










-
Lignin is the most abundant renewable aromatic polymer in nature and a key component of lignocellulosic biomass, accounting for 15%–30% of biomass content[1]. With a carbon content exceeding 60%, lignin contributes approximately 30% of Earth's organic carbon storage and serves as an important feedstock for producing renewable chemicals and biofuels[2]. Pyrolysis technology, as a core process for converting lignin into bio-oil and high-value chemicals, achieves efficient transformation. Elucidating the pyrolysis mechanism of lignin holds significant practical importance for optimizing thermochemical conversion processes, improving bio-oil quality, and enabling high-value utilization[3].
Lignin is composed of three basic structural units (p-hydroxyphenyl/H, guaiacyl/G, and syringyl/S) interconnected via ether linkages and C-C bonds, forming a complex three-dimensional polyaromatic structure[4]. This heterogeneity determines that its pyrolysis behavior is closely temperature-dependent[5]. Previous studies have demonstrated that bond cleavage during lignin pyrolysis (400–600 °C) is primarily governed by bond dissociation energies[6], and initiates with ether linkage scission to generate monomeric compounds such as phenol, anisole, guaiacol, and syringol derivatives with varying methoxy substitution. The primary products subsequently undergo secondary reactions of aromatic rings and substituent groups, forming complex reaction networks[7]. On one hand, they further decompose through cleavage and rearrangement reactions; on the other hand, they may repolymerize to form more thermally stable secondary products[8]. However, current experimental techniques cannot fully capture the dynamic reaction network due to limitations in tracking transient radicals and real-time reaction processes.
The ReaxFF MD approach effectively bridges this gap by extending the dynamic chemical processes of bond breaking and formation, thereby revealing the pyrolysis mechanism from reactants to products at the microscopic scale[9]. Typical feedstocks used in experimental and simulation studies of lignin pyrolysis include native lignin, isolated lignin, and lignin model compounds such as monomers and dimers. Zhang et al.[10] employed ReaxFF MD simulations to study a macromolecular softwood lignin model containing 15,920 atoms, investigating the initial bond cleavage pathways. The results indicated that different linkages exhibit markedly distinct initial reaction behaviors, with Cα/Cβ ether bond scission dominating in α-O-4 and β-O-4 structures, while ring-opening reactions govern the initial consumption of other linkages such as 4-O-5 and 5-5 and their associated monomer units. Sakurai et al.[11] examined the correlation between pyrolysis temperature and product yields and concluded that the distribution of gas, tar, and char is highly temperature-dependent, with higher temperatures favoring char carbonization, the formation of polycyclic structures, and gas production. Liu et al.[12] showed that in hardwood lignin, hydroxyl, methoxy, and benzene groups are the main hydrogen sources for H2, methoxy and hydroxyl groups and linkages contribute to CO, β-O-4 linkages and functional groups decrease continuously, and char grows via carbon chain extension and fragment bonding. The dimer model is often used as a model compound to study the pyrolysis mechanism of different linkages in lignin. Zhou et al.[13] used the ReaxFF MD method to investigate the fast pyrolysis of a lignin dimer DMPD with a representative β-O-4 linkage. The results showed that higher temperatures promote the formation of CO and C5-C12 products as well as the cleavage of C-O bonds. PPE is also a commonly used lignin dimer model for studying the depolymerization of β-O-4 linkages. Wang et al.[14] showed that in PPE models, the Cα-Cβ and Cβ-O bonds have significantly lower bond dissociation energies than other bonds, indicating that their homolytic cleavage dominates the pyrolysis mechanism. In actual pyrolysis, the high activation energy of unimolecular decomposition makes it difficult to generate sufficient H radicals[15]. Cui et al.[16] reported that the initial stage of β-O-4 lignin dimer pyrolysis is dominated by intermolecular radical reactions involving methyl radicals, in which -OCH3 and -OH substituents promote hydrogen transfer and CH4 formation, while -CH2OH substitution at the Cβ position reduces this hydrogen transfer. At the monomer level, phenol, anisole, guaiacol, and syringol are widely used as lignin-derived models to clarify substituent effects on bond cleavage, radical chemistry, and secondary transformations during pyrolysis. Hu et al.[17] demonstrated through ReaxFF MD that anisole undergoes initial decomposition at 1,976 K to form phenol, while small-molecule gases (H2, CO, and CH4) are primarily generated above 3,000 K through secondary reactions, with CO mainly derived from phenoxy radical decarbonylation. Nguyen et al.[18] demonstrated that guaiacol pyrolysis mainly follows the pathway 'guaiacol → hydroxyphenoxy radical → hydroxycyclopenta-dienyl radical + CO'. Furutani et al.[19] showed that the initial pyrolysis stage of syringol resembles that of anisole and guaiacol, all preferentially undergoing O-CH3 bond homolysis to generate methyl radicals and PhOH(OCH3)O radicals, which subsequently induce methoxy rearrangement and demethoxylation reactions. Hu et al.[20] found that phenolic hydroxyl groups maintain high stability during lignin pyrolysis, with significantly higher reaction barriers than various homolytic reactions involving methoxy groups. As an electron-donating substituent, phenolic hydroxyl groups affect the reactivity of other ring substituents and promote the elimination of para-side-chain functional groups[21].
While existing research has clarified the decisive role of inter-unit linkages in pyrolysis initiation and the distinct reactivity of methoxy versus phenolic hydroxyl groups in monomers, a systematic investigation into how multiple functional groups influence pyrolysis behavior remains notably absent. To address this scientific challenge, the present study employs ReaxFF MD simulations to systematically investigate the pyrolysis mechanisms of six lignin monomer model compounds with different aromatic substituents under various temperature conditions. The study focuses on the influence of substituents on the selectivity of initial bond cleavage, the evolution of radicals and intermediates, and the temperature-dependent molecular evolution and product distribution. By systematically analyzing these processes, this work aims to provide molecular-level insights into the role of substituents in high-temperature lignin pyrolysis and to lay a theoretical foundation for subsequent studies on selective lignin conversion.
-
The selected lignin model compounds (see Supplementary Fig. S1) cover key structural types, including H-type, G-type, and S-type monomers. Their aromatic rings feature varying numbers and positions of oxygenate functional groups (Caryl-OH and Caryl-OMe groups), while the C4 side-chain substituents (–CH3, –CH=CHCH3) provide an ideal comparative system for investigating structural effects on pyrolysis pathways and product distributions.
Structural optimization of monomer components was performed using Gaussian 16 software at the B3LYP/6-311+G(d,p) theoretical level[22,23]. The initial configurations for the model compounds were selected based on their electronic energies. The conformer with the lowest energy, which, according to the Boltzmann distribution, possesses the highest statistical population under equilibrium conditions, was chosen as the most representative starting structure for simulations. Subsequently, single-component models were constructed by randomly packing 100 molecules into a cubic periodic box using Packmol[24]. LAMMPS-format data files were generated using OVITO[25]. All ReaxFF MD simulations were completed using LAMMPS software[26]. The NPT ensemble with periodic boundary conditions was employed, using a timestep of 0.1 fs. Throughout the simulations, molecules and radical species were continuously identified based on connectivity defined by a bond order cutoff of 0.3 to monitor the reaction dynamics. The CHO-2016 reactive force field was selected, which has been validated for pyrolysis and combustion systems[27]. All simulations are repeated three times. The uncertainties for the primary quantitative trends (e.g., Time evolutions, product distributions) are within ± 15%. The simulation procedure consisted of three key steps. Firstly, energy minimization was performed to optimize molecular configurations by achieving local potential energy minima. Next, the system temperature was raised to 300 K, followed by a 20-ps relaxation simulation using the Nosé-Hoover thermostat to establish dynamic equilibrium of state parameters (temperature, pressure, density, kinetic and potential energies). Finally, a series of simulations was performed at five different target temperatures (2,000, 2,500, 3,000, 3,500, and 4,000 K), with an independent 300 ps run conducted at each temperature to obtain the respective reaction mechanisms and product distributions. The simulated temperatures were significantly higher than experimental conditions due to the vastly different timescales (picosecond-scale simulations vs second-scale experiments). Elevated temperatures accelerate atomic collisions within limited computational resources, thereby promoting chemical reactions and reducing required simulation time[13]. Previous studies have demonstrated that the reaction networks derived from high-temperature ReaxFF MD simulations are consistent with experimental observations[28,29]. Simulation results were analyzed using ChemTrayzer[30] integrated with custom Python scripts. This approach enabled the extraction of product distributions, elementary reaction pathways, and kinetic information, thereby elucidating the microscopic mechanisms of pyrolysis.
