









-
Durian (Durio zibethinus L.), a tropical plant belonging to the genus Durio in the family Malvaceae, originated in Borneo and Sumatra[1]. It is widely cultivated in Southeast Asian countries such as Malaysia, Brunei, and Thailand[2], with popular cultivars including Durio zibethinus cv. Musang King, Durio zibethinus cv. Monthong, and Durio zibethinus cv. KanYao. The fruit peel varies in color from green to brown, and the edible flesh consists of arils that range in hue from pale yellow and white to golden yellow. The first draft genome of durian was assembled, with a size of approximately 738 Mb, using PacBio HiFi reads and Chicago high-throughput chromosome conformation capture (Hi-C) scaffolding[3]. With the advancement of third-generation sequencing technologies, multiple cultivars have been sequenced to date. Nawae et al. generated chromosome-level genome assemblies for Durio zibethinus cv. Kradumthong, Durio zibethinus cv. Monthong, and Durio zibethinus cv. Puangmanee with assembled sizes of 832.7, 762.6, and 821.6 Mb, respectively, and their annotations covering 95.7%, 92.4%, and 92.7% of the embryophyta core proteins[4]. Li et al. integrated Illumina, PacBio HiFi, and Oxford Nanopore Technologies (ONT) ultra-long reads to generate a chromosome-level genome assembly of 777.8 Mb, which was further anchored to 28 chromosomes using Hi-C data, resulting in a chromosome-level assembly of 730.67 Mb. This assembly had a contig N50 of 14.23 Mb, and a scaffold N50 of 26.20 Mb, with 38,728 protein-coding genes annotated[5]. Ji et al. initially employed Illumina, PacBio HiFi, ONT reads, and Hi-C data to assemble a contiguous and complete chromosome-level haploid genome of D. zibethinus cv. KanYao[6]. While 19 chromosomes were assembled gap-free, nine chromosomes still contained residual gaps.
Telomeres are evolutionarily conserved fundamental structures in plant genomes, typically composed of short, tandemly repeated minisatellite sequences[7]. Telomerase is a ribonucleoprotein complex consisting of two core components: the telomerase RNA component and telomerase reverse transcriptase (TERT)[8]. As a key gene encoding a critical subunit of the telomerase complex, TERT serves to synthesize telomeric DNA at chromosome ends. This process compensates for the progressive shortening of telomere length during cell division, playing an indispensable role in maintaining chromosomal stability[9]. While TERT family genes have been extensively identified and characterized in various plant species, their systematic identification and analysis remain unexplored in durian.
Nowadays, genome annotation itself faces common challenges, including the only partial conservation of sequence patterns, highly variable intron lengths, inconsistent intergenic distances, prevalent alternative splicing, transposable element (TE) insertions, and the presence of pseudogenes[10]. In the durian genome, these challenges are compounded by its inherently high repetitive content and the numerous resulting assembly gaps. Together, they collectively hinder accurate gene model prediction, comprehensive variant detection, and in-depth exploration of functional elements within repetitive sequences. Therefore, to overcome these limitations and provide a foundational resource for reliable studies in species evolution, population genetics, and functional genomics, generating a telomere-to-telomere (T2T) genome assembly is crucial[11]. In recent years, T2T genomes have been successfully assembled for multiple species, including diverse plants such as Arabidopsis thaliana, Oryza sativa, Vitis vinifera, Zea mays, Brassica rapa, Citrullus lanatus, and Solanum lycopersicum. These achievements provide both a methodological blueprint and empirical support for tackling the challenges of fully assembling the complex durian genome. However, currently available durian genomes are not only fragmented but also primarily represent major tropical cultivars. In contrast, D. zibethinus cv. Chuongbo exhibits dwarf stature and enhanced cold tolerance, which are crucial for expanding durian cultivation. Thus, obtaining a high-quality, complete durian genome for D. zibethinus cv. Chuongbo addresses a key technical gap in durian genomics and enables the elucidation of the genetic basis for its adaptive traits, directly providing targets for molecular breeding.
By integrating PacBio HiFi reads, Oxford Nanopore ultra-long reads, and Hi-C chromatin conformation capture technology, we report the first gap-free genome of D. zibethinus cv. Chuongbo. This gap-free genome will serve as a reference resource of unprecedented precision, facilitating functional genomics, evolutionary studies, and the dissection of the genetic basis underlying key agronomic traits in durian.
