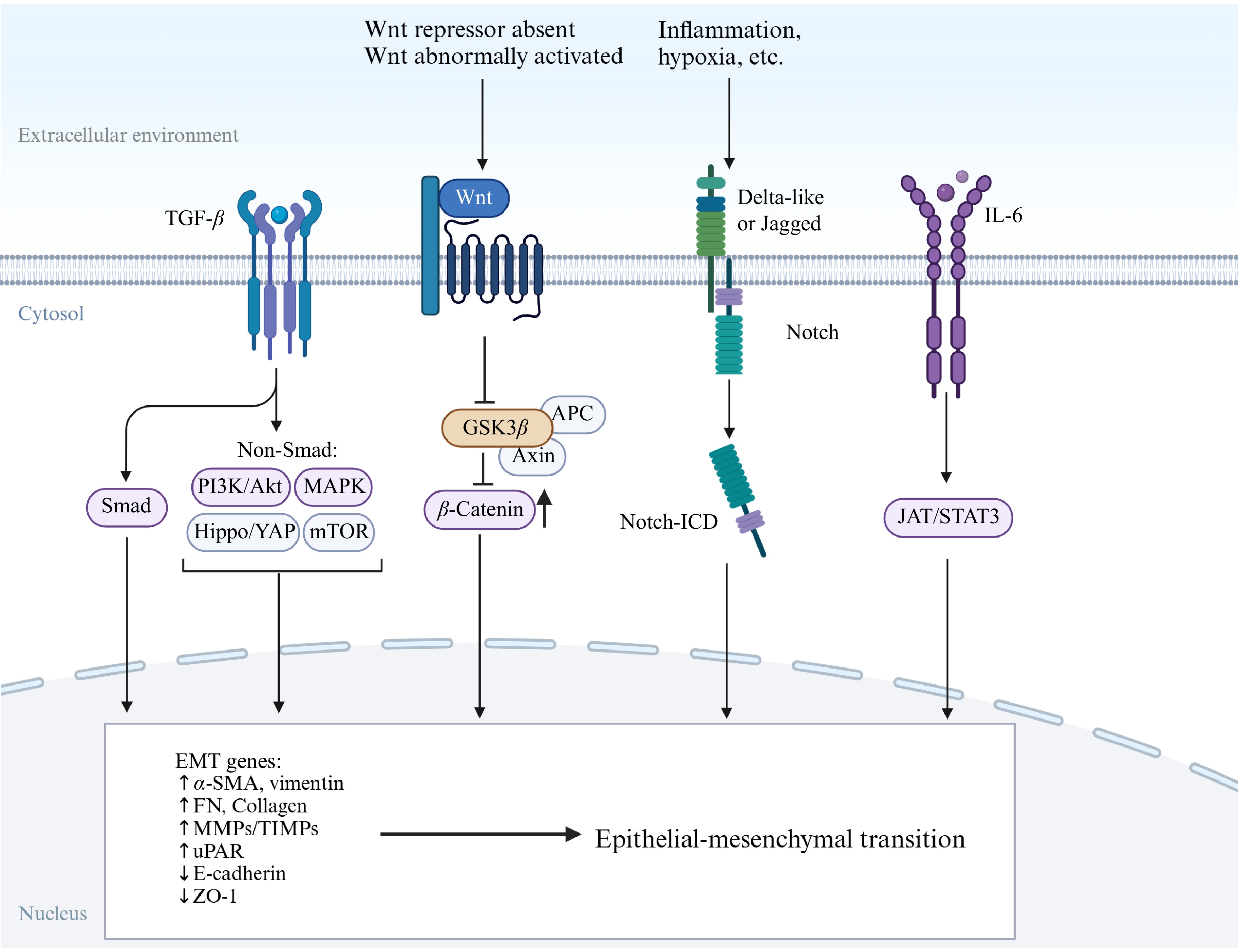
Figure 3.
Several main signaling pathways of EMT. The Wnt pathway is active because the Wnt/β-catenin pathway inhibitor is absent and β-catenin accumulates in the cytoplasm. The Notch pathway is activated by contact with neighboring cells, which comes from inflammation, ER stress, hypoxia, and others.
