

-
The broad range of beneficial biological activities exhibited by heterocyclic compounds and their derivatives makes them a crucial focus of pharmaceutical research. Several compounds used in medicine and pharmacy fields incorporate in their structure an heterocycle, in which extensive experimental and computational studies have been conducted to discover innovative and cost-effective methods to prepare these compounds[1]. The cycloaddition reactions, including the [3+2] cycloaddition reaction[2], represent a key method to synthesising a variety of heterocycles[3,4]. The 32CA reaction is a highly significant process for constructing heterocyclic five-membered rings. It offers a straightforward approach to preparing these important structures having several chiral centers, providing good control of stereoselectivity[5]. Today, the main challenge in organic chemistry is the control of stereoselectivity during the reaction. Among the developed methods[6], the most important are ones that employ the reagents and change the reaction conditions leading to obtaining different isomers. Other reagents[7], temperature[8], and solvent[9] were utilized to reverse the enantioselectivity, and thus both enantiomers could be prepared. Scientists have become interested in synthesizing heterocycles with enhanced biological activities due to the broad range of applications of these compounds. Heterocycles serve as vital frameworks in the modulation of drug polarity by replacing functional groups, thereby increasing bioavailability[10].
The structure of indole is a significant nucleus found in synthetic or natural compounds with important biological properties[11]. The thiopyranoindole heterocycles are also noteworthy due to their medicinal properties[12]. Among the important heterocycles, the isoxazolidines have a particular importance and are considered a useful class abundant in compounds[13]. Some isoxazolidine derivatives exhibit antifungal[14], anti-inflammatory[15], antimycobacterial[16], and cytotoxic activities[17], and they also act as DNA intercalators[18].
The C-S bond has shown promising potential as a target for the development of new drugs. Several studies have shown that compounds containing a C-S bond can exhibit interesting anticancer, antifungal, and antimicrobial properties[19]. In addition, current drugs such as sulfonamides and thiazolidinones contain C-S bonds and are used to treat several diseases, namely, diabetes, cardiovascular diseases, and bacterial infections. These examples suggest that the C-S bond may be an important fragment for the discovery of new biologically active molecules[20].
Over the past few years, there has been significant experimental and theoretical research carried out on intramolecular 32CA reactions that have been performed to investigate and discover the factors controlling the selectivity in this category of CA reactions[21−26].
The question of stereoselectivity is a consequence of the balance between steric and electronic effects. Thus, controlling the stereoselectivity at the cyclisation step is a great challenge in the organic synthesis field. This can be achieved by selecting appropriate reactants or by employing a molecular complex as a catalyst[27], especially in the case when cycloaddition reactions occur through a stepwise mechanism[28−30]. Molecular electron density theory (MEDT)[31] investigations revealed a connection between the reactivity of 32CA reactions, which involve the most basic three-atom components (TACs), and their electronic structure. This methodology has successfully provided a relation between the reactivity of the separated molecules and their electronic structure[32].
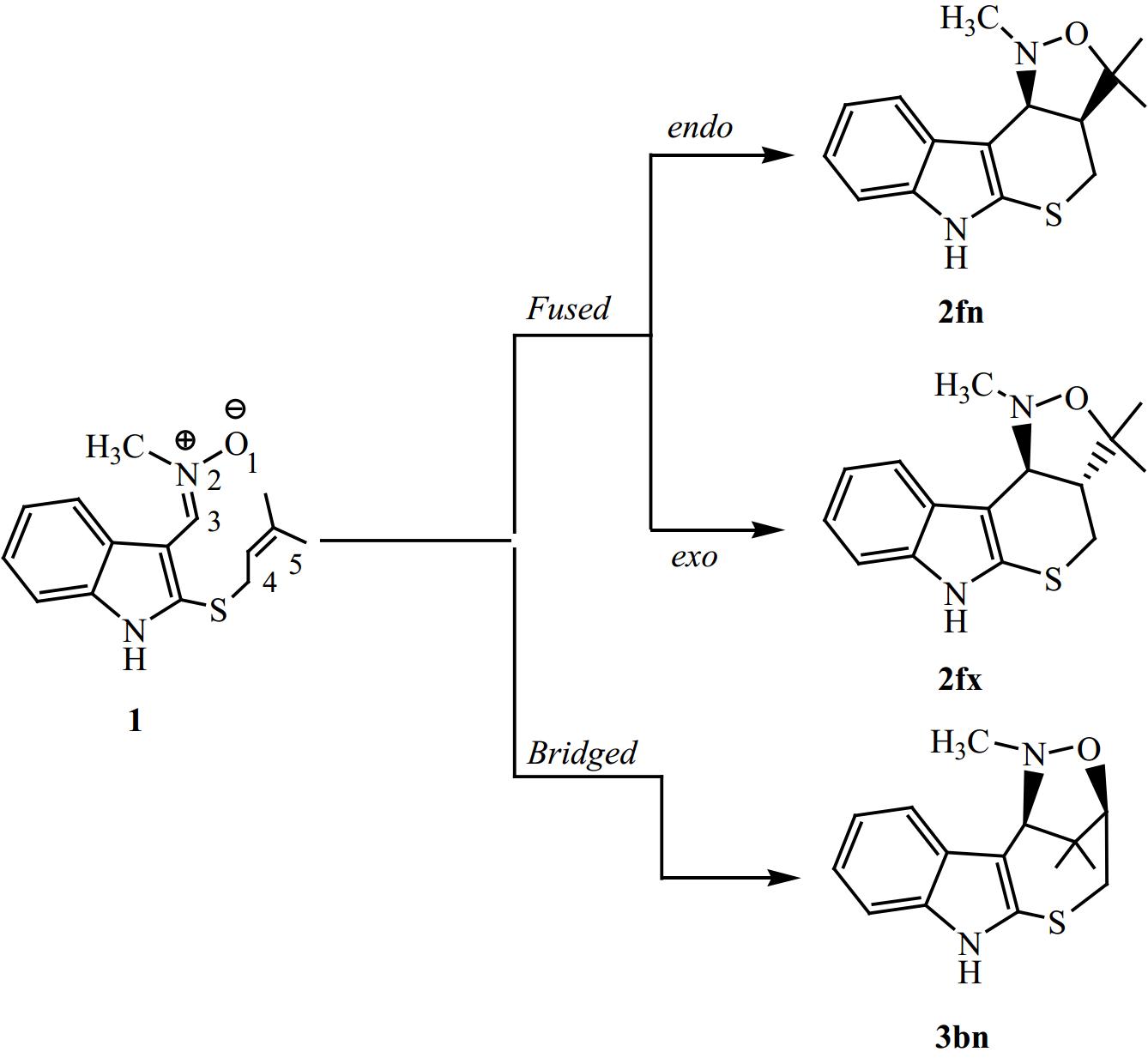
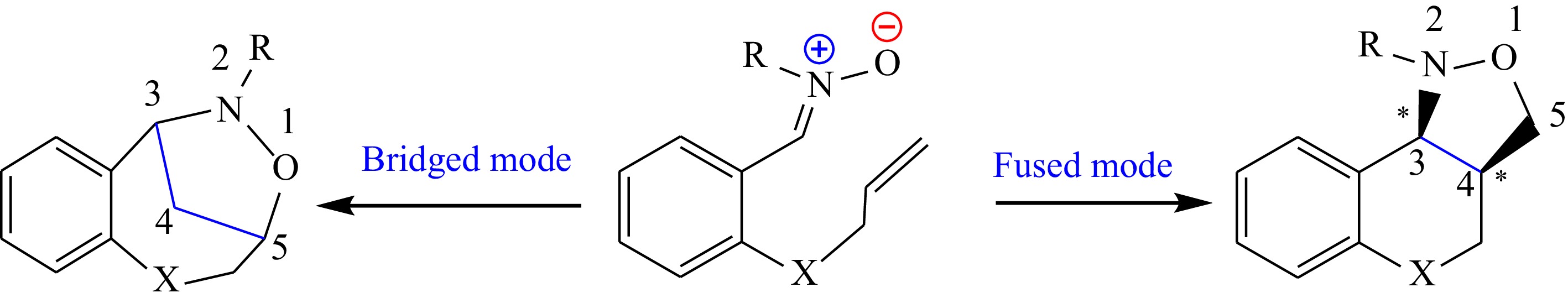
Nitrones are three-atom-components (TACs)[33] having a zwitterionic-type (zw-type) structure[32]. The intramolecular [3+2] cycloaddition (I32CA) reactions between TACs and alkenes involve heterocyclic structures through either bridged or fused modes. This method is highly versatile and efficient in creating complex heterocyclic architectures, which consist of oxygen and nitrogen atoms in a ring with five atoms (Fig. 1).
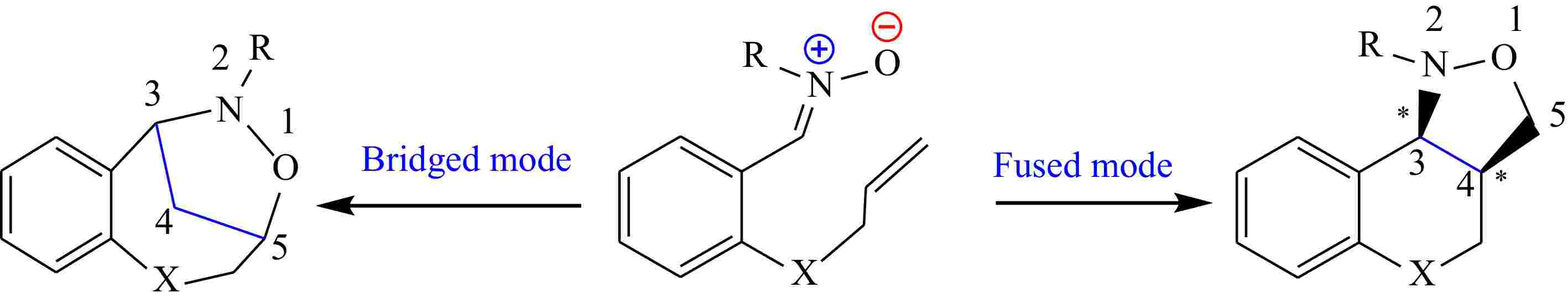
Figure 1.