-
Plant materials of the diploid durian cultivar D. zibethinus cv. Chuongbo were procured from Lingshui, Hainan (China). The plant is dwarf, cold-resistant, has delicate flesh with a milky aroma, and is suitable for cultivation in Hainan. Fresh young leaves were immediately frozen in liquid nitrogen, and stored at −80 °C for DNA extraction. For PacBio HiFi sequencing, High-molecular-weight genomic DNA was extracted from 0.5 g fresh young leaves using a CTAB[12] method and purified with a QIAGEN genomic DNA kit (cat. 13,323). After quality control, the DNA was sheared, size-selected (> 15 kb) using a PippinHT system, and used to construct a SMRTbell library (SMRTbell Prep Kit 3.0). Sequencing was performed on the PacBio Revio platform to generate HiFi reads. For Oxford Nanopore ultra-long sequencing, high-molecular-weight genomic DNA was separately extracted from 0.5 g of fresh young leaves using an SDS[13] method to maximize the recovery of ultra-long fragments. The DNA was purified and subjected to quality control, including visual inspection, agarose gel electrophoresis, NanoDrop spectrophotometry, and Qubit fluorometry. Following this, target DNA fragments were size-selected using a BluePippin system. A sequencing library was prepared by ligating adapters using the SQK-LSK109 kit. The final library was quantified with Qubit and sequenced on an Oxford Nanopore PromethION platform. To generate Hi-C libraries, chromatin from 0.5 g of formaldehyde-cross-linked fresh young leaves was digested with DpnII, and the ends were filled in with biotinylated nucleotides before proximity ligation. The DNA was then sheared to 300−700 bp, and interaction fragments were captured using streptavidin beads. Library quality was verified by Qubit 3.0, Agilent 2,100 Bioanalyzer, and qPCR. Qualified libraries were sequenced on the MGI platform with paired-end 150 bp reads.
To capture a comprehensive transcriptomic profile across different tissues of D. zibethinus cv. Chuongbo, fresh samples of root, stem, leaf, flower, and fruit were collected, immediately snap-frozen in liquid nitrogen, and stored at −80 °C to preserve RNA integrity. Total RNA was then extracted from these tissues using the RNeasy Plant Mini Kit (Qiagen, Germany) for subsequent RNA sequencing (RNA-Seq) analysis.
Estimation of durian genome size
-
The genome size of durian was estimated by flow cytometric analysis using maize as an internal reference standard. Briefly, nuclei isolated from durian leaf tissues were mixed with maize nuclei in a defined ratio and co-stained with propidium iodide. The mixed nuclear suspension was then analyzed on a BD FACScalibur flow cytometer. Samples were excited with a 488 nm blue laser, and PI fluorescence intensity was measured using an appropriate emission filter. The genome size of durian was calculated based on the ratio of the mean fluorescence intensities of durian and maize nuclei, using the known genome size of maize (approximately 2.3 Gb) as the reference.
The k-mer analysis was performed as part of a comprehensive genome survey. Initially, PacBio HiFi reads were filtered to retain sequences with a minimum length of 1,000 bp, and an average Phred quality score ≥ Q20. Following this, Jellyfish v2.2.10[14] was employed to conduct a frequency distribution analysis with k-mer size set to 21. Subsequently, GenomeScope v2.0[15] was used to estimate the genome size, heterozygosity, and duplication rate.
Genome assembly and quality assessment
-
The durian genome was assembled and analyzed using an integrated pipeline that combines long-read sequencing and chromatin conformation capture technologies. The initial assembly was performed with Hifiasm v0.16.1[16], utilizing both the filtered PacBio HiFi reads and the processed ONT reads. To ensure the purity of the initial assembly, the resulting contigs were screened against the NCBI non-redundant nucleotide (nt) database to identify and exclude any potential non-plant sequences (e.g., bacteria or fungi). Duplications were then removed using Purge_dups v1.2.6[17] with the -2 -T parameters to obtain a non-redundant assembly. Subsequently, the assembled contigs were polished iteratively using NextPolish v1.4.1[18] with the raw HiFi and ONT reads as references to enhance base-level accuracy. The polished contigs were scaffolded into chromosome-level assemblies using Hi-C data. The Hi-C data were processed through the Juicer v1.6[19] pipeline to generate chromatin interaction matrices. These matrices were used by the 3D-DNA v180114[20] software to perform chromatin conformation-guided assembly, anchoring, ordering, and orienting contigs onto chromosomes. The preliminary chromosomal models were manually reviewed and adjusted in the Juicebox v1.11.08[21] assembly visualization tool, where the contact maps were used to correct mis-joins and orientations, resulting in a high-quality chromosome-scale genome. However, this assembly still contained 18 gaps (represented by 'N's) within the sequences. To address this, we performed a gap-closing step using TGS-GapCloser v1.2.1[22], leveraging both HiFi reads and ONT reads. This process systematically filled the sequence gaps, effectively bridging intervals caused by complex repeats or regions of low coverage, and ultimately yielded a continuous, gap-free durian genome assembly.
A comprehensive quality assessment of the final genome assembly was conducted from multiple perspectives: Mapping rates of both ONT and HiFi reads to the final assembly were calculated using Minimap2 v2.1[23] to evaluate data utilization and assembly inclusiveness. The completeness of the genome assembly was assessed with BUSCO v5.7.1[24] (Benchmarking Universal Single-Copy Orthologs). The consensus quality value (QV) was evaluated using Merqury v1.3[25] to estimate sequence accuracy. Annotation quality was validated using OMArk[26], which assesses proteome completeness, consistency, and contamination relative to conserved gene families.
Telomere and centromere detection
-
For structural annotation of the genome, the QuarTeT[27] tool was employed to scan chromosomal termini, successfully identifying the canonical telomeric repeat pattern (CCCTAAA). The same tool was used to search for potential centromeric repeat sequences across the genome. Subsequently, the distribution of these candidate sequences was visualized with CentriVision v1.0.1 (minlength = 10, windows = 4,000)[28]. By analyzing the frequency distribution of these candidate repeat sequences along each chromosome, the approximate boundaries of the centromeric regions were inferred, providing crucial clues for subsequent studies.