Given its role in immune evasion, EMT represents a promising therapeutic target for preventing tumor resistance and metastasis. Current immunotherapy for CRLM is primarily effective in tumors with deficient mismatch repair and high microsatellite instability, indicating that these tumors can modulate their immune environment. Targeting EMT could potentially enhance the efficacy of CRLM treatment[51]. It has been demonstrated that NP137-mediated inhibition of netrin-1 reduces the proportion of cancer cells undergoing EMT, decreases the number of metastatic foci, and increases the sensitivity of tumor cells to chemotherapy[106]. More research on EMT and its interplay with the immune system may be beneficial in providing more effective treatment modalities for CRLM patients.
-
The 5-year overall survival (OS) rate for patients with advanced stage IV CRC (CRC with distant organ metastasis) remains a sobering 14%. Liver metastasis (LM), is the most common site for CRC spread and the primary driver of cancer-related deaths in these patients. It is estimated that as many as 20% of CRC patients have LM at the time of initial diagnosis 50%−60% of CRC patients will develop LM, underscoring the critical need to enhance treatment strategies focused on CRLM to improve survival outcomes[107].
Surgery
-
According to recent studies, the 5-year survival rate for patients with CRLM can range from 30% to 57% in resectable patients, while the 5-year survival rate for unresectable patients is less than 5%, which indicates that hepatectomy for CRLM remains the most recommended treatment currently[108,109]. Hepatectomy was indicated for CRLM when the following three conditions were met: (1) the primary CRC was curatively resected or can be synchronously resected; (2) metastasis located only in the liver; and (3) the hepatic metastatic lesions can be completely resected with preservation of more than 30% of functional liver tissue[110]. Synchronous resection means resecting primary colorectal cancer and liver metastases at the same time. For patients who cannot meet the conditions of synchronous resection, the primary colorectal cancer lesion can be surgically resected first, and then the liver metastasis can be resected three months later. Surgical resection of liver metastases first, followed by resection of colorectal primary lesions has also been carried out and applied, and there is no statistically significant difference between the efficacy, complications, and mortality rate and the traditional staged resection[111,112].
However, the 5-year survival rate after hepatectomy in patients with CRLM is only about 50%, not to mention the fact that many patients do not fulfill the indications for hepatectomy[113]. Therefore, the development of other therapies for the treatment of CRLM is significant.
Chemotherapy
-
For patients with non-resectable CRLM, chemotherapy is effective in extending survival and enhancing quality of life[114]. Moreover, chemotherapy can convert non-resectable lesions into resectable ones which is known as conversion therapy or downstaging therapy. Recent studies have shown that downstaging chemotherapy holds promise for liver-limited metastatic disease initially deemed technically non-resectable, with up to 44% of such patients becoming candidates for successful, potentially curative surgical resection[115]. Additionally, for patients with resectable liver metastases from colorectal cancer, it is recommended that they undergo chemotherapy before and after surgery[116].
Treatment for metastatic CRC encompasses a range of active pharmaceuticals, used in combination or as monotherapy. In standard patient cases, the integration of chemotherapy with targeted agents effectively manages disease progression and achieves high conversion resection rates[117]. According to the National Comprehensive Cancer Network (NCCN), first-line chemotherapy for intensive treatment candidates include FOLFOX, which pairs 5-fluorouracil with leucovorin and oxaliplatin; CAPEOX, combining capecitabine with oxaliplatin; FOLFIRI, which uses 5-fluorouracil with leucovorin and irinotecan; and injectable 5-fluorouracil/leucovorin or capecitabine[118]. Based on the data from several clinical trials, FOLFIRI significantly increased the R0-transformed resection rate in CRLM patients alone compared with FOLFOX conventional chemotherapy (36% vs 12%)[119]. The addition of cetuximab significantly increased the R0-transformed resection rate (25.7% vs 7.4%) and significantly improved the objective response rate (ORR) and OS[120]. However, this intensive chemotherapy resulted in more peripheral neuropathy (19% vs 0) and neutropenia (50% vs 28%)[121]. It is important to note that chemotherapy's side effects must be weighed against patient tolerance.
Immune therapy
-
Currently, immune checkpoint blockade (ICB) is a well-established immunotherapy that has shown significant efficacy in various cancers, especially CRC. However, the response to ICB in CRC is contingent upon the tumor's microsatellite instability (MSI) or mismatch repair (MMR) status. MSI is a genetic phenomenon, in which microsatellites are prone to insertion or deletion mutations during DNA replication, due to dysfunction of the DNA mismatch repair (dMMR) system. Microsatellites are regions of short tandem repeats in the genome, typically found in non-coding areas or non-functional parts of genes. MSI-H (MSI-high) tumors tend to respond better to certain types of immunotherapies because they typically have a higher tumor mutational burden, making them more recognizable and susceptible to immune system attack[122].