The possible reactive channels of the I32CA reaction.
Furthermore, these modes allow for the construction of stereo- and regio-selective compounds. The resulting heterocyclic architectures are highly valuable in many applications, namely, pharmaceutical chemistry, where, the specific arrangement of atoms can greatly impact the compound's properties and potential applications. Overall, the I32CA reactions of nitrones provide a valuable tool for synthesizing complex, highly specific heterocyclic structures with diverse applications[34,35].
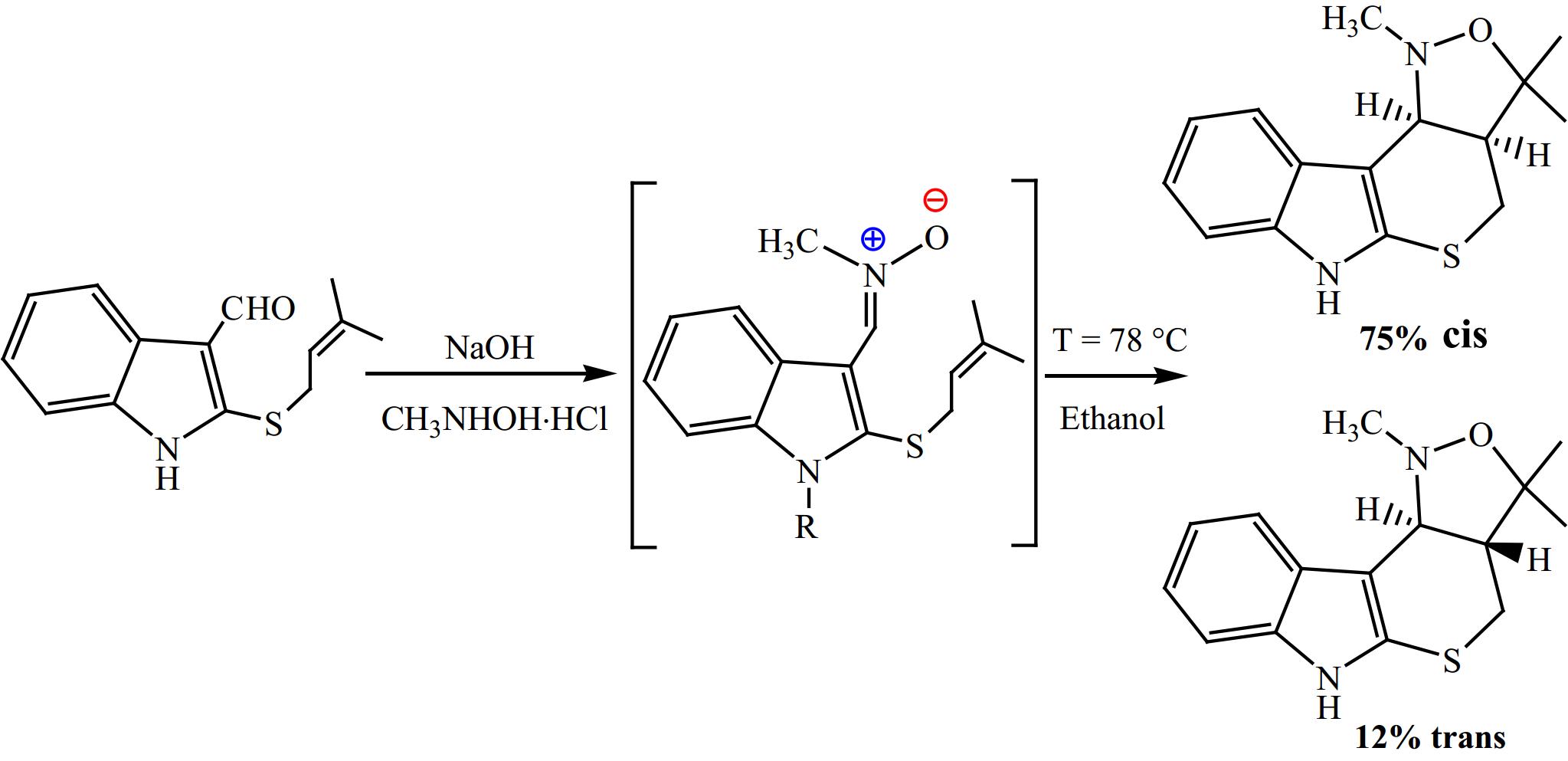
Majumder & Bhuyan[36] described the preparation of new dihydroisoxazole/tetrahydroisoxazole annulated thiopyrano[2,3-b]indole obtained from oxindole through I32CA reaction (Fig. 2). This synthetic method has allowed for the production of innovative compounds that may have potential applications in the fields of medicinal or pharmaceutical chemistry.
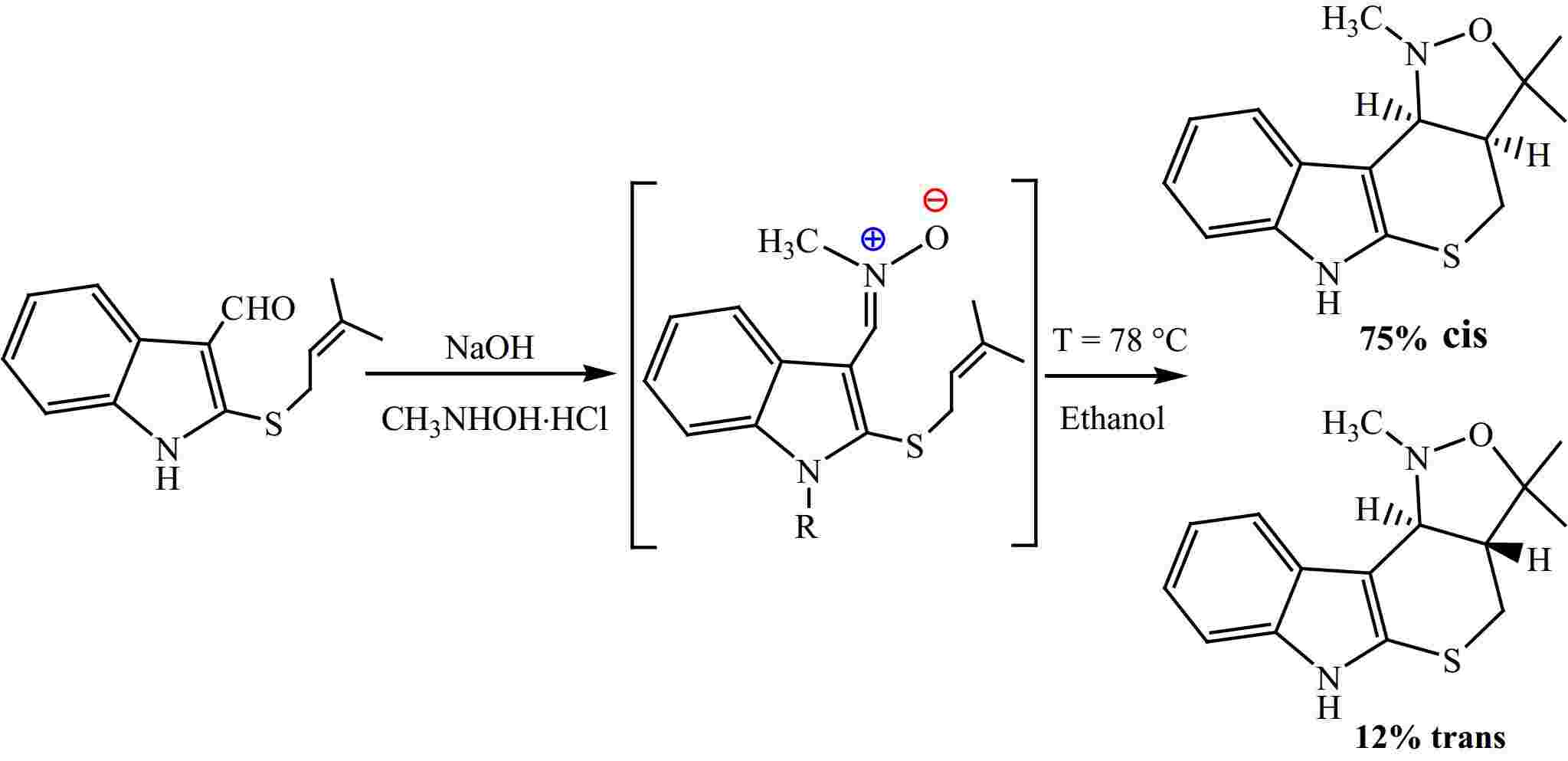
Figure 2.
The synthesis of tetracyclic isoxazolidines from a simple oxindole.
Herein, we aimed to conduct a computational analysis with the goal of analysing the selectivity observed in the experimental results reported by Majumder & Bhuyan[36], as well as to understand in a deep manner the factors governing the selectivity and to elucidate their influence on the mechanism of this I32CA reaction.
-
Previous DFT investigations indicate that the use of the B3LYP/6-31G(d,p) method explained correctly the experimental observations of cycloaddition reactions[37−39]. Thereby, in this work, all calculations were performed within the B3LYP/6-31G(d,p) computational level[40]. The frequency calculations of the optimized geometry of stationery points, namely, reactants, transition states (TSs), and cycloadducts (CAs) were characterized to ensure that only TSs possessed a single imaginary frequency (for more details, refer to the ESI). Thermodynamic functions were computed in toluene at 383.15 K and 1 atm, based on the optimized structures in the gas phase[41], then scaled by a factor of 0.96[42]. The atomic charges of TS structures were computed within the natural bond orbital (NBO) method[43]. For the study of toluene solvation effects, single-point calculations were performed through the optimized gas phase structures using self-consistent reaction fields (SCRF)[44] within the polarisable continuum model (PCM)[45]. These calculations have been realized using the Gaussian 09 package[46].