Gene prediction and annotation
-
For repetitive sequence analysis, RepeatModeler v2.0.3[29] was utilized to cluster repeats through the construction of a de novo repeat library. Subsequently, RepeatMasker v4.1.2[30] was employed to identify repetitive sequences. For coding gene prediction, HISAT2 v2.1.0[31] was then used to align all transcriptome data to the genome. The resulting SAM files were converted to BAM format using SAMtools v1.22[32]. Subsequently, BRAKER3 v3.0.3[33] was used for de novo gene prediction, which automatically trains species-specific parameters and annotates gene structures by integrating transcriptomic alignments (from root, stem, leaf, flower, and fruit tissues), and protein homology evidence from five closely related Malvaceae species: Theobroma cacao (GCF_000208745.1), Hibiscus cannabinus (GCA_047302245.1), Gossypium arboreum (GCF_025698485.1), Corchorus olitorius (GCA_001974825.2), and Bombax ceiba (
https://figshare.com/articles/dataset/Genome_of_B_ceiba_and_C_pentandra/21708509 ). For functional annotation, the predicted protein-coding genes were queried against the eggNOG[34], InterPro[35], NR[36], Swiss-Prot[37], and Pfam[38] databases. The program cmscan in Infernal[39] was used to identify ribosomal RNA (rRNA), small nuclear RNA (snRNA), and microRNA (miRNA) sequences using the Rfam database[40]. tRNAscan-SE[41] was used to predict transfer RNA (tRNA) sequences.Genome evolution analysis
-
The protein sequences of 12 other species were extracted from public databases. The sequences for the following nine species were obtained from the NCBI databases under the provided accession numbers: Oryza sativa (GCF_001433935.1), Arabidopsis thaliana (GCF_000001735.4), Vitis vinifera (GCF_000003745.3), Corchorus olitorius (GCA_001974825.2), Corchorus capsularis (GCA_001974805.1), Theobroma cacao (GCF_000208745.1), Herrania umbratica (GCF_002168275.1), Gossypium barbadense (GCA_008761655.1), and Gossypium raimondii (GCA_000327365.1). Additionally, the protein sequences for Bombax ceiba and Ceiba pentandra were sourced from the figshare repository (
https://figshare.com/articles/dataset/Genome_of_B_ceiba_and_C_pentandra/21708509 ), while those for Durio zibethinus cv. KanYao were obtained from another figshare dataset (https://figshare.com/articles/dataset/Durian_genome_annotation/25237591 ). Orthologous gene families were clustered using OrthoFinder v2.5.5[42] under default parameters. A maximum-likelihood phylogenetic tree was then constructed from the aligned single-copy genes with IQ-TREE v2.2.3[43], employing 1,000 ultrafast bootstrap replicates. Divergence times were estimated using R8s v1.81[44] in conjunction with calibration points obtained from the TimeTree[45] website (www.timetree.org ). The fossil calibration points used were O. sativa vs A. thaliana at 142.1−163.5 million years ago (MYA), A. thaliana vs V. vinifera at 109.8−124.4 MYA, C. olitorius vs. T. cacao at 19.1−59.4 MYA, and B. ceiba vs H. umbratica at 30.3−42.0 MYA. Gene family expansion and contraction analyses were performed with CAFE5[46]. The gene families of T. cacao, H. umbratica, B. ceiba, C. pentandra, and D. zibethinus cv. Chuongbo were clustered using jvenn (http://jvenn.toulouse.inra.fr/app/example.html )[47]. The protein sequences of durian-specific gene families were screened for GO and KEGG enrichment analyses.TERT gene family analysis and comparison
-
The characteristic protein domain of the TERT family (PF12009) was downloaded from the Pfam database (
http://pfam.xfam.org ). An initial hidden Markov model (HMM) profile was built using the retrieved domain sequence. Potential TERT homologs were searched against the genome assemblies and annotated proteomes of the 27 Malvaceae species using hmmer v3.3.2[48]. The screened sequences were compared with ClustalW, and the hidden Markov model of these verified sequences was constructed by hmmbuild. Finally, 38 TERT family members were screened. All TERT protein sequences were aligned with MUSCLE v5.3[49], and the comparison results were uploaded to MEGA v12.0.13[50]. The Neighbor-Joining (NJ) phylogenetic tree was constructed with 1,000 bootstrap replicates and the Maximum Composite Likelihood model. The TERT family protein motifs were analyzed using MEME v5.5.7[51]. -
We selected D. zibethinus cv. Chuongbo for genome assembly. Flow cytometry of fresh young leaf tissues estimated its genome size to be approximately 790 Mb (Supplementary Table S1), while k-mer analysis (k = 21) from genome survey sequencing predicted a genome size of 706.79 Mb and a genome heterozygosity of 1.08% (Fig. 1b). These results provided preliminary insights into the genomic characteristics of D. zibethinus cv. Chuongbo. A high-quality genome of D. zibethinus cv. Chuongbo was assembled using a hybrid long-read sequencing strategy combining PacBio HiFi and ONT Ultra-long data. The PacBio HiFi sequencing yielded 97.18 Gb of data (118× coverage) with a read N50 of 16.80 kb, while the ONT Ultra-long sequencing provided 31 Gb (37× coverage) with a read N50 of 150 kb (Supplementary Table S2). These data were jointly assembled using Hifiasm. The resulting assembly comprised 1,190 contigs, with a contig N50 of 21.64 Mb. Hi-C sequencing clean data were used to anchor and order the contigs onto 28 pseudochromosomes, representing the chromosome-scale genome assembly of D. zibethinus cv. Chuongbo (Supplementary Tables S3 and S4).