To investigate the pyrolysis kinetic characteristics, the consumption rates of parent molecules within 500 K intervals across the 2,000–4,000 K temperature range were used. The initial decomposition rate constants k at each temperature were obtained by exponential fitting of the decomposed parent molecule counts. The k is calculated as Eq. (1):
$ \begin{matrix}\mathrm{ln}{\mathrm{N}}_{0}-\mathrm{ln}{\mathrm{N}}_{\mathrm{t}}=k\mathrm{t}\end{matrix} $ (1) where, N0 is the initial number of parent molecules (N0 = 100), t is the reaction time, and Nt is the number of parent molecules at time t. The Arrhenius equation is expressed as Eq. (2):
$ k=A\;{\exp}\left(-\dfrac{{E}_{a}}{RT}\right) $ (2) The values of A and Ea can be determined by fitting the Arrhenius equation. Taking the logarithm yields the relationship between the reaction rate constant k and activation energy Ea as the following Eq. (3):
$ \ln \left(k\right)=\ln \left(A\right)-\dfrac{{E}_{a}}{RT} $ (3) where, Ea is the activation energy, T is the pyrolysis temperature, R is the universal gas constant, and A is the pre-exponential factor.
-
The evolution of parent molecule decomposition and fragment generation was analyzed across 2,000–4,000 K to characterize temperature and structural effects on pyrolysis characteristics. According to Fig. 1a–c, the simulation results demonstrate that increasing temperature significantly accelerates the pyrolysis process of parent molecules. Molecular complexity was found to substantially influence decomposition kinetics, with more labile structures exhibiting faster degradation rates. At 2,000 K, phenol showed negligible decomposition, maintaining a dynamic equilibrium between decomposition and recombination. Other compounds decomposed relatively slowly, with syringol exhibiting the fastest decomposition rate and highest conversion (though < 1/3 ultimate conversion). The negligible decomposition of phenol is attributed to the high stability of the Caryl-OH bond, whereas the fastest decomposition of syringol originates from the presence of two low-bond-energy O-CH3 bonds. At 3,000 K, all compounds except phenol underwent complete decomposition within 300 ps, with reaction rates increasing significantly. This trend became more pronounced at 4,000 K, where full decomposition occurred within just 30 ps. This demonstrates that an increase in the number of such labile methoxy groups provides more initiation sites, thereby accelerating the overall decomposition process. The evolution of fragment quantities in the simulated system at different temperatures is shown in Fig. 1d–f, revealing that the total number of fragments increases with rising temperature. At 2,000 K, limited thermal decomposition occurs due to the relatively low temperature. As temperatures increase, the pyrolysis rates accelerate significantly, leading to substantial growth in fragment production. The molecular complexity shows a positive correlation with fragment generation across all temperatures, where compounds containing more substituents consistently produce greater quantities of fragments during pyrolysis.
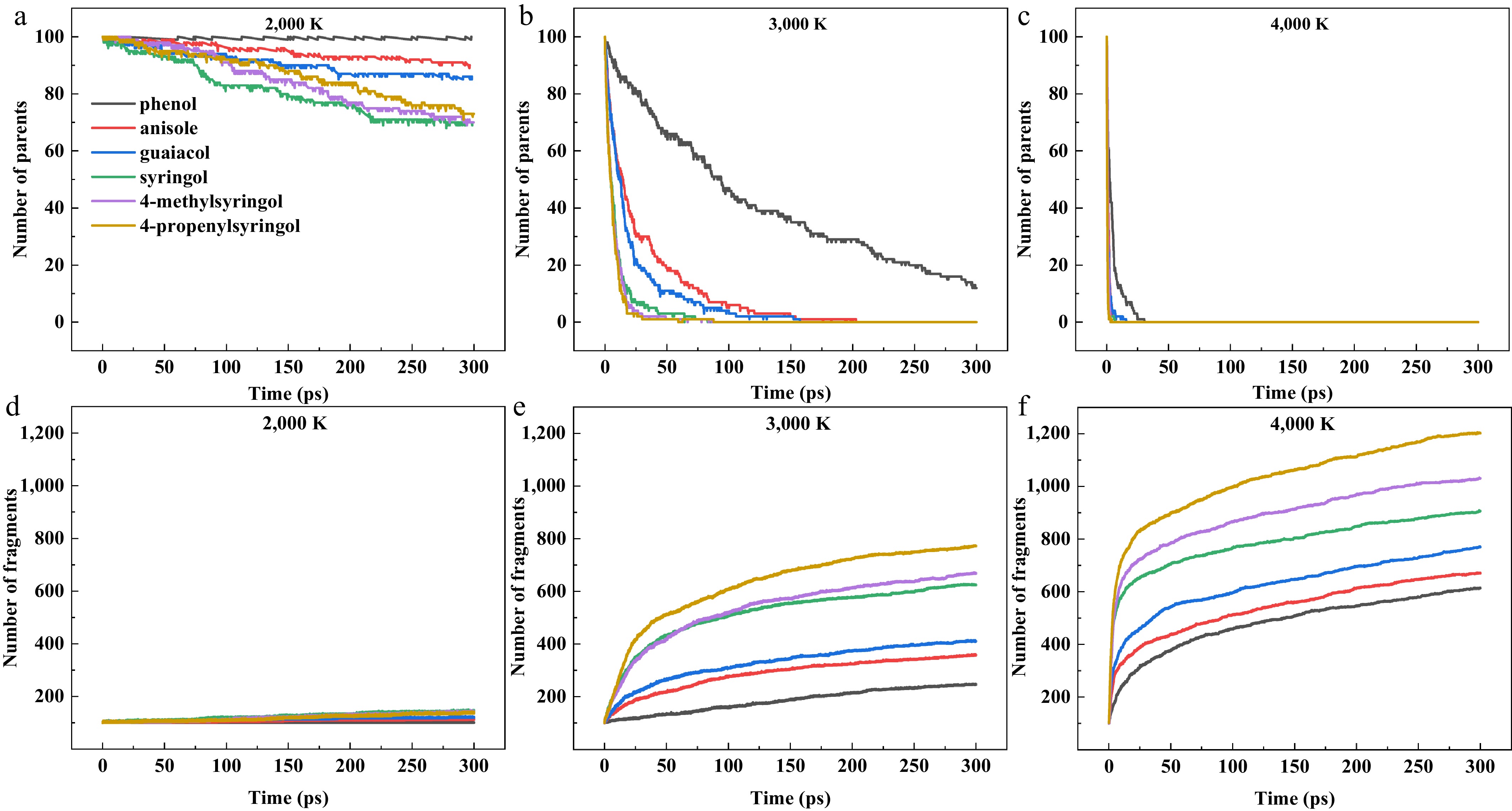
Figure 1.
(a)–(c) Time evolution of parent molecule and fragment. (d)–(f) Quantities for six model compounds at pyrolysis temperatures of 2,000, 3,000, and 4,000 K.
The kinetic data, including the pre-exponential factors (A) obtained from the Arrhenius fitting (see Supplementary Fig. S2 and Supplementary Table S1) show that phenol exhibits the highest Ea (350.6 kJ/mol), while syringol shows the lowest (204.9 kJ/mol). The activation energies of 4-methylsyringol (222.1 kJ/mol) and 4-propenylsyringol (230.6 kJ/mol) are intermediate between these two compounds. These results clearly demonstrate that additional methoxy groups effectively reduce activation energy, whereas C4 substituents (propenyl/methyl groups) increase Ea. The observed trend can be explained by fundamental chemical principles. The relatively weak O-CH3 bond readily undergoes homolytic cleavage, generating phenoxy radicals and thereby lowering the effective activation barrier for pyrolysis initiation. In contrast, alkyl substituents such as methyl or propenyl groups stabilize the resulting phenoxy radical through σ-π hyperconjugation or extended π-π conjugation, respectively. This radical stabilization consequently raises the overall measured activation energy. This conclusion is consistent with the data on the evolution of parent molecule decomposition, as shown in Fig. 1a.