The electron localization function (ELF)[47] analysis has been conducted using Multiwfn software[48]. The non-covalent interaction (NCI) study has been realized through the analysis of the reduced density gradient together with the low-gradient isosurfaces[49], which was obtained from the NCI plot[50]. QTAIM analysis[51] were conducted using Multiwfn software[48].
-
Previous theoretical studies have shown that the majority of 32CA reactions occur via a mechanism of one step[52−54]. Therefore, in this study, we have exclusively focused on this mechanism for the I32CA reaction of (E)-N-((2-((-methylbut-2-en-1-yl)thio)-1H-indol-3-yl)methylene)methanamine oxide 1. Thereby, we have identified and characterized three TSs and their corresponding CAs, in relation to the potential regio- and stereo-selective approaches. Therefore, the present I32CA reaction can proceed through two regioisomeric channels, the fused or bridged one (Fig. 3).
Figure 3.
Reactive pathways associated with the I32CA of 1.
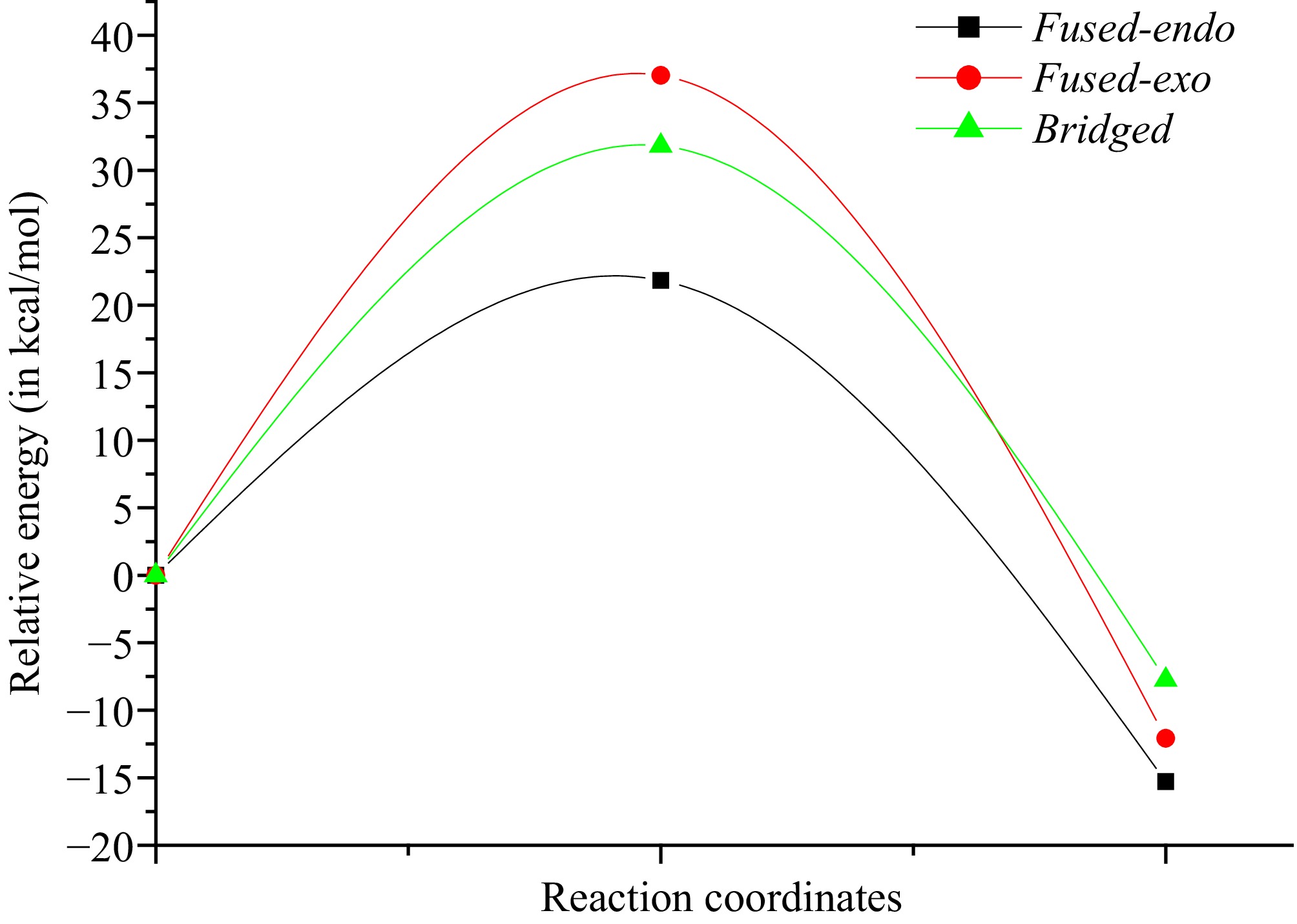
The Cartesian coordinates of 1, TSs, and CAs are shown in Supplementary Table S1 together with the values of imaginary frequencies of TSs. Values of relative energies corresponding to (E)-N-((2-((-methylbut-2-en-1-yl)thio)-1H-indol-3-yl)methylene)methanamine oxide 1, and TSs and CAs are shown in Table 1, whereas absolute energy values are given in Supplementary Table S2. The energetic profiles in the toluene solvent of the three corresponding pathways are given in Fig. 4.
Table 1. Values of relative energies of (E)-N-((2-((-methylbut-2-en-1-yl)thio)-1H-indol-3-yl)methylene)methanamine oxide 1, TSs, and CAs in gas-phase and solution of toluene.
System ΔEGas phase (kcal/mol) ΔEToluene (kcal/mol) TS2n 18.7 21.8 TS2x 34.9 37.0 TS3n 30.5 31.8 CA2n −17.5 −15.3 CA2x −13.3 −12.1 CA3n −9.1 −7.7 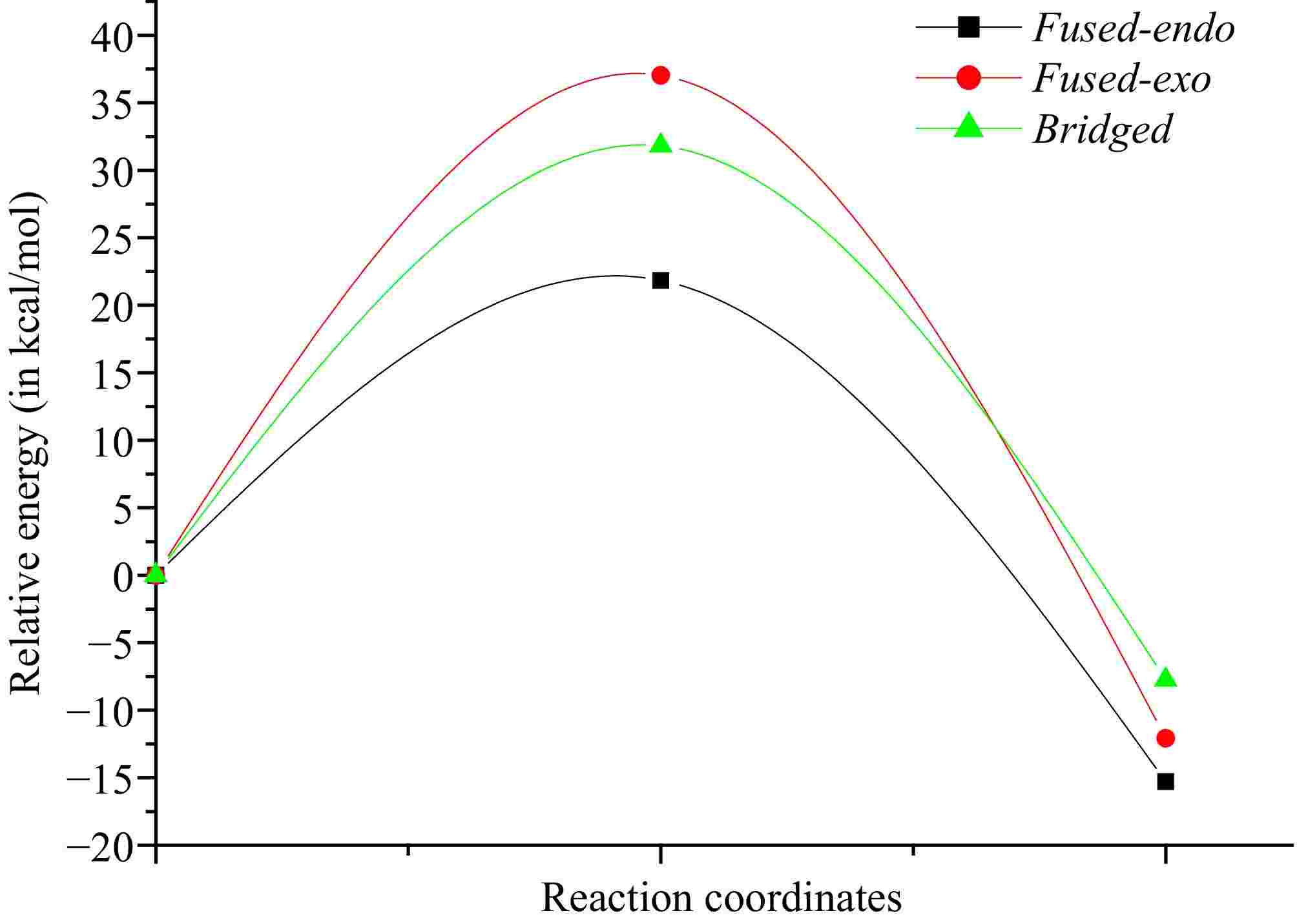
Figure 4.