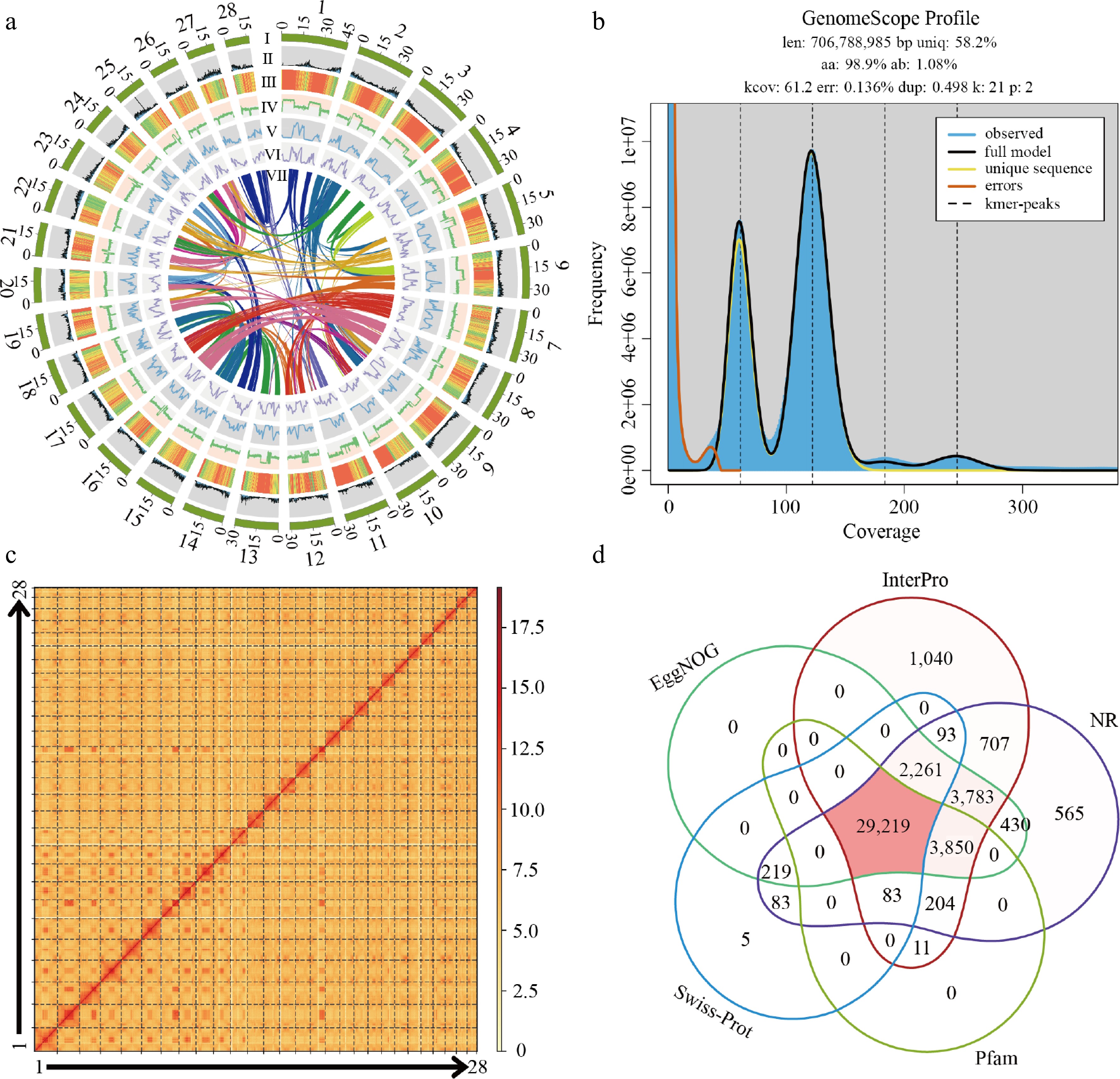
Figure 1.
The genome of D. zibethinus cv. Chuongbo. (a) Circos plot showing the genome details. Labels I−VII indicate: (I) 28 chromosomes of D. zibethinus; (II) gene density; (III) repeat sequence content; (IV) GC content density; (V) density of Copia LTR-RTs; (VI) density of Gypsy LTR-RTs; (VII) syntenic blocks (all window sizes = 50 kb). (b) Distribution profiles of 21-mer analysis of short reads. (c) Hi-C heatmap of D. zibethinus cv. Chuongbo. (d) Venn diagram of function annotations from various databases.