Pyrolysis products classification
-
The distribution of pyrolysis products at different temperatures is shown in Fig. 2. The products can be classified into aryl oxygenates (Fig. 2a), non-aryl oxygenates (Fig. 2b), C1–C5 hydrocarbons (Fig. 2c), and C6 aliphatic hydrocarbons (Fig. 2d), respectively. C6 aliphatic hydrocarbons refer to highly reactive, non-aromatic intermediates with six carbon atoms, represented by highly unsaturated linear or cyclic C6Hx structures such as cyclohexa-1,2,4,5-tetraene and hexa-1,3,5-triyne. At low temperatures (2,000–2,500 K), oxygenated compounds (both aryl and non-aryl, as shown in Fig. 2a, b, respectively) account for a high proportion. This trend is primarily attributed to the facile homolytic cleavage of the O-CH3 bond in methoxy groups, which exhibits a relatively low bond dissociation energy. This cleavage directly yields oxygenated radical intermediates, a point further corroborated in a subsequent section of this study, and such radicals subsequently drive the formation of the observed oxygenated products. Additionally, a conversion relationship exists between aryl and non-aryl oxygenates at the elevated temperatures (2,500–4,000 K) as shown in Fig. 2a, b. This conversion occurs mainly through the elimination of oxygenate groups from aromatic rings and the ring-opening reactions. In the high-temperature region above 3,000 K, C1–C5 hydrocarbons increase significantly. This significant increase results from the thermally induced opening of aromatic rings. A general reaction pathway leading from the parent molecule to C1–C5 species involves sequential steps of demethylation, demethoxylation, ring-opening via decarbonylation, isomerization, dehydroxylation, and recombination. A representative formation route for penta-1,2-dien-4-yne (C5H4) is illustrated in Supplementary Fig. S3. The yield of C6 aliphatic hydrocarbons increases slowly with temperature, reaching a maximum at 3,500 K before declining. This peak, followed by a reduction, suggests that higher temperatures promote further cracking of six-carbon skeletons into C1–C5 products.
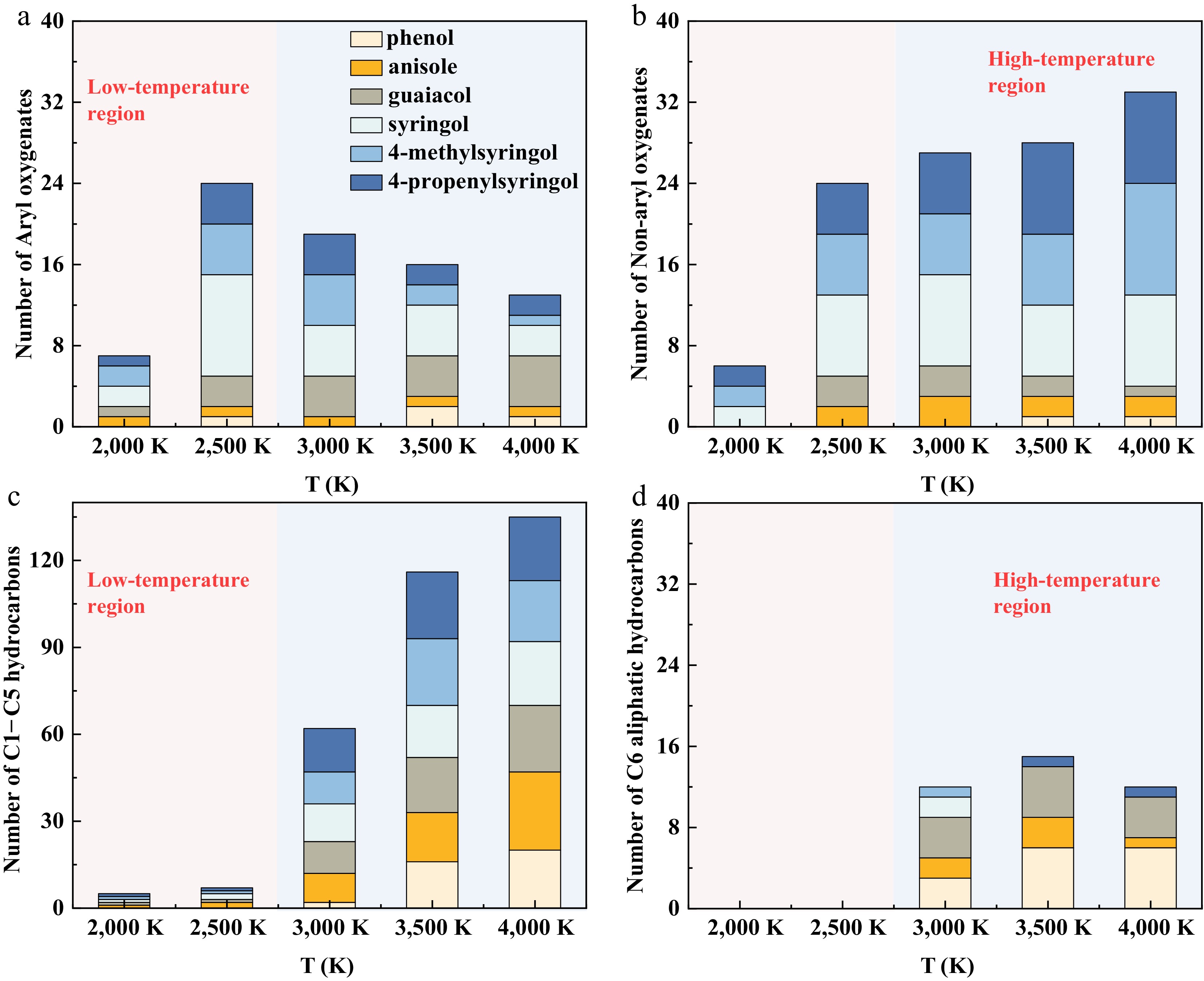
Figure 2.
Distribution of pyrolysis products for six model compounds at five temperatures. (a) Aryl oxygenates. (b) Non-aryl oxygenates. (c) C1−C5 hydrocarbons. (d) C6 aliphatic hydrocarbons.
During pyrolysis, the yield of aromatic oxygenates strongly correlates with the number of oxygenate groups in the parent molecule. Consequently, S-type compounds produce more aryl oxygenates than others. Syringol generates higher quantities of aryl oxygenates compared to 4-methylsyringol and 4-propenylsyringol. The alkyl side chains in S-type monomers undergo competitive cleavage during pyrolysis, thereby suppressing aryl oxygenate compound generation. The formation of non-aryl oxygenates is likewise influenced by the number of oxygen functional groups. During pyrolysis, oxygenate groups (e.g., ·OCH3) readily undergo cleavage to form non-aryl oxygenates (such as formaldehyde). S-type monomers, containing three oxygen functional groups, exhibit significantly enhanced yields of non-aryl oxygenates. Aromatic ring opening, as the critical step for deep decomposition, directly determines subsequent product formation pathways. Anisole produces the highest yield of C1–C5 hydrocarbons at 4,000 K, primarily because its single methoxy group readily cleaves to form ·CH3 while the absence of other groups on the aromatic ring enables efficient ring-opening reactions. In contrast, the multifunctional structures of S-type monomers disperse carbon sources, resulting in lower C1–C5 hydrocarbon production efficiency. Phenol demonstrates the highest C6 aliphatic hydrocarbon yield at 3,500–4,000 K due to its single ·OH group and intact six-carbon structure. Anisole and guaiacol exhibit moderate yields, while S-type monomers show the lowest C6 aliphatic hydrocarbon production.
Major initial pyrolysis pathways
-
To investigate the influence of temperature and molecular structure on the selectivity of pyrolysis reaction pathways, the branching ratios of the initial pyrolysis reactions for each parent molecule at various temperatures were plotted in Fig. 3a–c. In the temperature range of 2,000–4,000 K, the methoxy group cleavage in anisole primarily generates ·CH3, whereas in guaiacol, the methoxy group cleavage tends to produce ·OCH3. This may be due to the significant reduction in the demethoxylation energy barrier caused by ·CH3 produced during guaiacol pyrolysis, which promotes the demethoxylation reaction[31]. S-type compounds contain two methoxy groups, and the proportions of their demethylation and demethoxylation reactions exhibit a roughly opposite trend. The branching ratio for demethylation follows the order of syringol > 4-methylsyringol > 4-propenylsyringol, while the branching ratio for demethoxylation shows the reverse order. The presence of hydrocarbon side chains facilitates the demethoxylation reaction. This observation is further corroborated by the diminished yield of aryl oxygenates, as illustrated in Fig. 2a. Phenol, which lacks a methoxy group, consistently shows the highest probability of dehydroxylation. Its initial pyrolysis primarily proceeds through the cleavage of the Caryl-OH bond, generating phenyl radicals (·C6H5). The branching ratio indicates that at lower temperatures, the proportion of the Caryl-OH bond cleavage reaction is higher; as temperature increases, competing pathways such as aromatic ring cleavage become more prominent, reducing the proportion of this reaction. Comparative analysis with other compounds shows that guaiacol has a higher probability of dehydroxylation (Fig. 3c). For S-type compounds, methoxy group decomposition is prioritized, resulting in a lower dehydroxylation probability. This observation can be explained by the significant difference in the bond dissociation energies of the Caryl-OH, O-CH3, and Caryl-OCH3 bonds. Among these, syringols undergo demethylation and demethoxylation, further inhibiting Caryl-OH bond cleavage.