Curves of energy profiles in toluene solvent of the I32CA reactive paths of (E)-N-((2-((methylbut-2-en-1-yl)thio)-1H-indol-3-yl)methylene)methanamine oxide 1.
According to the values in Table 1, we notice from an analysis between the ΔE values in gas-phase for the possible reactive routes in the studied I32CA reaction that the fused-endo mode (Ea = 18.8 kcal/mol) is the kinetically favoured cycloadduct. The relative energy difference (ΔΔE) of 11.8 kcal/mol between Ea of fused-endo and the second most favorable pathway, the bridged-endo pathway. Additionally, it is observed that all reactive routes are irreversible because the corresponding cycloadducts are more stable than reactant 1 (ΔEcycloaaducts < 0). Moreover, the bridged cycloadduct is less stable compared with the fused ones, which may be attributed to the ring strain presented in the bridged ring. Thus, the bridged ring that is characterized by a seven-membered ring that is unstable than one having six atoms formed from the fused channel. Note that the obtained large activation energies also suggest that these I32CA reaction pathways proceed via a low-polar mechanism[55,56].
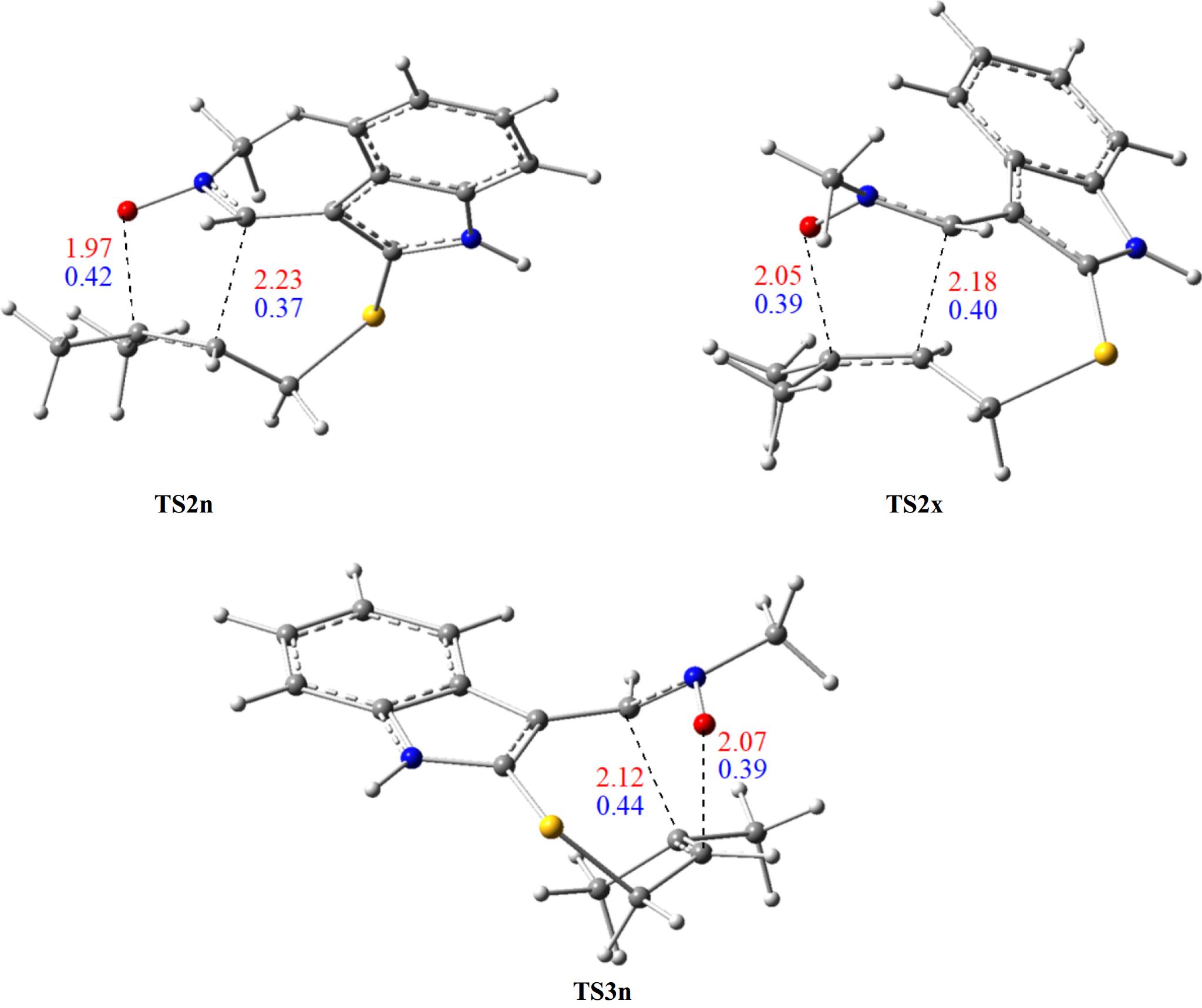
On the other hand, we notice that the cycloadduct favoured kinetically is stable, confirming that the studied I32CA is under kinetic control. These findings are in perfect accordance with the data observed experimentally. The optimised structures of TSs associated with the I32CA of (E)-N-((2-((methylbut-2-en-1-yl)thio)-1H-indol-3-yl)methylene)methanamine oxide 1 are depicted in Fig. 5, as well as the distances and the Wiberg bond order (BO) of the bonds under formation.
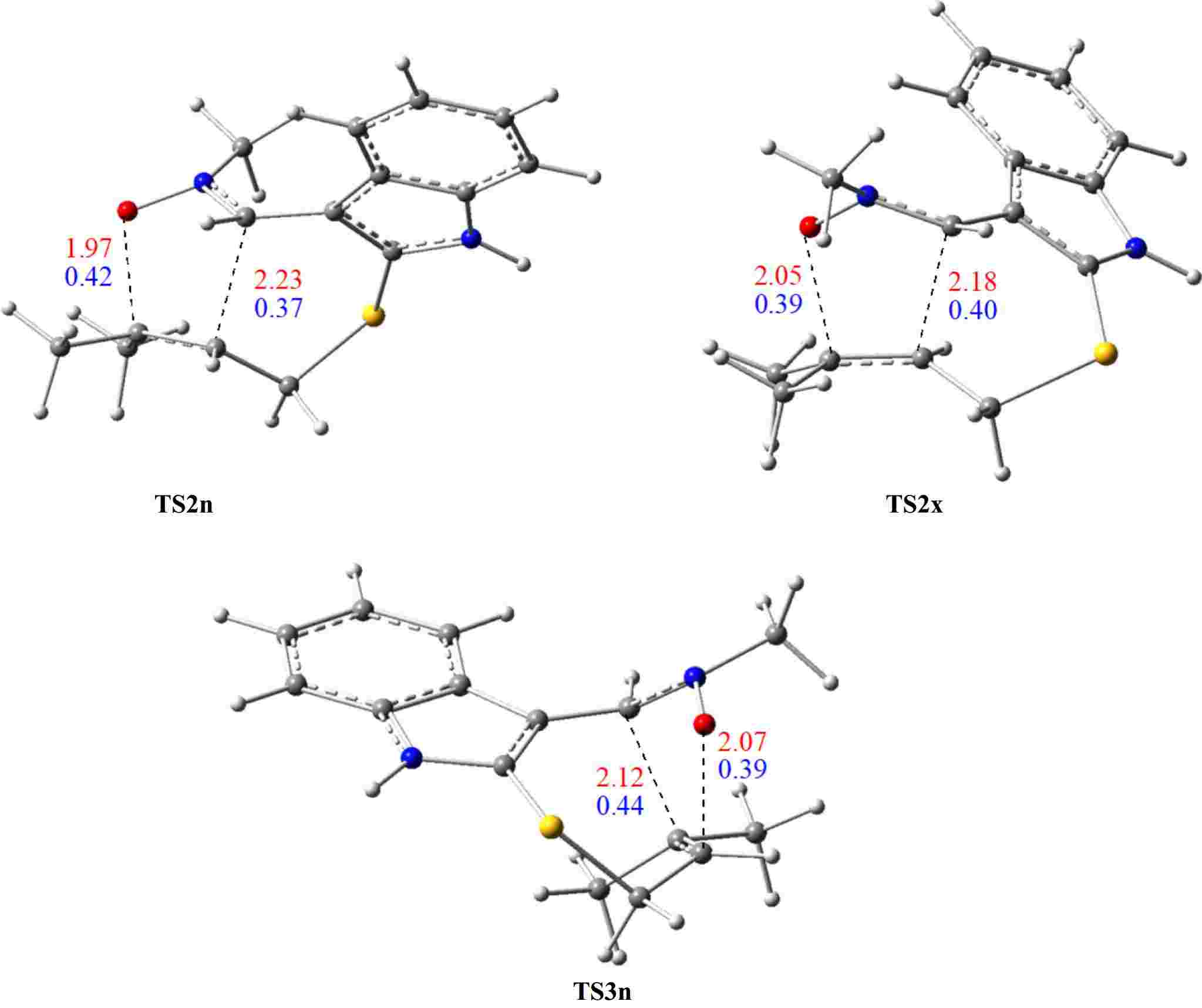
Figure 5.
TSs optimised structures of the I32CA of (E)-N-((2-((methylbut-2-en-1-yl)thio)-1H-indol-3-yl)methylene)methanamine oxide 1, showing the lengths (in Å, in red), and BO (in blue) of bonds under formation.