Following gap closure and corrections via the hybrid assembly approach, a chromosome-level genome of D. zibethinus cv. Chuongbo was obtained, consisting of 28 pseudomolecules with only 18 gaps remaining. These residual gaps were manually resolved using extended HiFi and ONT reads, resulting in a gap-free D. zibethinus cv. Chuongbo genome assembly. The final assembly has a total length of 824.78 Mb and scaffold N50 of 30.88 Mb (Supplementary Table S1). The assembled genome size is slightly larger than the earlier flow cytometry estimate, which may reflect systematic bias in cytometric genome size prediction. Distributions of gene density, repeat content, and GC content across the genome are presented in Fig. 1a. The accuracy, completeness, and continuity of the D. zibethinus cv. Chuongbo genome was evaluated using multiple validation approaches. Hi-C interaction maps displayed strong diagonal patterns, confirming proper intra-chromosomal spatial organization (Fig. 1c). To comprehensively evaluate the quality of the genome assembly, we assessed both its accuracy and completeness. HiFi and ONT reads were mapped back to the assembled genome using Minimap2, yielding high mapping rates of 99.16% and 99.88%, confirming strong consistency between the assembly and the original sequencing data (Supplementary Table S5). Additionally, BUSCO analysis against the embryophyta_odb10 dataset revealed that 99.0% [C: 99.0% (S: 71.0%, D: 28.0%), F: 0.8%, M: 0.2%, n: 1614] of conserved plant single-copy orthologs were complete, indicating high gene-space completeness. The quality value scores calculated using Merqury indicated an error rate of 0.0001 for the genome of D. zibethinus cv. Chuongbo with a QV score of 40 (Table 1). Furthermore, OMArk assessment of the annotated proteome demonstrated robust quality, with 97.1% completeness, 91.0% lineage consistency, and a total absence of detectable contamination. To validate the structural integrity of the telomere-to-telomere (T2T) genome assembly, we scanned the assembled sequence for telomeric and centromeric regions. Using the canonical conserved plant telomere repeat motif (CCCTAAA) as a query, we identified a total of 53 telomeres across the 28 pseudochromosomes. Telomeres were detected at both ends in most chromosomes; however, only one telomere was identified on chr1, chr4, and chr23 (Supplementary Table S6). With Quartet software, we detected 28 centromeres of 28 chromosomes, with sizes ranging from 0.22 to 16.4 Mb of the gap-free genome (Supplementary Table S7). Visualization of these regions with CentriVision v1.0.1 revealed characteristic parallel diagonal patterns in self-alignment dot plots, confirming the presence of long-range tandem repeat arrays typical of functional centromeres (Supplementary Fig. S1). These results collectively validate the structural accuracy, sequence continuity, and overall completeness of our gap-free genome.
Table 1. Genomic statistics of Durio zibethinus.
Genome Durio zibethinus Chuongbo KanYao[5] Ploidy 2n = 56 2n = 56 Estimated genome size (Mb) 790 808.9 Assembled genome size (Mb) 824.78 777.8 Genomic heterozygosity (%) 1.08 1.4 Largest contig (Mb) 40.32 35.2 Contig N50 (Mb) 21.64 14.23 Number of scaffold 28 111 Largest scaffold (Mb) 46.4 36.3 Scaffold N50 (Mb) 30.88 22.7 Repeat sequence content (%) 62.18 60.85 GC content (%) 33.3 32.69 Number of genes 44,024 38,728 Gaps 0 83 QV 40.0 37.5 Genome BUSCOs (%) 99.0 99.06 Completeness OMArk (%) 97.1 94.45 A total of 512,871,105 bp repeat elements were identified, which accounted for 62.18% of the genome of D. zibethinus cv. Chuongbo. We identified interspersed repeats accounting for 59.29% of the genome of D. zibethinus cv. Chuongbo, with a total length of 488,980,373 bp. Notably, the unclassified fraction dominated at 40.84%, indicating the genome has accumulated a large number of ancient, divergent, and fragmented transposable element (TE) sequences. Among the classified TEs, long terminal repeat (LTR) retrotransposons were the predominant component at 15.46%, with the Gypsy family being the most abundant at 12.21% and the Copia family at 2.92% (Supplementary Table S8). This finding suggests that D. zibethinus cv. Chuongbo experienced large-scale LTR retrotransposon bursts during its evolutionary history.
We annotated the genome of D. zibethinus cv. Chuongbo using BRAKER3, with key inputs to ensure annotation accuracy: protein sequences from reference species, including Theobroma cacao, Hibiscus cannabinus, Gossypium arboreum, Corchorus olitorius, and Bombax ceiba for homology reference, transcriptome sequencing data for transcriptome-guidance, and de novo gene structure prediction enabled by the tool. A total of 44,024 protein-coding gene structures were successfully identified through this annotation process. Functional annotation results showed that 39,762, 41,251, 41,497, 31,963, and 33,367 genes were annotated in EggNOG, InterPro, NR, Swiss-Prot, and Pfam, respectively, with 29,219 genes receiving annotations from all five databases (Fig. 1d, Supplementary Table S9). BUSCO assessment of gene annotation was 99.0%. Additionally, tRNAscan-SE was used for de novo prediction of tRNAs, while other types of non-coding RNAs (ncRNAs) were identified using the Rfam database. In total, we detected 1,029 tRNAs, 2,979 rRNAs, 188 miRNAs, and 698 snRNAs, with rRNAs being the most abundant ncRNA class.