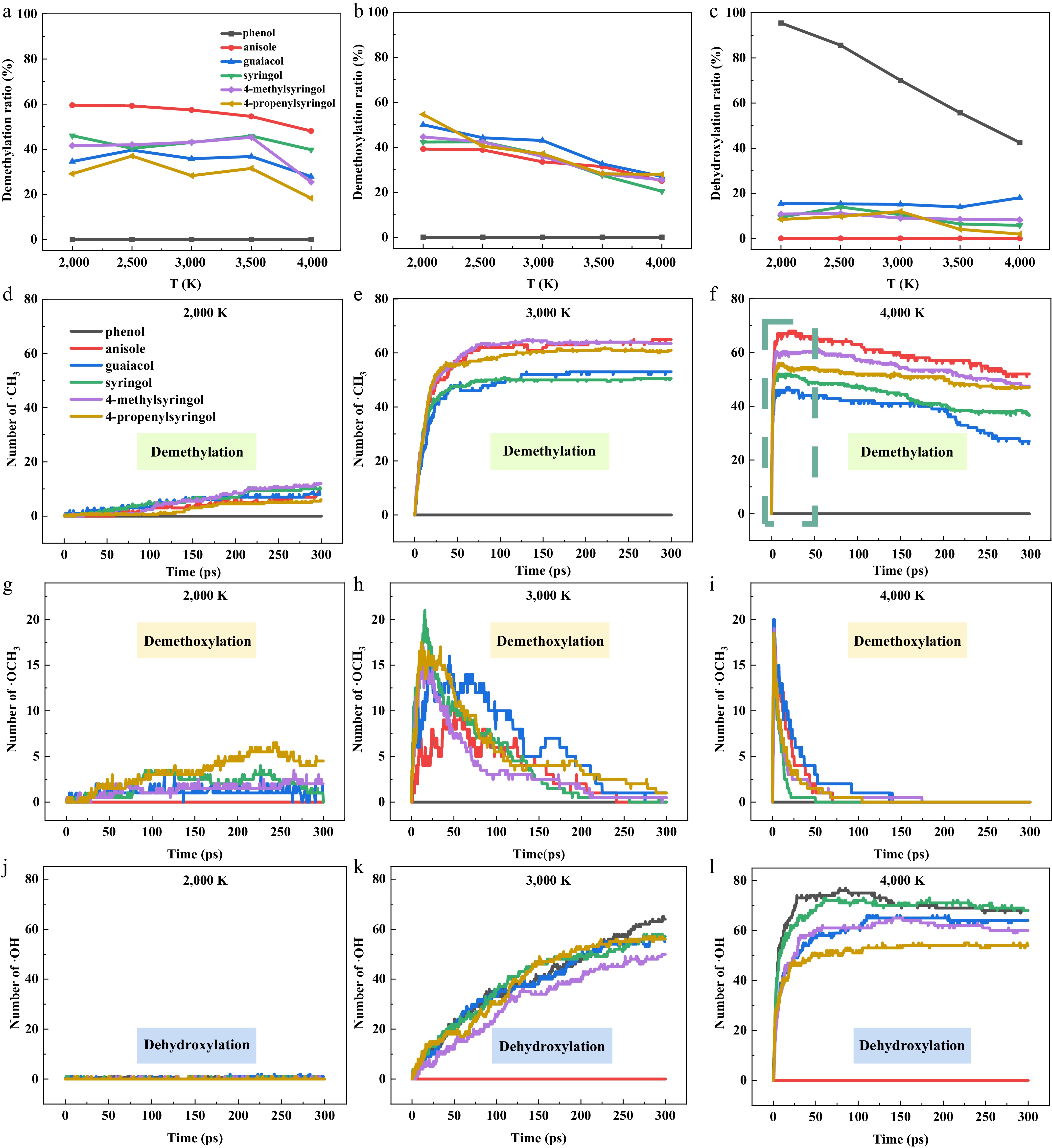
Figure 3.
Temperature dependence of initial pyrolysis reaction branching ratios and key radical numbers for six model compounds. (a)–(c) Branching ratios of the initial pyrolysis reaction pathways. (d)–(l) The number of ·CH3, ·OCH3, and ·OH radicals obtained at 2,000, 3,000, and 4,000 K. The number of ·CH3 and ·OCH3 radicals for the syringol derivatives (syringol, 4-methylsyringol, and 4-propenylsyringol) was divided by 2.
To further quantify the contributions of the above reactions to the production of radicals (·CH3, ·OCH3, and ·OH), the evolution trends of radical generation from six parent molecules during pyrolysis at different temperatures were analyzed as shown in Fig. 3d–l. At 2,000 K, the ·CH3 generation yields of the six compounds were similar, with no significant differentiation. When the temperature increased to 3,000 K (Fig. 3e), all five compounds exhibited a similar two-stage evolution pattern, with a rapid generation phase in the initial stage (< 50 ps), followed by a dynamic equilibrium plateau. Notably, 4-methylsyringol and anisole maintained similar CH3 generation yields throughout the reaction time scale. At 4,000 K (Fig. 3f), the methyl yields are accompanied by a noticeable slow decrease due to methyl decomposition into methylene via losing ·H. The O-CH3 bond cleavage is the primary pathway for methyl radical formation.
At 2,000 K (Fig. 3g), 4-propenylsyringol produces a relatively high quantity of methoxy radicals. The branching ratio plot (Fig. 3b) indicates that the demethoxylation proportion of 4-propenylsyringol is higher at low temperatures. When the temperature increases to 3,000 K (Fig. 3h), the methoxy radical yield reaches a peak after an initial rapid generation phase, followed by a slow decline. Other compounds exhibit similar generation trends. The methoxy radical yield drops to zero rapidly for all compounds at 4,000 K (Fig. 3i), indicating that at high temperatures, methoxy radicals, as transient intermediates, are readily consumed.
Due to the delocalization effect of oxygen's lone pair electrons into the π-bond orbitals of the aromatic ring, the Caryl-OH bond in phenolic compounds is strengthened, with a bond dissociation energy as high as 468 kJ/mol[32], resulting in a very low yield of ·OH at 2,000 K (Fig. 3j). At 3,000 K (Fig. 3k), the ·OH yields continue to increase and approach similar levels for all compounds. At the highest temperature of 4,000 K (Fig. 3l), the ·OH generation curves exhibit an initial rise followed by a plateau. Phenol, which preferentially generates ·OH through phenolic hydroxyl dissociation, shows distinct yield peaks at 3,000 K and in the early stage at 4,000 K.
Initial pyrolysis mechanisms
-
To deeply investigate the thermal decomposition process of parent molecules, the detailed pyrolysis mechanism at 3,500 K was extracted. Homolytic cleavage of oxygenate groups and subsequent isomerization reactions are the two primary types of unimolecular initial pyrolysis pathways. The pyrolysis of phenol primarily proceeds through three reaction pathways, including dehydroxylation, dehydrogenation, and isomerization, as shown in Fig. 4. The hydroxyl group of phenol dissociates to produce ·C6H5 and ·OH. The dehydrogenation reactions generate ortho-hydroxyphenyl radical and an H atom. Phenol also undergoes isomerization, forming unsaturated enol compounds. The primary initial pyrolysis pathway for anisole is the loss of ·CH3, forming phenoxy radicals. Phenoxy radicals can rearrange to form epoxide intermediates, which further convert into more stable oxygenated conjugated systems. Another pathway involves direct removal of ·OCH3, forming ·C6H5. The primary initial pyrolysis pathway for guaiacol is the homolytic cleavage of the O-CH3 bond, forming ortho-hydroxyphenoxy radical and ·CH3. From the perspective of bond dissociation energy, the O-CH3 bond cleavage has the lowest energy compared to other bonds in typical phenolic monomers, making it more prone to cleavage[33]. Subsequently, homolytic cleavage of the Caryl-OCH3 bond and the Caryl-OH bond occur. These bond cleavage reactions produce methoxy and ortho-hydroxyphenyl radicals, as well as ·OH and ortho-methoxyphenyl radicals, respectively. The isomerization reactions of ortho-methoxyphenyl radical and ortho-hydroxyphenyl radical were also observed, giving epoxide and ring-opening intermediates. These epoxide intermediates primarily undergo further re-isomerization to form corresponding hydroxyphenoxy radicals. From the initial pyrolysis reactions of the three compounds, it is evident that homolytic cleavage reactions dominate in the primary reaction, while secondary reactions are primarily characterized by isomerization.