In the fused structures, the distances of the O–C and C–C bonds are respectively 1.97 and 2.23 Å at TS2n, and 2.05 and 2.18 Å at TS2x. In contrast, in the bridged structure (TS3n), these bond lengths are 2.07 and 2.12 Å. Also, the distance of the C–C bond is larger than that of the O–C bond in all three TS structures, suggesting a synchronicity mechanism in both modes.
Using Wiberg bond indices[57], the BO analysis for the two regioisomeric pathways indicates that the C–C bond formation is generally advanced slightly compared to that of the O–C, with the exception of the favorable transition state, TS2n, we notice the opposite, in which the formation of O–C bonds is slightly advanced compared with the other C–C. This highlights a slight asynchronous bonding process. These BO values support a previously predicted asynchronous mechanism.
Since this I32CA reaction of (E)-N-((2-((methylbut-2-en-1-yl)thio)-1H-indol-3-yl)methylene)methanamine oxide 1 was conducted in toluene and because of solvation effects that may influence on the energy values, a supplementary single-point calculation taking into account the toluene solvent through the gas-phase optimized structures were performed (see Table 1). Also, we noticed that by inclusion of toluene solvent effects in the calculations, the reactant, TSs, and CAs become more stable in comparison with gas-phase results. Including solvent effects led to increases in activation energies by 3.1, 2.1, and 1.3 kcal/mol for TS2n, TS2x, and TS3n respectively. In addition, the solvation effects also reduced slightly the reaction’s exothermicity by 2.2, 1.2, and 1.4 kcal/mol for CA2n, CA2x, and CA3n respectively. These results may be explained by the improvement of solvation of (E)-N-((2-((methylbut-2-en-1-yl)thio)-1H-indol-3-yl)methylene)methanamine oxide 1 relative to the TSs and CAs in polar solvents[58]. Despite these changes, the regioselectivity and stereoselectivity patterns observed in the gas phase stay the same.
Table 2 provides the values of thermodynamic functions for the I32CA reaction of (E)-N-((2-((methylbut-2-en-1-yl)thio)-1H-indol-3-yl)methylene)methanamine oxide 1, while absolute values are shown in Supplementary Table S3.
Table 2. Relative thermodynamic function values for the TSs and CAs of the I32CA of (E)-N-((2-((methylbut-2-en-1-yl)thio)-1H-indol-3-yl)methylene)methanamine oxide 1.
System ΔH (kcal/mol) ΔS (cal/mol/K) ΔG (kcal/mol) TS2n 20.8 −22.2 28.6 TS2x 35.7 −20.3 42.8 TS3n 30.8 −23.3 38.9 CA2n −14.5 −26.6 −5.2 CA2x −11.3 −24.8 −2.6 CA3n −7.1 −27.4 2.5 From the values of the activation enthalpies corresponding to the three competing reactive paths, we remark that the fused-endo path (TS2n) has the lowest value (20.7 kcal/mol), which is considered the most favourable reactive pathway, leading to the construction of the tetracyclic isoxazolidine CA2n. In addition all reactive pathways are characterised by a negative sign of relative enthalpy associated to the cycloadducts, which account for an exothermic character for this I32CA reaction.
Given the intramolecular nature of the studied reaction, these pathways exhibit low entropy values. Adding entropic contributions increases the value of relative Gibbs free energy to reach 28.6 kcal.mol-1 for the most favoured cycloadduct CA2n. The activation enthalpies for TS2x and TS3n are 35.7 and 30.8 kcal/mol, respectively, in which their free activation energies reaching respectively, 42.8 and 38.9 kcal/mol. Therefore, the fused-exo and bridged modes are less favorable pathways in this I32CA reaction that produce exclusively the CA2n, in great agreement with what observed experimentally[36]. Likewise, the relative free energy of the cycloadducts show that the pathways associated with fused mode are exergonic character but that of the bridged one is endergonic.
Analysis of the natural molecular mechanism
-
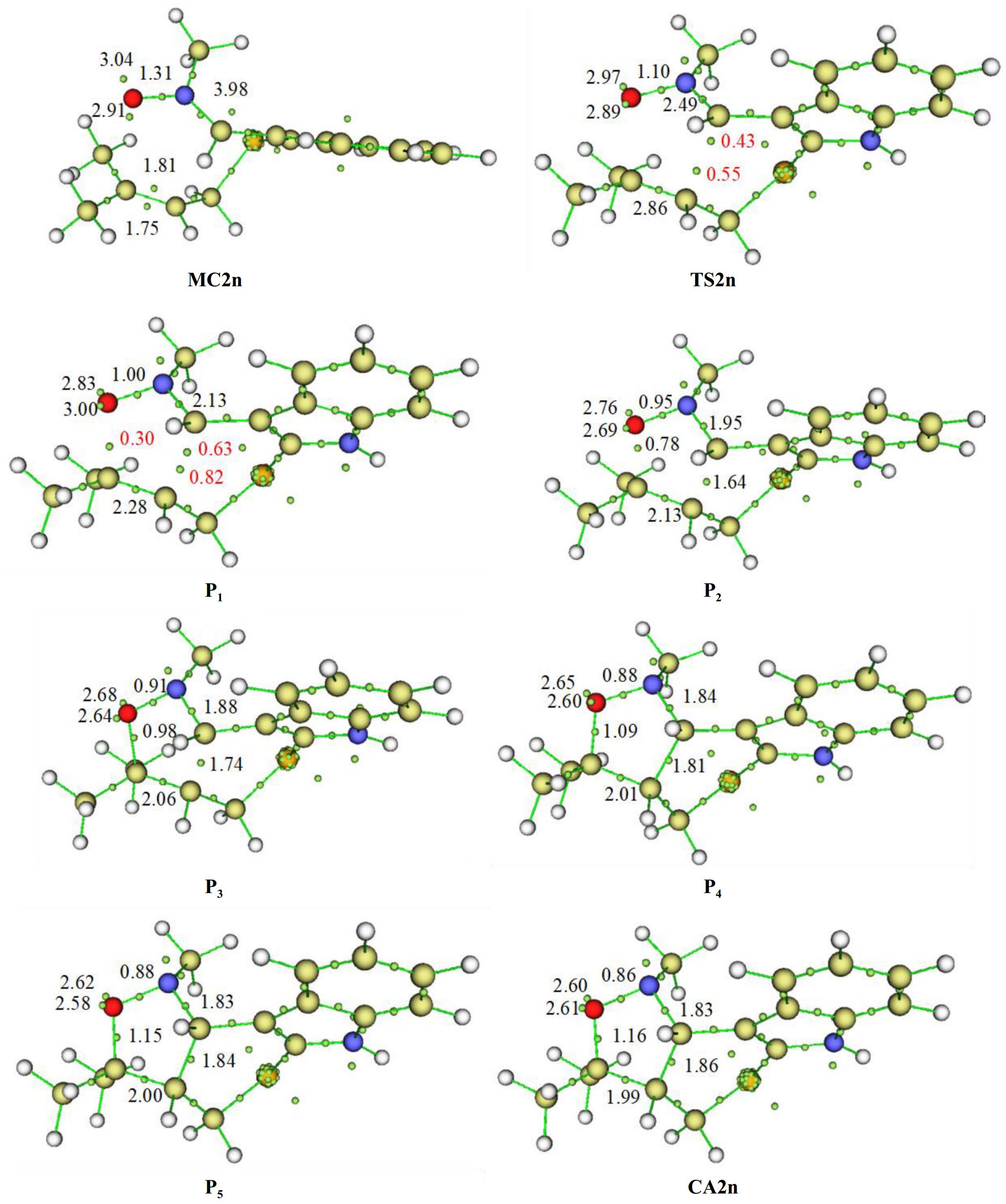
To study the molecular mechanism type of the present I32CA, we opted for the ELF topological analysis[47]. Thereby, after deep analysis of the IRC curve of the favorable fused-endo channel, we have selected some pertinent channels for this analysis. The curve IRC of the favourable TS2n as well as the selected point positions, MC2n, TS2n, P1, P2, P3, P4, P5, and CA2n associated with this pathway are illustrated in Supplementary Fig. S1. The electron density of the pertinent electronic basins obtained from ELF analysis on the structure corresponding to studied points on the IRC curve are collected in Table 3. Figure 6 shows the structure of different selected points together with the position of the ELF valence basins attractor.
Table 3. Electron density of the pertinent basins of the chosen points on the reactive fused-endo pathways of the I32CA of 1.