Orthologue and phylogenetic analyses
-
We analyzed the expansion and contraction of homologous gene families across 13 accessions, including Oryza sativa, Arabidopsis thaliana, Vitis vinifera, Corchorus olitorius, Corchorus capsularis, Theobroma cacao, Herrania umbratica, Bombax ceiba, Ceiba pentandra, Gossypium barbadense, Gossypium raimondii, and two cultivars of Durio zibethinus (cv. KanYao and cv. Chuongbo). To specifically elucidate the evolutionary relationships within Malvaceae, a phylogenetic tree was subsequently constructed using protein sequences of single-copy genes, with O. sativa designated as the outgroup. Phylogenetic analysis revealed that D. zibethinus cv. Chuongbo diverged from H. umbratica approximately 35 MYA, and from its conspecific cultivar D. zibethinus cv. KanYao around 2 MYA (Fig. 2a). Gene family expansion and contraction are key hallmarks of adaptive evolution. In D. zibethinus cv. Chuongbo, 2,252 gene families underwent expansion, while 265 exhibited contraction. Across five representative Malvaceae species (T. cacao, H. umbratica, B. ceiba, C. pentandra, and D. zibethinus cv. Chuongbo), a total of 15,104 gene families were identified. Notably, the number of species-specific gene families varied among these taxa: 28 in T. cacao, 13 in H. umbratica, 505 in B. ceiba, 573 in C. pentandra, and 761 in D. zibethinus cv. Chuongbo (Fig. 2b).
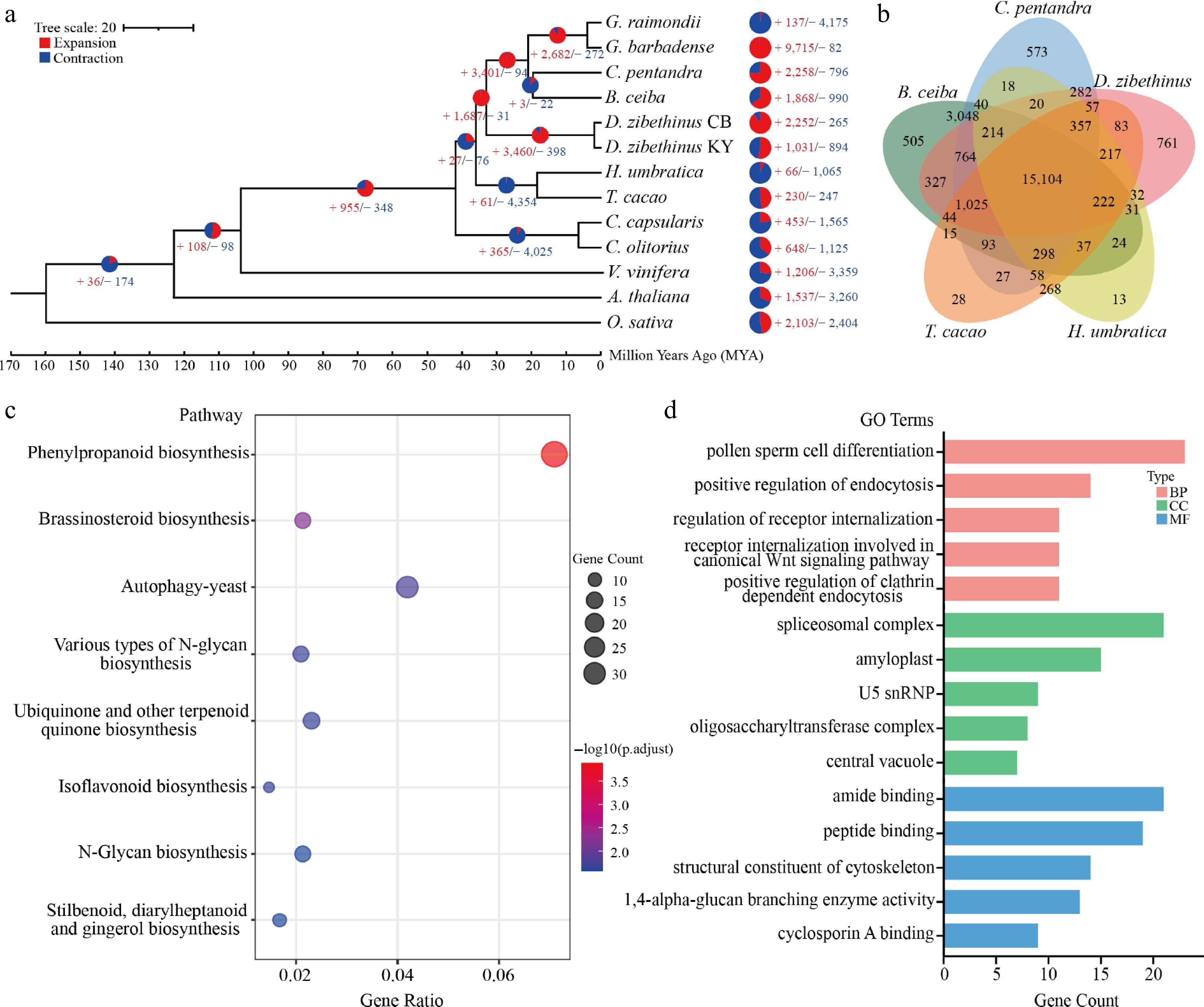
Figure 2.