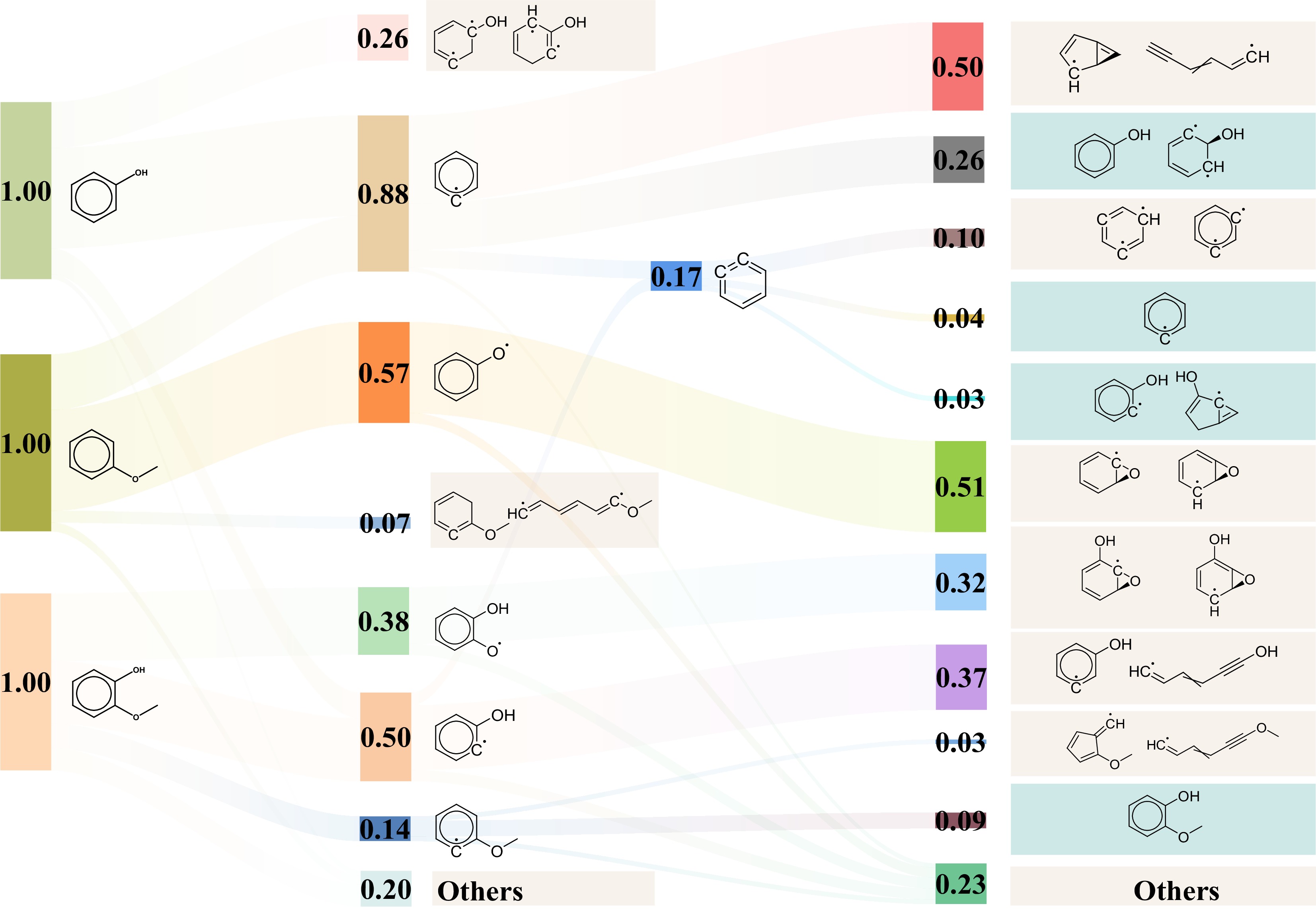
Figure 4.
Sankey diagram of the initial pyrolysis mechanisms of phenol, anisole, and guaiacol at 3,500 K. The isomerization products are highlighted in blue, and the recombination reaction products in cyan, both located in the far right.
To further elucidate the influence of additional methoxy groups and substituents at the 4-position on initial pyrolysis reactions, the following analyzes the initial pyrolysis mechanism of syringol as shown in Fig. 5. The unimolecular cleavage of the O-CH3 bond, generating ·CH3 and the PhOH(OCH3)O radical, is the primary initial pathway for syringol, accounting for 47% of the reactions. This pathway can lead to interconversion between epoxide isomers. Further demethoxylation produces 2-hydroxybenzenone. Subsequent homolytic cleavage reactions, including demethylation and dehydroxylation, also occur. The second primary pyrolysis reaction of syringol involves the cleavage of the methoxy group to form Ph(OH)(OCH3) radical, contributing 28%. The Ph(OH)(OCH3) radical primarily undergoes an isomerization reaction via the 1,2-shift of the hydroxyl group. The third primary pyrolysis reaction is dehydroxylation, followed by demethoxylation, yielding 1-methoxycyclohexa-1,3-dien-5-yne. The proportion of 'other' products from the primary reaction of syringol is notably high at 19%. It suggests that the reaction pathways of this compound involve more complex radical cleavage mechanisms. Both 4-methylsyringol and 4-propenylsyringol undergo similar initial pyrolysis pathways (see Supplementary Figs. S4, S5), beginning with O-CH3 bond demethylation followed by epoxide isomerization and methoxy group elimination. In contrast, hydroxyl groups, methyl groups, and propenyl groups associated with direct side-chain elimination rarely participate in reactions[34]. Compared to structurally simpler H-type and G-type compounds, the multiple methoxy groups and side chains in S-type monomers generate a greater diversity of intermediates during pyrolysis.
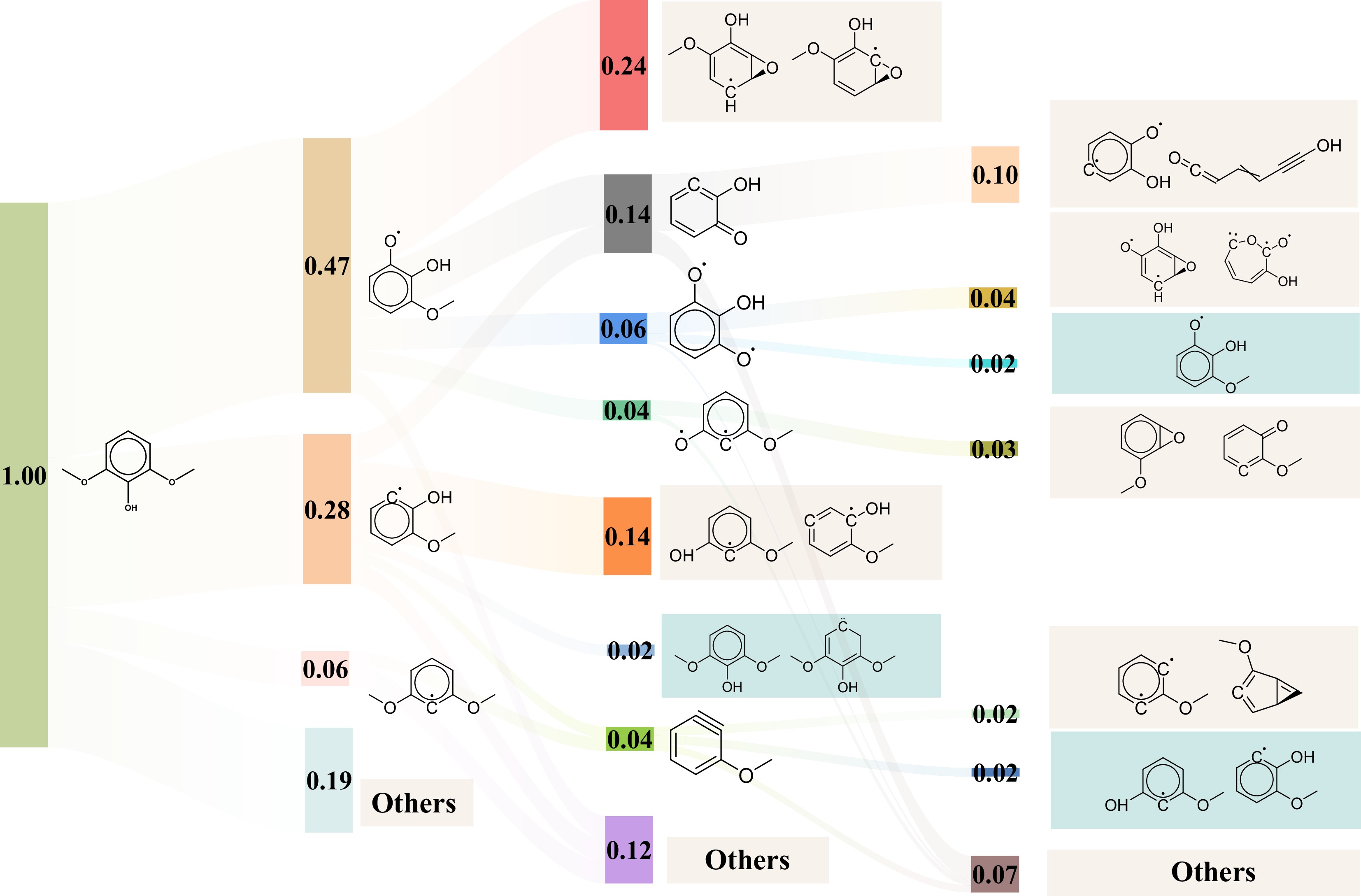
Figure 5.