MC2n TS2n P1 P2 P3 P4 P5 CA2n d(C3−C4) 2.91 1.97 1.77 1.66 1.57 1.54 1.48 1.47 d(O1−C5) 3.01 2.23 2.00 1.87 1.75 1.71 1.62 1.58 V(O1−N2) 1.31 1.10 1.00 0.95 0.91 0.88 0.88 0.86 V(N2−C3) 3.98 2.49 2.13 1.95 1.88 1.84 1.83 1.83 V(C4−C5) 1.81, 1.75 2.86 2.28 2.13 2.06 2.01 2.00 1.99 V(C3…C4) − − − 1.64 1.74 1.81 1.84 1.86 V(O1…C5) − − 0.30 0.78 0.98 1.09 1.15 1.16 V(C3) − 0.43 0.63 − − − − − V(C4) − 0.55 0.82 − − − − − V(O1) − − − − − − − − V(C5) − − − − − − − − 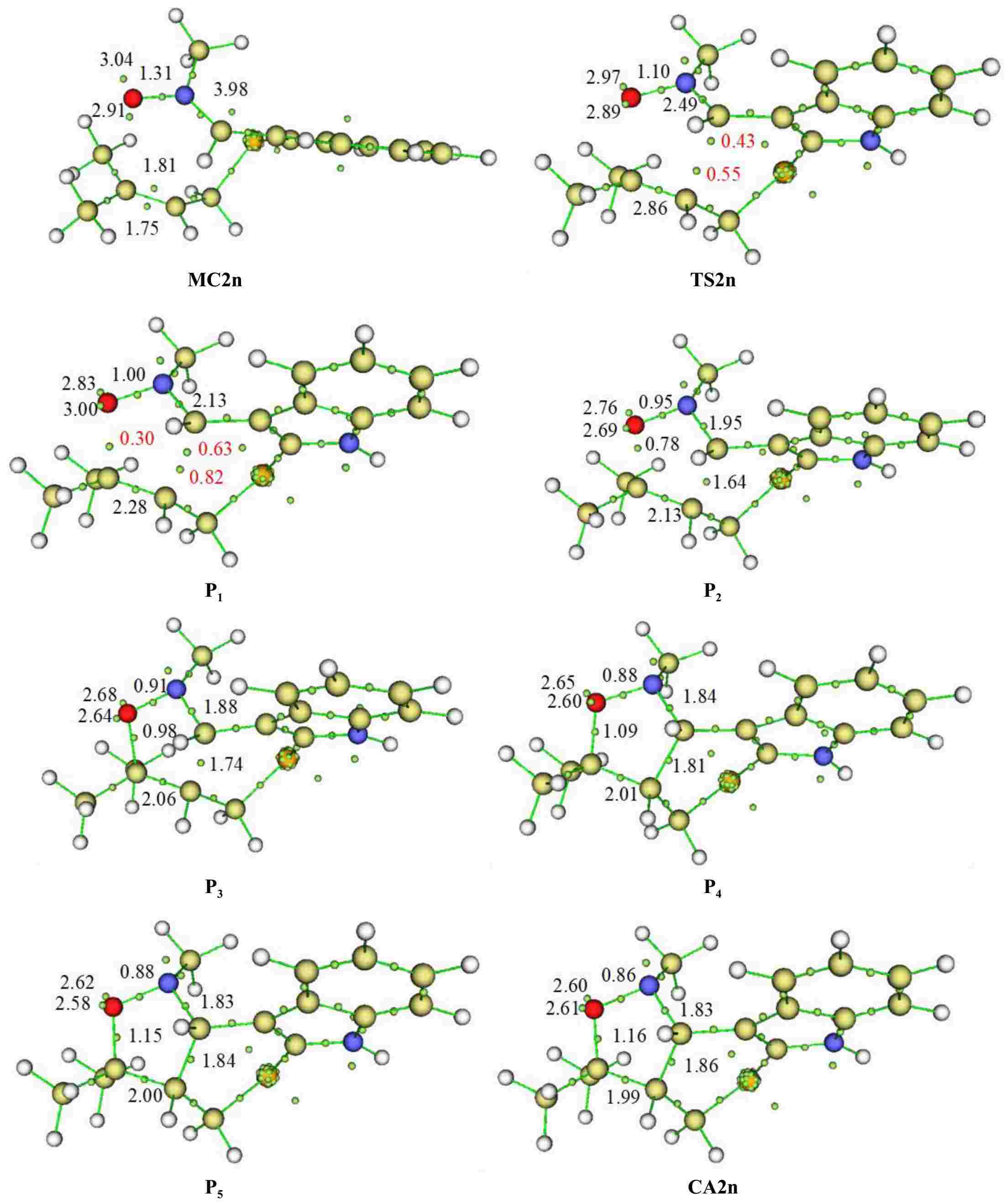
Figure 6.
Structure of different selected points together with the position of the ELF valence basin attractors of the chosen structures corresponding to the fused-endo pathways of the I32CA of 1.
The most important basins in this molecular system corresponding to the reactive regions of 1, are the (C3−N2 and N2−O1), and C4−C5 (for atom numbering see Fig. 3).
The structure of MC2n is characterized by V(N2,C3) disynaptic basin with 3.98 e of population, accounting for the double-bond character of this region. Also, the O1−N2 shows disynaptic basin V(O1,N2) which integrating of 1.31 e. Moreover, the reactive region C4−C5 has two disynaptic basins, V(C4,C5) and V'(C4,C5) characterised by a total electronic population equal to 3.56 e, indicating a double bond character of this reactive region.
At the second point TS2n, in which d(C3…C4) = 2.23 Å and d(O1…C5) = 1.97 Å, the population of the C3−N2 region that is has a disynaptic basin V(C3−N2) decreased to becomes 2.49 e, leading to the appearance of one new monosynaptic basin V(C3), with a 0.43 e. Another new monosynaptic basin V(C4) with 0.55 e appeared at this point produced from the disappearance of V'(C4,C5) disynaptic basin at this point, which leads to C4–C5 only having a monosynaptic basin integrating 2.86 e.
At P1, where the distances of the C3…C4 and O1…C5 are respectively 2.00 and 1.77 Å, we remark an apparition of new disynaptic basin V(O1,C5) integrate 0.30 e. In addition to this change, we notice an increase of the electronic population of V(C3) and V(C4) monosynaptic basins to become, 0.63 and 0.82 e respectively, generating from the depopulation of the disynaptic basins V(C4,C5) and V(C3,N2) which become 2.28 and 2.13 e respectively.
At P2, characterised by d(C3…C4) = 1.87Å and d(O1…C5) = 1.66 Å, the important evolution is the generation of a new V(C3,C4) disynaptic basin characterized by 1.64 e, accounting that the second new C3−C4 single bond is formed. Also, we notice an increase in the electron density of V(O1,C5) disynaptic basin which becomes 0.78 e.
At P3, in which d(O1−C5) = 1.57Å and d(C3−C4) = 1.75Å, the remarkable changes are the augmentation in the population of the new disynaptic basins V(C3,C4) and V(O1 C5) by slight values of 0.10 and 0.20 e to reach respectively 1.74, and 0.98 e. This increase in electron density of the newly forming disynaptic basins is accompanied by a decrease of that associated to the V(C4,C5) and V(N2,C3) which become 2.06 and 1.88e respectively.
At P4, in which d(O1−C5) = 1.54 Å and d(C3−C4) = 1.71 Å, the main development in electronic mechanism is only a slight increase in the population of the disynaptic basins V(O1,C5) and V(C3,C4) to become 1.09 and 1.81 e respectively.
At P5, in which d(O1–C5) = 1.47 Å and d(C3–C4) = 1.62 Å, there is no noticeable topological change except the slight augmentation in the population of disynaptic basins V(C3,C4) and V(O1,C5) by 0.03 and 0.06 e, to become 1.84 and 1.15 e, respectively.
At the cycloadduct CA2n, where d(O1–C5) = 1.47 Å and d(C3–C4) = 1.62 Å, the population associated with disynaptic basins V(O1,C5) and V(C3,C4) continue to increase by 0.01 and 0.02 e to reach 1.86 and 1.16 e respectively.
From this analysis, we can conclude that the mechanism of this I32CA reaction is two-stage one-step[59] , where, the O1–C5 single bond formation begins first, followed by that of the second C3–C4.
Origin of the fused-endo stereoselectivity
NCI analysis
-
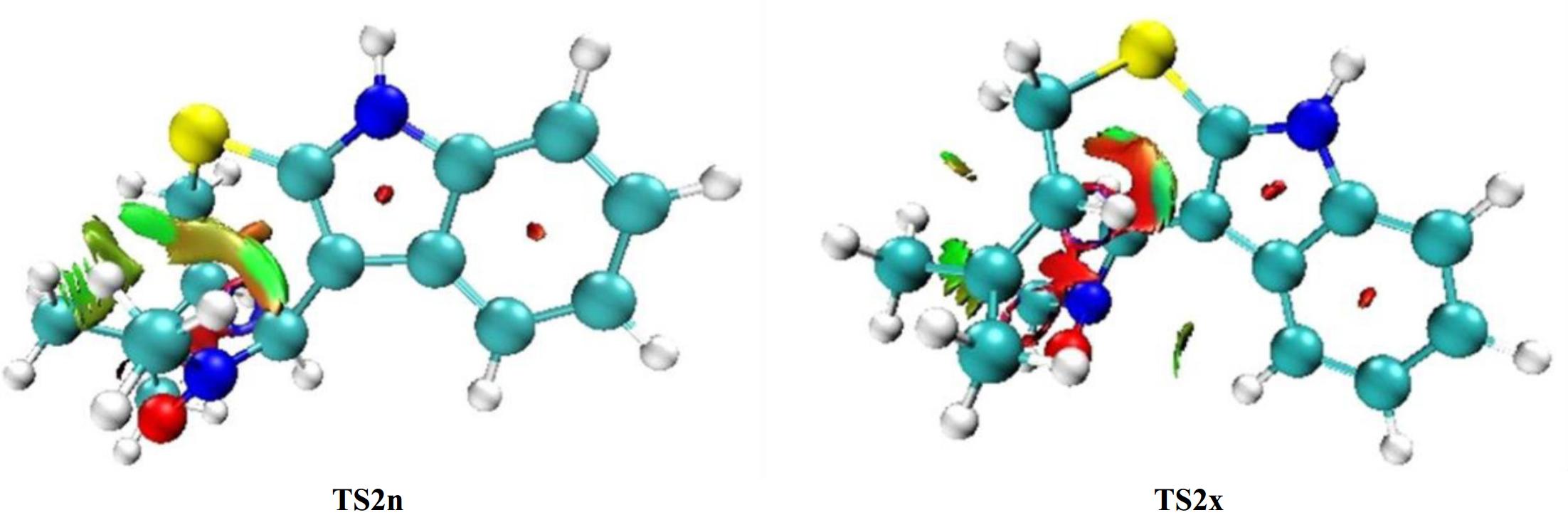
Based on our prior computational studies showing that NCIs are the main cause for the stereoselectivity observed in the 32CA reactions[60−63] thereby, a supplementary analysis of the structure associated to TS2n and TS2x is necessary to discover the main reason of the fused-endo selectivity. Figure 7 illustrates the NCI density gradient isosurfaces for TS2n and TS2x.