Comparative genomic analysis of D. zibethinus cv. Chuongbo. (a) Expansion and loss of gene orthogroups leading to D. zibethinus cv. Chuongbo (CB). (b) Venn diagram showing orthogroups shared by D. zibethinus cv. Chuongbo and related genomes. (c) KEGG analysis of unigenes in D. zibethinus cv. Chuongbo. (d) GO analysis of unigenes in D. zibethinus cv. Chuongbo.
KEGG enrichment analysis of the species-specific gene families in D. zibethinus cv. Chuongbo showed that these families were predominantly enriched in metabolism-related biological pathways, such as Phenylpropanoid biosynthesis, Brassinosteroid biosynthesis, and Autophagy-yeast (Fig. 2c). Furthermore, GO enrichment analysis revealed that the core functions of these species-specific gene families were primarily involved in signal transduction regulation, stress responses, specialized metabolism, and fundamental cellular processes (Fig. 2d).
Genome-wide identification of TERT genes
-
In the field of plant biology, the TERT gene has been a focus of extensive research due to its crucial roles in cellular lifespan and related physiological processes. However, the evolutionary history and characteristics of TERT genes within the Malvaceae family remain largely unexplored. Here, we identified 38 TERT family members from 27 Malvaceae species, including two durian cultivars that provide insights into intra-specific diversity (Supplementary Table S10). Notably, TERT is generally maintained as a single-copy gene in most of the examined genomes. Two copies were detected in G. barbadense, G. darwinii, G. hirsutum, G. mustelinum, G. tomentosum, H. umbratica, and H. sabdariffa. TERT copy number varies between durian cultivars. D. zibethinus cv. Chuongbo harbors a single copy, whereas D. zibethinus cv. KanYao possesses four copies distributed one on hap1, and three on hap2, the highest among all examined accessions (Fig. 3). Motif conservation analysis revealed that all TERT family members share highly conserved motifs, with motif3 and motif10 present in every gene, suggesting that these two motifs may play functionally important roles in the TERT family (Supplementary Table S11).

Figure 3.
Phylogeny and conserved motifs of the TERT gene family in Malvaceae.
-
The field of de novo genome assembly has recently undergone a paradigm shift, transitioning from fragment-based drafts to telomere-to-telomere completeness[52]. This shift was catalyzed by the maturation around 2019 of high-fidelity long-read sequencing and ultra-long read technologies[53]. These advancements, when combined with Hi-C data and advanced assemblers like Hifiasm, Verkko, and Canu, have enabled the assembly of near-complete genomes for many complex eukaryotic species. This evolution marks the official entry of genome assembly into the T2T era, addressing long-standing challenges in resolving repetitive regions and structural complexities[54].
Durian has long presented genome assembly challenges due to persistent gaps, particularly in repetitive regions such as telomeres and centromeres. These limitations have significantly hindered advancements in durian genomics and molecular breeding[55]. This study reported a gap-free genome of D. zibethinus cv. Chuongbo, covering all 28 chromosomes with 53 telomeres and 28 centromeres. The genome assembly of D. zibethinus cv. Chuongbo has a total length of 824.78 Mb, a scaffold N50 of 30.88 Mb, and a GC content of 33.30%, reflecting high contiguity and structural representativeness. The high BUSCO completeness (99.0%) and QV score of 40 indicate that the assembly is complete and suitable for comparative genomics and gene function analysis[56]. The identification of 512.87 Mb of repeat elements, accounting for 62.18% of the genome, aligns with similar genome analyses in other durian cultivars, where repetitive elements contribute significantly to genome size and structural complexity. Through genome annotation, we predicted 44,024 protein-coding genes. The quality of the annotated proteome was rigorously validated using OMArk, which revealed a high completeness of 97.1% and a lineage consistency of 91.0%, with no detectable contamination. Furthermore, over 94% of these genes received functional assignments across multiple public databases, underscoring the reliability of D. zibethinus cv. Chuongbo gene models for downstream functional genomics. The completion of a gap-free D. zibethinus cv. Chuongbo genome provides a critical resource for elucidating genomic structure and gene function across the Malvaceae family[57].
Comparative genomics analysis provides important insights into the evolutionary history of D. zibethinus cv. Chuongbo. Our divergence time estimation indicates that D. zibethinus cv. Chuongbo and H. umbratica diverged approximately 35 MYA. Meanwhile, the split between D. zibethinus cv. Chuongbo and its conspecific cultivar, D. zibethinus cv. KanYao, was estimated at ~2 MYA. The antiquity of this divergence suggests that the separation between these two cultivars likely reflects a pre-domestication split of their ancestral wild lineages, rather than variation generated during or after human cultivation. This pattern, in which deep phylogenetic splits between intraspecific lineages predate domestication, is not unique to durian and has also been documented in other domesticated species such as apple (Malus domestica)[58] and lettuce (Lactuca sativa)[59], where modern cultivars retain genomic signatures of ancestral divergence among multiple wild progenitors that diverged long before domestication. Collectively, these estimated timeframes offer a temporal perspective on key speciation and diversification events within the lineage. Durian exhibits a strong bias toward gene family expansion, with 2,252 expanded vs only 265 contracted families in D. zibethinus cv. Chuongbo, highlighting genomic innovation as a key evolutionary driver. Notably, this cultivar harbors 761 species-specific gene families, a number that far exceeds those of its close Malvaceae relatives. Functionally, these lineage-specific genes show enrichment in signal transduction and stress responses, with notable enrichment in specific metabolism-related pathways like phenylpropanoid and brassinosteroid biosynthesis.