Sankey diagram of the initial pyrolysis mechanisms of syringol at 3,500 K.
Evolution of primary radical intermediates
-
The temporal evolution of dominant radical intermediate quantities from initial pyrolysis reactions-specifically, the primary radicals generated through homolytic cleavage of the weakest bond in each parent molecule was analyzed across temperatures, as shown in Fig. 6. Phenol exhibits limited radical generation till 3,000 K due to insufficient thermal activation for dehydroxylation, while other compounds show obvious radical formation via demethylation at 2,500 K. In general, primary radicals exhibit both greater yields and extended lifetimes at mild pyrolysis temperatures. However, as the pyrolysis temperature increases, the population of primary radicals declines sharply due to their further decomposition and competition from alternative initiation pathways. The formation of primary radicals needs an induction period (> 50 ps) at 2,000 K (2,500 K for phenol) for all compounds except syringol. The demethylation of syringol exhibits continuous progression without an induction period, demonstrating its highest pyrolytic reactivity among the studied compounds. This observation aligns with the pyrolysis characteristics analysis presented earlier, further confirming the activating effect of methoxy groups on the parent molecular structure, as well as the stabilizing influence of alkyl substituents. Comparative analysis reveals that S-type compounds, with their multi-substituted structures, exhibit accelerated radical consumption kinetics relative to simpler analogues (phenol, anisole, and guaiacol). This enhanced reactivity stems from kinetically favorable side-group reactions, as evidenced by the complete depletion of primary radicals in S-type compounds within 100 ps at 3,000 K, whereas residual primary radicals persist beyond 300 ps in other compounds. These structural effects created distinct temperature-dependent radical evolution patterns, where increased substituent complexity both lowered activation requirements and enhanced radical termination processes[35].
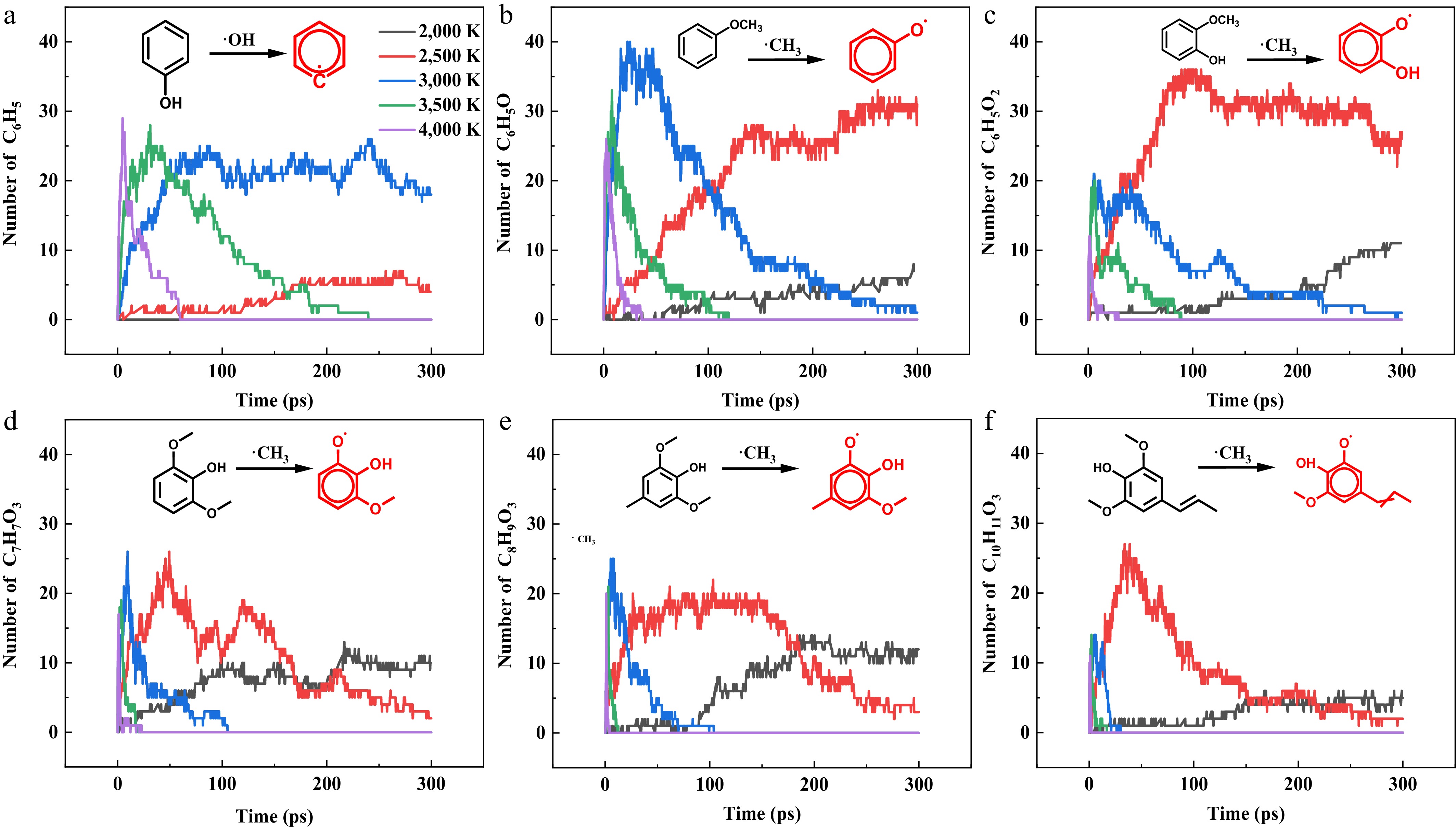
Figure 6.
Temporal evolution of six characteristic initial radicals across five pyrolysis temperatures (2,000, 2,500, 3,000, 3,500, and 4,000 K).