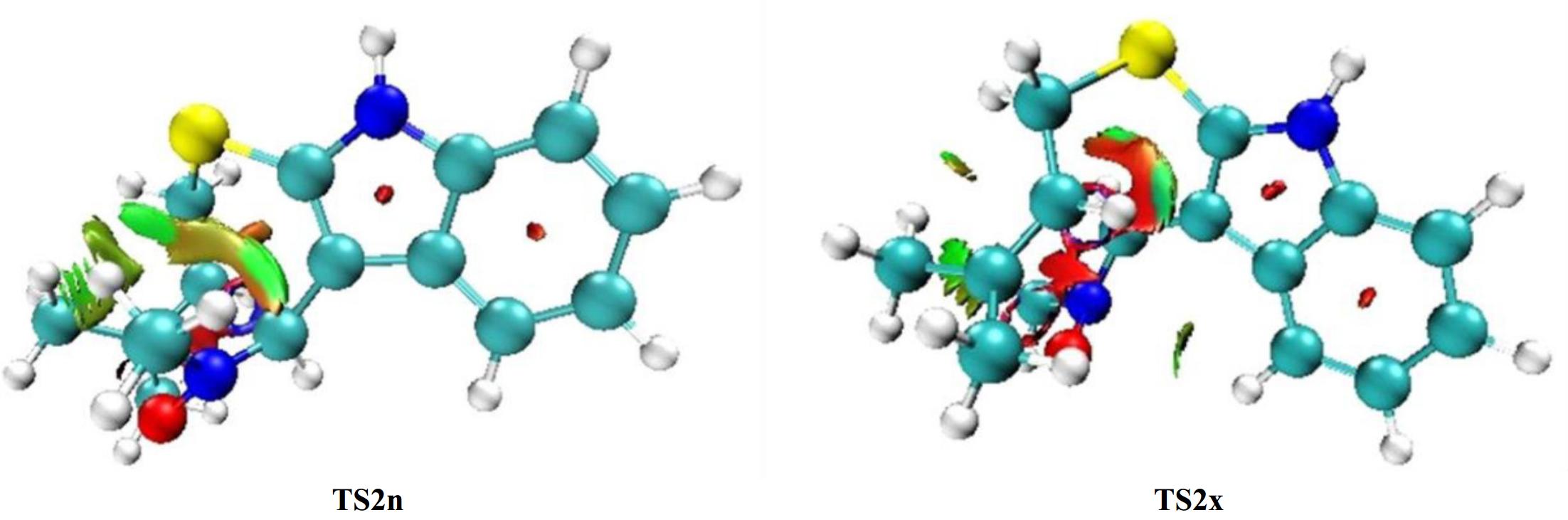
Figure 7.
NCI analysis of TS2n and TS2x structures with isosurfaces = 0.40.
From Fig. 7 we observe the existence of green surface in the zone located between the carbon atom of the alkene fragment and the oxygen of nitrone framework 1 in both TS2n and TS2x that may be due to the interactions of the newly forming O1…C5 and C3…C4 bonds. On the other hand, we observe the existence of several green surfaces in TS2n structure that are considered as indices for the existence of weak NCIs. Otherwise, we note that TS2x is characterized by a small green surface and large red surface that are associated with steric hindrances that decrease the stability of this structure.
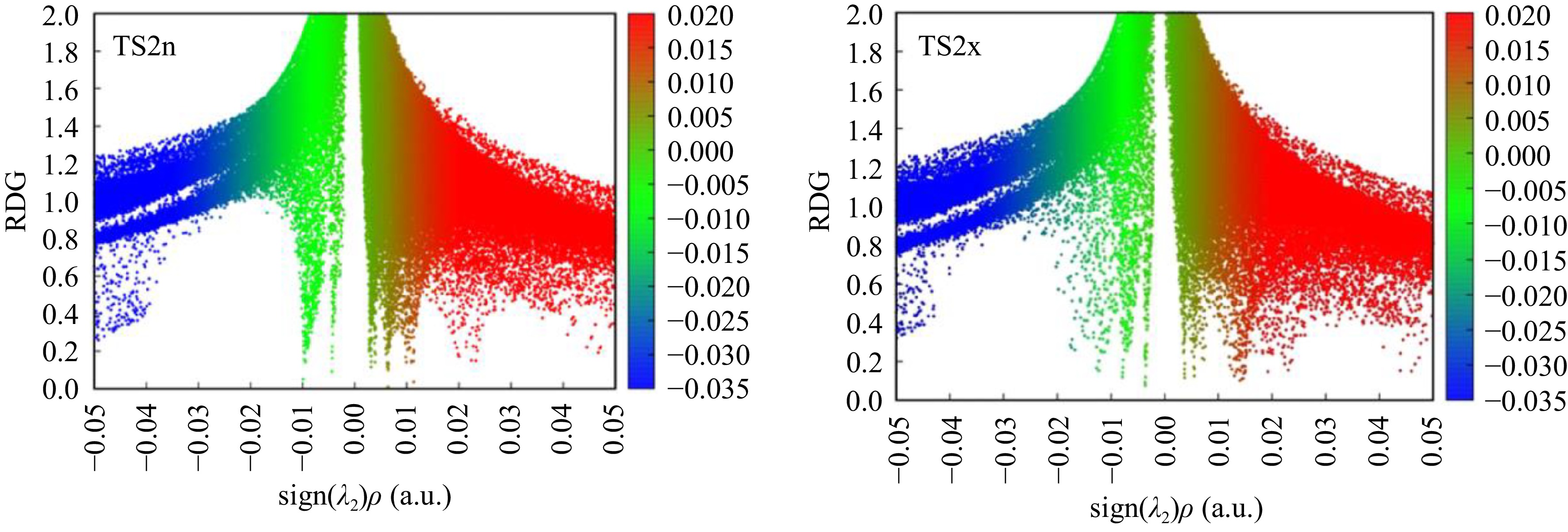
To confirm the existence of NCIs at these structures, we have realized a supplementary analysis based on the reduced density gradient, in which Fig. 8 shows the low-gradient isosurfaces for TS2n and TS2x. The low-gradient isosurfaces associated with TS2n show the existence of four low-density gradient spikes at −0.005, −0.008, −0012, and −0.020 a.u, account for the formation of four favourable NCIs. On the other hand, the low-gradient isosurfaces corresponding to TS2x shows the existence of only two low-density gradient spikes at −0.005 and −0.010 a.u. The presence of several favorable NCIs in TS2n is the main reason of its stability in comparison with TS2x that have only two NCIs.
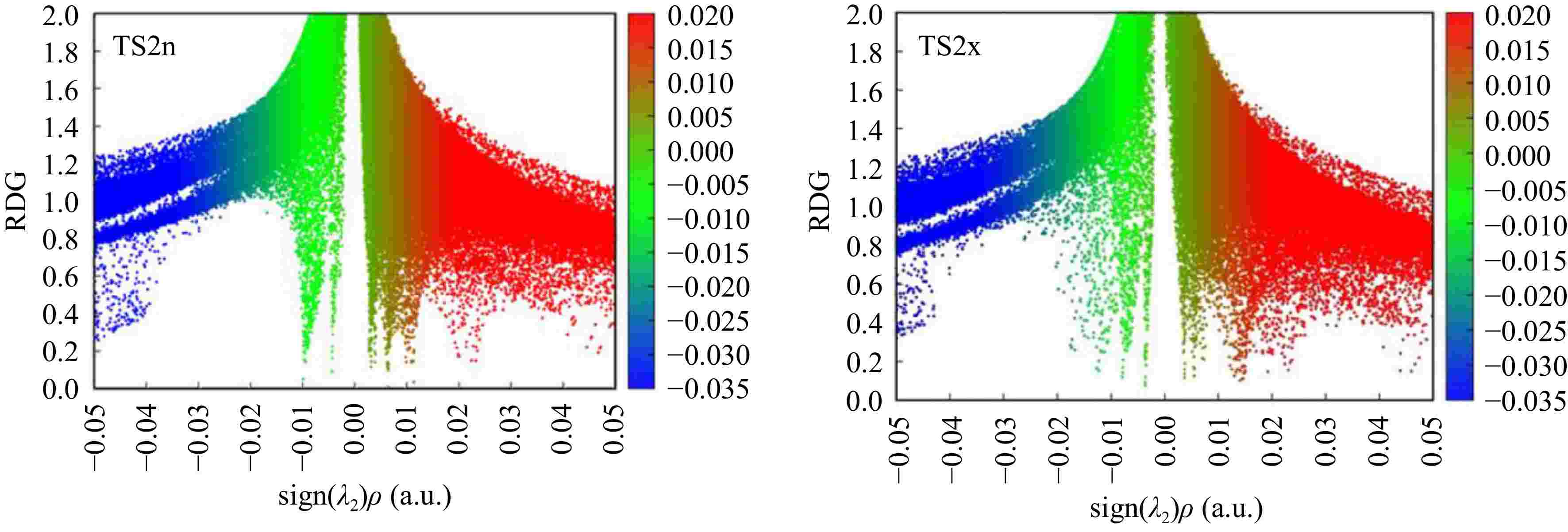
Figure 8.
Plots of RDG for TS2n and TS2x structures.