It is now recognized that dMMR/MSI-H is an independent good prognostic factor for CRC, and such patients tend to be highly efficacious for ICB (e.g., PD-1/PD-L1 antibody) therapy[123]. Currently MSI and MMR protein tests are recommended by guidelines for all CRC patients[118,124]. The FDA approved pembrolizumab for dMMR/MSI-H metastatic CRC in 2017, and nivolumab was later approved, both as monotherapy and in combination with ipilimumab, a kind of CTLA-4 antibodies, for patients who have failed other treatments[125]. As a promising anti-tumor therapy, ICB holds significant potential for the treatment of CRC. Further research is necessary to confirm the efficacy and safety of ICB agents in patients with CRLM, regardless of their MSI or MMR status. Additionally, the combination therapy targeting PD-1 with other therapies, such as CTLA-4 antibodies and anti-angiogenic agents shows great promise, aiming to address the lack of treatment options for patients with dMMR/MSI-H[30].
Targeted therapy
-
The advent of bevacizumab, which targets VEGF, and cetuximab, which targets the epidermal growth factor receptor (EGFR), in 2004 heralded a new era in molecular targeted therapy for metastatic CRC. The results of a randomized clinical study by Wang et al. (NCT03493048) showed that the difference in efficacy between cetuximab plus FOFOLFOXIRI and FOLFOX was insignificant, suggesting that cetuximab plays a vital role in combination with chemotherapy[126]. Regorafenib and furaquintinib are also now widely used as multi-target tyrosine kinase inhibitors. These monoclonal antibodies have achieved significant success in clinical use. The combination of cetuximab with fluorouracil, leucovorin, and irinotecan (FOLFIRI) has been shown to enhance outcomes for patients with both primary and metastatic CRC[127,128]. In patients having RAS gene mutations, including KRAS and NRAS, anti-EGFR therapy has not demonstrated survival benefits over chemotherapy. This is because these mutations occur in the RAS pathway, which lies downstream of growth factor signaling, rendering anti-EGFR monoclonal antibodies ineffective[118]. Consequently, determining the RAS gene status has become a key step in predicting the efficacy of EGFR-targeted therapy for metastatic CRC. Now bevacizumab has become the preferred targeted therapy for patients with RAS-mutant metastatic CRC[129,130]. The early detection of RAS gene mutations is crucial for guiding drug selection and assessing treatment efficacy.
In addition to the commonly used EGFR and VEGF inhibitors, a variety of other targeted therapies can be employed in the treatment of metastatic CRC. Vemurafenib, a BRAF inhibitor, together with Cobimetinib, a MEK inhibitor, is specifically utilized for patients with mCRC harboring the BRAF V600E mutation[131,132]. For mCRC patients with HER2 overexpression, Trastuzumab, a HER2 inhibitor, is applied[133].
Recently, therapies that target stromal-tumor interactions have gained more and more attention, potentially benefiting a broad range of cancers that metastasize to the liver. Stromal cells have a stable genome compared to unstable cancer cells, suggesting that targeting them might be more effective. However, targeting hepatic myofibroblasts alone might only halt cancer progression, not kill tumor cells. A combination approach with cytotoxic therapies could be more effective, but this depends on a clearer understanding of these interactions in liver metastasis, an area that is gaining research interest.
Local therapies
-
In the landscape of CRLM management, local therapies play a pivotal role in augmenting the effectiveness of systemic treatments. These approaches, encompassing interventional therapies and radiation treatment, are designed to target tumors with precision, minimizing damage to healthy tissue while maximizing the destruction of cancer cells.
For solitary liver metastases not exceeding 3 cm in diameter, stereotactic body radiotherapy (SBRT) can achieve favorable therapeutic outcomes. SBRT is an accurate form of external radiation therapy that utilizes a single high dose (5−30 Gy) and large fractionated doses (1−5 sessions) to deliver an effect akin to surgical resection, also known as stereotactic ablative radiotherapy (SABR)[134]. Other commonly employed radiation therapy approaches include selective internal radiation therapy, among others. Other methods of locally destroying tumors include anhydrous ethanol injections, microwave ablation, and freezing[135,136]. It is worth noting that local treatment of tumors is of little significance in isolation and should function as a component of systemic therapy.