Evolution of major oxygenated species
-
Figure 7 illustrates the statistical distribution of three characteristic neutral oxygenates (CO, Formaldehyde, ethynol) generated from six models during pyrolysis, with these components exhibiting relatively significant yield advantages in the systems. Within the temperature range examined over 3,000–4,000 K, CO is the predominant gaseous oxygenate product. Its formation exhibits significant temperature dependence and structural selectivity. Specifically, the CO yield increases continuously with temperature (Fig. 7a, d), but different kinetic behaviors are observed in different temperature intervals. At 3,000 K, it shows a stable quasi-first-order reaction growth pattern, while at 4,000 K, it exhibits a two-stage characteristic of rapid initial formation followed by a deceleration in the later stages. CO formation mainly follows the decarbonylation pathway of formyl radicals (·CHO) (Fig. 7g), which is significantly activated under high-temperature conditions[36]. The ·CHO primarily originates from the effective utilization of oxygen atoms from the OH group. S-type compounds consistently exhibit higher CO yields than mono-methoxylated compounds at both 3,000 and 4,000 K. Formaldehyde (HCHO) formation initiates at 2,000 K, with significantly higher yields compared to CO and ethynol (HC≡COH) (Supplementary Fig. S6). Its production rises sharply at 3,000 K, peaks rapidly at 4,000 K, and then gradually declines. HCHO yields positively correlate with the methoxy group content in parent molecules, which readily cleave at lower temperatures. Reaction pathway analysis reveals that HCHO primarily forms through single-step hydrogenation of the ·CHO, while its dominant consumption occurs via hydrogen recombination to regenerate ·OCH3 (Fig. 7h). HC≡COH production remained limited, exhibiting a stepwise increase pattern at 3,000 K (Fig. 7c). This suggests the reaction had initiated but proceeded slowly, likely due to its high activation energy barrier that only a fraction of molecules could overcome even at 3,000 K. When temperature reaches 4,000 K (Fig. 7f), HC≡COH yields initially increased before gradually declining. The bell-shaped profile reflects the reaction pathway complexity. High temperatures promoted HC≡COH formation but also accelerated competing consumption pathways. HC≡COH predominantly forms and decomposes through isomerization reactions, including its interconversion with: C=CHOH (Fig. 7i).
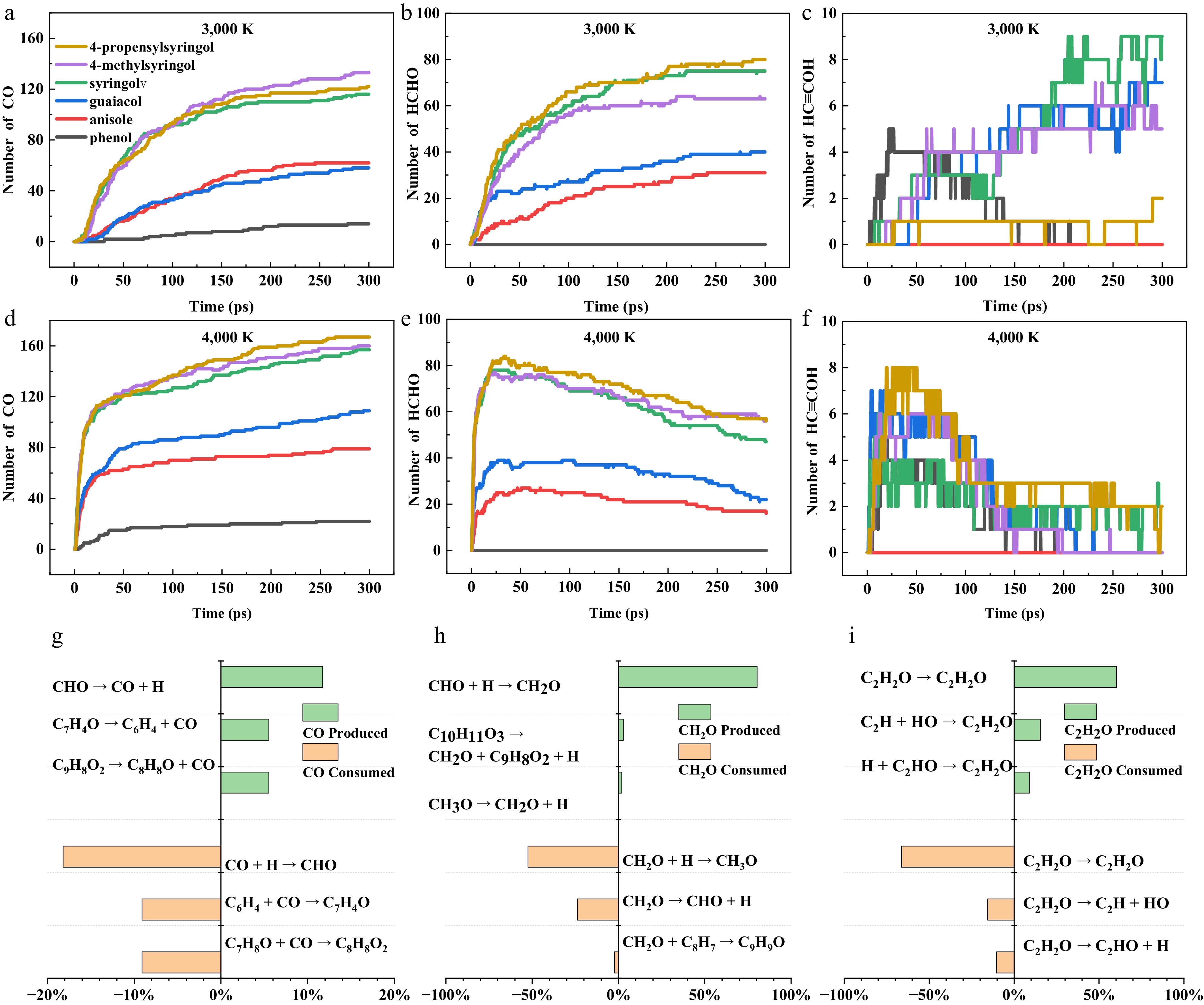
Figure 7.
Quantitative evolution and pathway analysis of key oxygenated small molecules during the pyrolysis of six model compounds. (a)–(c) Quantity-time profiles of CO, HCHO, and HC≡COH generated from the six model compounds at 3,000 K. (d)–(f) Quantity-time profiles of the same three products at 4,000 K. (g)–(i) Generation and consumption pathways of CO, HCHO, and HC≡COH during the pyrolysis of 4-propenylsyringol at 3,500 K.
Evolution of major alkyl species
-
Representative variations in hydrocarbon quantities during pyrolysis at high temperatures is shown in Fig. 8. Acetylene accumulates progressively as a thermodynamically stable end product in Fig. 8a, d (3,000 and 4,000 K, respectively), showing a significant temperature and structure dependence. At 4,000 K, 4-propenylsyringol achieves the highest acetylene yield, while syringol produces the least acetylene. Acetylene generation occurs both by hydrogen addition to ethynyl radicals and through decomposition of polycarbon compounds, with the reverse reactions constituting the main consumption mechanism as shown in Fig. 8h. C4+ intermediates, butadiyne, and penta-1,2-dien-4-yne, exhibit characteristic transient evolution patterns, displaying bell-shaped concentration profiles (initial increase followed by decrease) that are typical of reactive intermediates. At 3,000 K, all model compounds maintain butadiyne yields at single-digit levels (Fig. 8b). Similarly, penta-1,2-dien-4-yne reaches maximum yield in the guaiacol system at 3,000 K under formation-depletion equilibrium (Fig. 8c), but undergoes complete consumption at 4,000 K due to thermally enhanced decomposition. Isomerization reactions are identified as the main source of both butadiyne and penta-1,2-dien-4-yne (Fig. 8i, j), with reverse pathways accounting for their consumption. This isomerization tendency fundamentally stems from π-bond rearrangement propensity at high temperatures, ultimately forming more stable products with conjugated or delocalized electron structures. Aromatic ring opening serves as the prerequisite for C4+ hydrocarbon formation, explaining the generation of butadiyne and penta-1,2-dien-4-yne at high temperatures.