QTAIM study
-
QTAIM study of the electron density ρ produces some critical points (cps), that are points on the molecular system characterized by
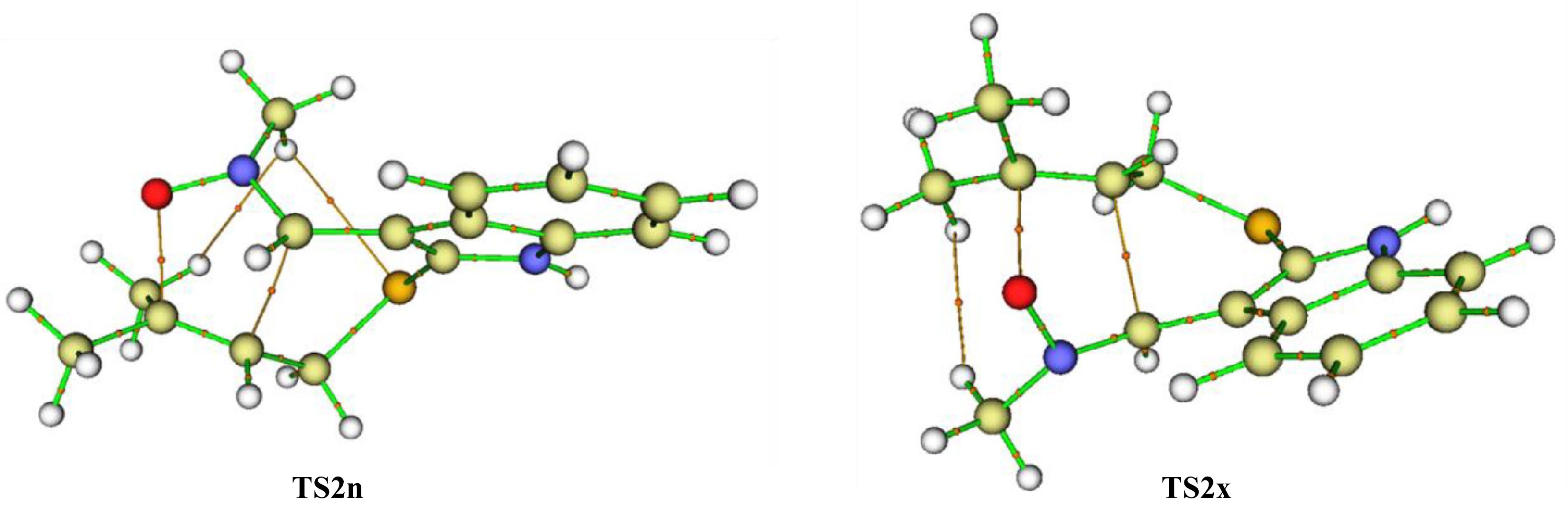
∇ ∇ ∇ ∇ For distinguishing between the NCS presented at both TS2n and TS2x, a QTAIM study[51] at these structures have been realized, in which the values of total electron density, total electron energy density and Laplacian of the (3,−1) cps are given in Table 4. Figure 9 illustrates the schematisation of the QTAIM molecular graphs associated to TS2n and TS2x that are obtained from the QTAIM study.
Table 4. QTAIM parameters associated with the (3,−1) cps associated with TS2n and TS2x.
Interaction ρcbcp ∇2ρbcp Hbcp TS2n O1…C5 0.79 0.12 -0.15 C3…C4 0.58 0.43 0.13 H…H 0.44 0.16 0.99 H…S 0.99 0.32 0.15 TS2x O1…C5 0.69 0.18 -0.97 C3…C4 0.65 0.33 -0.16 H…H 0.30 0.81 0.30 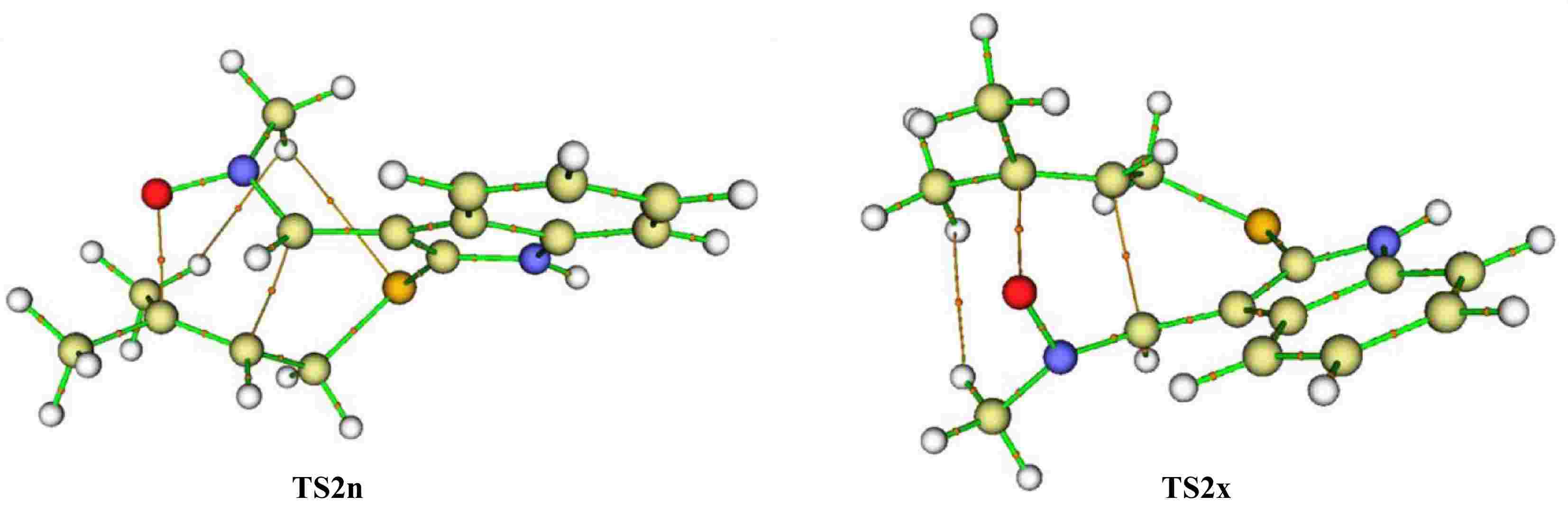
Figure 9.
QTAIM molecular schematisation of TS2n and TS2x together with bond paths and (3,−1) critical points.
Figure 9 shows that except the interactions associated with the forming bonds, TS2n has two NCIs that are H…H and H…S, while TS2x has only one H…H NCI type. Thereby, the stabilisation of TS2n may be attributed to the presence of a supplementary H…S NCI.
Table 4 shows that all (3,-1) bcps associated with H…H and H…S interactions presented both in TS2n and TS2x are characterized by
∇ -
In this study, we conducted a theoretical analysis of the molecular mechanism, regioselectivity, and stereoselectivity of the I32CA reaction of (E)-N-((2-((methylbut-2-en-1-yl)thio)-1H-indol-3-yl)methylene)methanamine oxide 1, derived from 2-((2-methylprop-1-en-1-yl)thio)-1H-indole-3-carbaldehyde through MEDT within the B3LYP/6-31G(d,p) DFT method. Our main findings are as follows:
(i) The B3LYP/6-31G(d,p) computational level, accurately explains the regio- and stereo-selectivity observed experimentally.
(ii) The I32CA reaction occurs under kinetic control leading to formation of a single cycloadduct obtained from compound 1.
(iii) Analysis of the TSs as well as bond order indices indicates that this I32CA reaction occurs through a one-step slightly asynchronous mechanism.
(iv) The effect of solvation increases the activation energies but does not modify the gas phase selectivity patterns.
(v) The studied I32CA is characterized by exothermic behaviour for all reactive pathways and exergonic behaviour for the fused paths and an endergonic for the bridged one.
(vi) Bond distance analysis of the forming bond at the TSs shows that this I32CA reaction precedes through a slightly asynchronous mechanism.
(vii) ELF study shows that this I32CA occurs through a non-concerted two stage one-step mechanism.
(viii) NCI and QTAIM topological analyses indicate that the stabilisation of the favoured TS2n is attributed to the presence of several weak NCIs.
This study was carried out within the framework of the PRFU project Code: B00L01EN210120220001 of the Ministry of Higher Education and Scientific Research of the Algerian Government.
-
The authors confirm contribution to the paper as follows: study conception and design: Hellel D, Bouabbaci H, Chafaa F; data collection: Hellel D, Chafaa F, Khorief Nacereddine A; analysis and interpretation of results: Hellel D, Chafaa F, Bouabbaci H, Khorief Nacereddine A; draft manuscript preparation: Hellel D, Chafaa F, Khorief Nacereddine A. All authors reviewed the results and approved the final version of the manuscript.
-
All data generated or analyzed during this study are included in the published article and its supplementary information files.
-
The authors declare that they have no conflict of interest.
- Supplementary Table S1 Cartesian coordinates for the stationary points involved in the I32CA reaction of (E)-N-((2-((methylbut-2-en-1-yl)thio)-1H-indol-3-yl)methylene)methanamine-oxide 1 together with the imaginary frequencies of transition states.
- Supplementary Table S2 Values of total and relative energies in gas phase and in toluene solvent of (E)-N-((2-((-methylbut-2-en-1-yl)thio)-1H-indol-3-yl)methylene)methanamine oxide 1, TSs and CAs.
- Supplementary Table S3 Enthalpies (H), relative enthalpies (ΔH), entropies (S), relative entropies (ΔS), free Gibbs energies (G), and relative Gibbs free energies (ΔG) for the TSs and CAs involved in the I32CA of (E)-N-((2-((methylbut-2-en-1-yl)thio)-1H-indol-3-yl)methylene)methanamine oxide 1.
- Supplementary Fig. S1 The IRC profile of the favourable TS2n together with the position of the selected points of the I32CA reaction of 1.
- Copyright: © 2025 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Hellel D, Chafaa F, Bouabbaci H, Khorief Nacereddine A. 2025. A MEDT investigation of mechanism and selectivities of the intramolecular [3+2] cycloaddition reaction of (E)-N-((2-((methylbut-2-en-1-yl)thio)-1H-indol-3-yl)methylene)methanamine-oxide. Progress in Reaction Kinetics and Mechanism 50: e005 doi: 10.48130/prkm-0025-0005 

Figure 1.
The possible reactive channels of the I32CA reaction.