For patients with a large tumor burden of liver metastases and limited response to systemic chemotherapy, hepatic arterial infusion chemotherapy (HAI), radiofrequency ablation (RFA), and transarterial chemoembolization (TACE) can be appropriately combined at the right time. It has been reported that this combined interventional treatment approach, when used in conjunction with immunotherapy, may offer greater therapeutic benefits[137]. According to a study, interventional therapies such as TACE can be used in the follow-up of chemotherapy-resistant patients with liver metastatic colorectal cancer. The retrospective study reported response rates between 40% and 60%[138].
In conclusion, amazing progress has been made in CRLM treatment. Surgical resection remarkably improves 5-year survival in patients with resectable CRLM. Chemotherapy extends survival for non-resectable CRLM patients and can convert non-resectable lesions to resectable. Immune therapy, particularly for MSI-H/dMMR CRC, shows significant efficacy. Targeted therapies, such as anti-VEGF and anti-EGFR agents, play a crucial role in treatment. Local therapies augment systemic treatments' effectiveness. Figure 4 provides a visual guide to the recommended treatment strategies for CRLM as outlined in the clinical guidelines, emphasizing the importance of multi-therapy to improve patient outcomes. Moreover, new therapies based on the liver immune microenvironment are also developing rapidly.
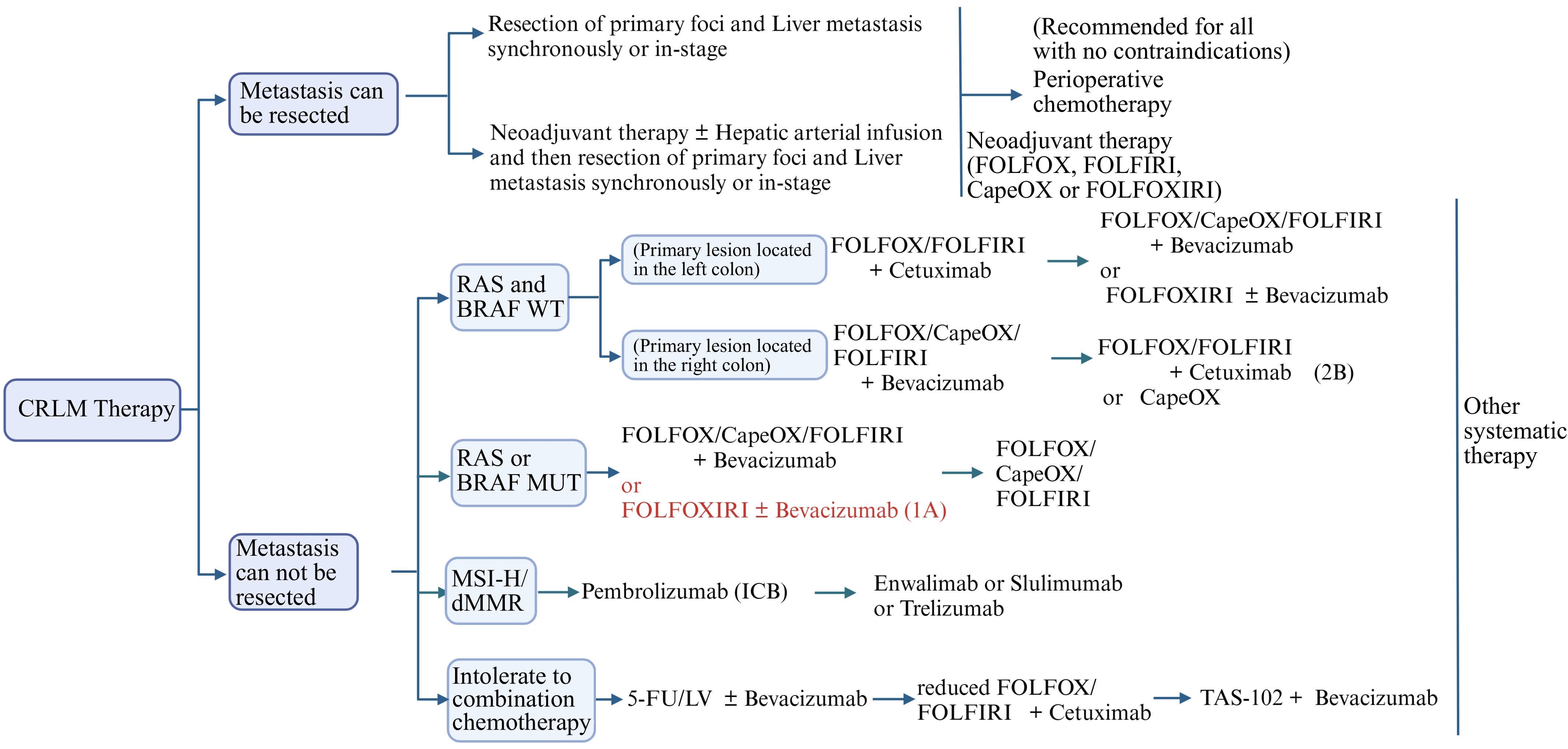
Figure 4.