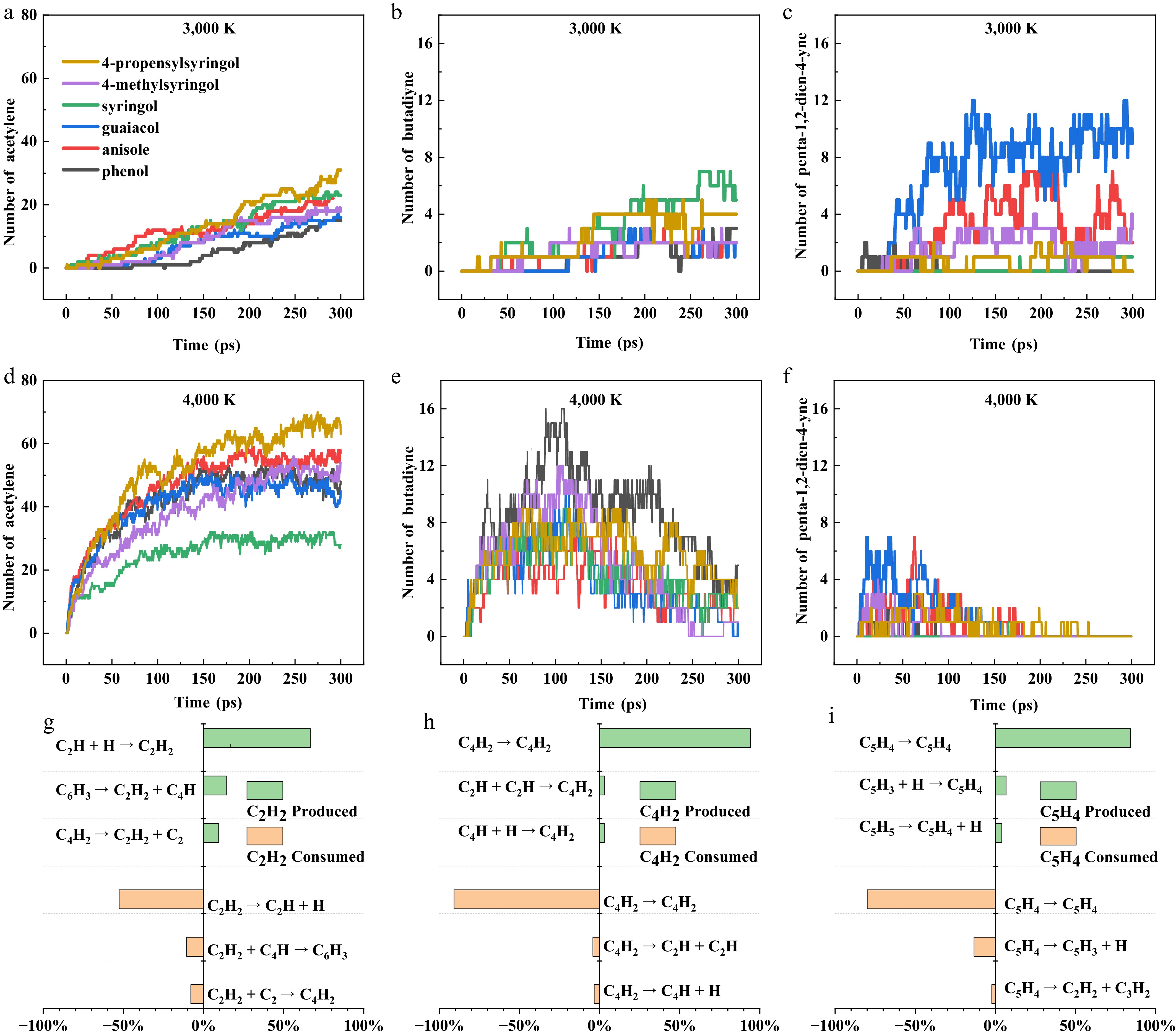
Figure 8.
Quantitative evolution and pathway analysis of key hydrocarbons during the pyrolysis of six model compounds. (a)–(c) Number-time evolution profiles of acetylene (C2H2), butadiyne (C4H2), and penta-1,2-dien-4-yne (C5H4) generated from the six model compounds at 3000 K. (d)–(f) Number-time evolution profiles of the same three products at 4000 K. (g)–(i) Generation and consumption pathways of C2H2, C4H2, and C5H4 during the pyrolysis of 4-propenylsyringol at 3,500 K.
-
This study employed ReaxFF MD simulations to systematically investigate the high-temperature pyrolysis (2,000–4,000 K) mechanisms and kinetics of representative lignin-derived phenolic model compounds, elucidating the critical roles of temperature and molecular structure. Activating methoxy groups substantially lowered activation energies and accelerated parent molecule decomposition and fragment generation, while stabilizing alkyl chains hindered decomposition and suppressed aryl oxygenate formation. Product distribution shifted from aryl/non-aryl oxygenates at low temperatures to C1–C5 hydrocarbons via ring opening at high temperatures, with C6 aliphatics peaking at 3,500 K. Initial pyrolysis was dominated by homolytic cleavage of oxygenate groups (O-CH3, Caryl-OH). Increasing temperature promoted ring cleavage and other competing pathways. Primary radical yields and lifetimes decreased sharply with rising temperature, with complex S-type structures exhibiting both accelerated formation due to methoxy groups and rapid consumption facilitated by diverse substituents. Secondary reactions were governed by the isomerization of initial radicals to produce epoxide intermediates. Among oxygenates, CO emerged as the dominant stable gaseous product above 3,000 K, with its yield strongly dependent on temperature and methoxy group count. Formaldehyde formation/consumption reflected methoxy reactivity, while ethynol production was kinetically limited. For hydrocarbons, acetylene accumulated as the dominant stable end-product, its yield enhanced by temperature and hydrocarbon side chains but reduced by methoxy-rich structures. Transient C4+ intermediates are formed via ring opening and isomerization. The findings provide fundamental insights into the complex pyrolytic decomposition mechanisms of lignin model compounds, establishing clear structure-reactivity-temperature relationships essential for optimizing the thermochemical conversion of lignocellulosic biomass into targeted fuels and chemicals.
This work was supported by the National Natural Science Foundation of China (Grant No. 22269007), and the Hainan Provincial Natural Science Foundation of China (Grant Nos 323RC418, 221QN175).
-
The authors confirm their contributions to the paper as follows: study conception and design, methodology, writing – review & editing, funding acquisition: Liu X; investigation: Zeng H, Ge M, Sun M, Sun R, Meng H; data curation: Zeng H, Ge M; draft manuscript preparation: Zeng H. All authors reviewed the results and approved the final version of the manuscript.
-
All data generated or analyzed during this study are included in this published article.
-
The authors declare that they have no conflict of interest.
- Supplementary Table S1 Pre-exponential factors for the pyrolysis of the six model compounds.
- Supplementary Fig. S1 Molecular structures of six lignin model monomers and a schematic of the simulation model constructed from 100 4-propenylsyringol molecules.
- Supplementary Fig. S2 Pyrolysis kinetic parameters at different temperatures.
- Supplementary Fig. S3 Reaction pathway for C5H4 formation from syringol via aromatic ring-opening.
- Supplementary Fig. S4 Sankey diagram of the initial pyrolysis mechanisms of 4-methylsyringol at 3500 K.
- Supplementary Fig. S5 Sankey diagram of the initial pyrolysis mechanisms of 4-propenylsyringol at 3500 K.
- Supplementary Fig. S6 Quantity-time profiles of CO, HCHO, and HC≡COH generated from the six model compounds at 2000 K.
- Copyright: © 2026 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Zeng H, Ge M, Sun M, Sun R, Meng H, et al. 2026. Influence of molecular structure and temperature on the pyrolysis mechanism of lignin model compounds: insights from ReaxFF MD simulations. Progress in Reaction Kinetics and Mechanism 51: e009 doi: 10.48130/prkm-0026-0010 

Influence of molecular structure and temperature on the pyrolysis mechanism of lignin model compounds: insights from ReaxFF MD simulations
- Received: 23 December 2025
- Revised: 06 February 2026
- Accepted: 17 March 2026
- Published online: 30 April 2026
Abstract: This study employs ReaxFF MD simulations to systematically investigate the pyrolysis behaviors of six lignin monomer model compounds with varying methoxy group numbers and C4-position substituents over 2,000 to 4,000 K. The results indicate that methoxy groups are the key structural units governing initial reactivity, lowering activation energies, and accelerating the fragmentation of parent molecules. While alkyl groups increase activation energies and hinder decomposition. As temperature rises, dominant reaction pathways undergo significant shifts. At lower temperatures (2,000–2,500 K), reactions primarily involve homolytic cleavage and removal of methoxy groups, leading to oxygen-containing products; under high-temperature conditions, ring-opening reactions of aromatic structures are markedly enhanced, promoting the substantial formation of C1–C5 hydrocarbons. Among oxygen-containing products, carbon monoxide gradually evolves into the dominant stable product above 3,000 K, and its generation is synergistically promoted by both elevated temperature and higher methoxy group abundance. Formaldehyde exhibits distinct dynamic evolution patterns at 3,000 and 4,000 K. Regarding hydrocarbon products, acetylene emerges as the predominant stable end-product with continuous accumulation, facilitated by both high-temperature conditions and hydrocarbon side-chain structures. These results clarify, at the atomic scale, the intrinsic relationships among 'molecular structure-reaction pathway-temperature effects' during the pyrolysis of lignin monomer model compounds, providing theoretical foundations for the in-depth understanding of lignin pyrolysis mechanisms and the optimization of thermochemical conversion processes.