CRLM's recommended treatment flowchart. FOLFOX: Calcium folinate + Fluorouracil (5-FU) + Oxaliplatin; CapeOX: Oxaliplatin + Capecitabine; LV: Leucovorin (folinate); FOLFIRI: Calcium folinate + Fluorouracil + Irinotecan; FOLFOXIRI: Calcium folinate + Fluorouracil + Oxaliplatin + Irinotecan.
-
Despite advances in understanding the biology of CRLM, treatment outcomes are still suboptimal. While targeted therapies like EGFR and VEGF inhibitors show promise, the impact of other angiogenesis inhibitors are less clear. There is a growing interest in therapies that address the complex interactions between tumors and the surrounding stroma, particularly the role of hepatic myofibroblasts in cancer progression. These interactions may be more effectively managed in tandem with cytotoxic therapies, but this requires a deeper comprehension of their specific roles in metastasis.
Immunotherapy, particularly immune checkpoint blockade, has emerged as a promising approach for CRLM. Its effectiveness, however, is contingent upon the tumor's microsatellite instability (MSI) or mismatch repair (MMR) status, and its broader application in CRC is still under investigation. How to deal with the suppressive immune microenvironment of CRLM to rejuvenate the immune cells, and how to expand the applicability of immunosuppressive agents are the focus of current immunotherapy research.
In summary, the treatment of CRLM requires a comprehensive consideration of various treatment methods, including surgical resection, chemotherapy, radiotherapy, interventional therapy, molecular targeted therapy, and immunotherapy. Future research must delve into the intricacies of the liver's immune microenvironment to devise innovative treatment strategies aimed at enhancing survival rates and the quality of life for CRLM patients.
This study was supported by grants from the National Natural Science Foundation of China [Grant Nos U23A20451 (L.X.) and 82273310 (L.X.)], the Natural Science Foundation of Hubei Province [Grant No. 2022CFA016 (L.X.)], and the Basic Research Support Program of Huazhong University of Science and Technology [Grant No. 2023BR038 (L.X.)].
-
Not applicable.
-
The authors confirm contribution to the paper as follows: draft manuscript preparation, figure design: Wen J, Jiang J; manuscript revision for intellectual content and commented: Zhang J, Wu Z, Hu D, Wei W, Zhang Z; study conception and design: Xia L. All authors reviewed the results and approved the final version of the manuscript.
-
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.
-
The authors declare that they have no conflict of interest.
-
# Authors contributed equally: Junjie Wen, Junqing Jiang
- Copyright: © 2025 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Wen J, Jiang J, Zhang J, Wu Z, Hu D, et al. 2025. Colorectal cancer liver metastases: immune environment and therapeutic perspectives. Gastrointestinal Tumors 12: e003 doi: 10.48130/git-0025-0002 

Colorectal cancer liver metastases: immune environment and therapeutic perspectives
- Received: 05 November 2024
- Revised: 17 December 2024
- Accepted: 02 January 2025
- Published online: 12 February 2025
Abstract: Colorectal cancer is one of the most prevalent malignant tumors, and liver metastasis is the main cause of its poor prognosis. Although many therapies for colorectal cancer are available, most of them are much less effective after liver metastasis occurs. Since the complex tumor environment in the liver is crucial for the formation of pre-metastatic ecological niche and for the colonization and metastasis of colorectal cancer, further study of the mechanism of colorectal cancer liver metastasis and the immune microenvironment should be helpful to find new therapeutic methods. This review explores the role of various cells in the hepatic immune microenvironment in different stages of early colorectal cancer liver metastasis, evaluates current treatment strategies, and provides new insights for therapeutic approaches targeting the immune microenvironment.
-
Key words:
- Colorectal cancer /
- Liver metastasis /
- Pre-metastatic niche /
- Targeted therapy /
- Immune microenvironment