A total of 38 TERT genes were identified across 27 Malvaceae species. To ensure the reliability of the evolutionary analysis, only intact sequences harboring both the Telomerase_RBD (PF12009) and RT domains were retained, and no pseudogenes were detected in the final dataset. The predominant single-copy status of the TERT gene in most species underscores strong evolutionary constraint, likely due to its indispensable role in maintaining telomere integrity[60]. Notably, TERT copy number differs dramatically between durian cultivars. Durio zibethinus cv. KanYao harbors four copies distributed asymmetrically across haplotypes (one on hap1 and three on hap2), while D. zibethinus cv. Chuongbo contains only one. This copy number variation may represent the genetic basis for adaptive divergence between cultivars. Additionally, two TERT copies were found in several polyploid Gossypium species and a few others, suggesting that lineage-specific whole-genome duplications may have facilitated rare retention of paralogs. Conserved motif analysis further highlights functional importance, with motif3 and motif10 present in all identified TERT protein sequences. These motifs likely represent core functional domains. Together, these findings establish an evolutionary framework for studying how TERT gene dosage and sequence conservation relate to telomere dynamics and genome stability across Malvaceae.
Overall, this study presents the first gap-free reference genome of durian, which demonstrates high contiguity, completeness, and accuracy. It provides a key genomic resource for in-depth investigation of durian genome architecture, functional gene evolution in Malvaceae plants, and the molecular basis of unique traits.
This work was supported by the Project of National Key Laboratory for Tropical Crop Breeding (NO. NKLTCB202337), Hainan Province Science and Technology Special Fund (ZDYF2023XDNY050), Hainan Provincial Natural Science Foundation of China (325QN234).
-
The authors confirm their contributions to the paper as follows: designed this research: Wang W, Chen F; collected the plant samples: Wang S, Zhang J, Guo G; conducted experiments, and performed analyses: Wang S, Li Z; wrote the manuscript: Wang S. All authors reviewed the results and approved the final version of the manuscript.
-
Raw data from PacBio HiFi, Oxford Nanopore, Hi-C, and transcriptome sequencing are available online at the National Genomics Data Center (https://ngdc.cncb.ac.cn) with the project ID PRJCA053033. The genome, coding sequences, proteins, and gff3 files could be found at FigShare (https://doi.org/10.6084/m9.figshare.30814499).
-
The authors declare that they have no conflict of interest.
-
accompanies this paper online at: https://doi.org/10.48130/tp-0026-0003.
-
Received 8 December 2025; Accepted 12 February 2026; Published online 13 March 2026
- Supplementary Fig. S1 Self-alignment dot plots of the Chromosome 1 centromere. Plots shown are a systematic sample(windows 100-104, 500-504, 1000-1004, 1500-1504, 2000-2004) uniformly show the long parallel diagonals diagnostic of centromeric tandem repeats.
- Supplementary Table S1 Flow Cytometric Analysis.
- Supplementary Table S2 Summary of HiFi, ONT, and Hi-C reads results.
- Supplementary Tables S3 Length of each pseudo-chromosome by Hi-C.
- Supplementary Tables S4 Statistics of updated assembly by Hi-C sequencing in scaffold level.
- Supplementary Table S5 Statistics of HiFi Reads and ONT Reads Alignment Results.
- Supplementary Table S6 Telomere motif counts.
- Supplementary Table S7 Centromeres motif counts.
- Supplementary Table S8 Repeat sequence statistics.
- Supplementary Table S9 Gene models and Gene function annotation.
- Supplementary Table S10 The TERT gene family in Malvaceae.
- Supplementary Table S11 Conserved motif sequences of the TERT gene family in Malvaceae.
- Copyright: © 2026 by the author(s). Published by Maximum Academic Press on behalf of Hainan University. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Wang S, Zhang J, Guo G, Li Z, Chen F, et al. 2026. Gap-free genome of Durio zibethinus cv. Chuongbo. Tropical Plants 5: e004 doi: 10.48130/tp-0026-0003 

Gap-free genome of Durio zibethinus cv. Chuongbo
- Received: 08 December 2025
- Revised: 23 January 2026
- Accepted: 12 February 2026
- Published online: 13 March 2026
Abstract: Durian (Durio zibethinus L.) is a tropical fruit of substantial nutritional and economic value from the family Malvaceae. Several genome assemblies for durian have been reported previously, but these assemblies contain gaps that have restricted their completeness and hindered their practical utility for downstream research. Here, we present the first gap-free genome assembly of Durio zibethinus cv. Chuongbo by integrating PacBio HiFi, Oxford Nanopore, and Hi-C sequencing data. The assembled genome is 824.78 Mb across 28 chromosomes, with a scaffold N50 of 30.88 Mb and 44,024 protein-coding genes. Comparative genomic analyses dated the divergence between D. zibethinus cv. Chuongbo and Herrania umbratica to 35 million years ago, and between two durian cultivars to 2 million years ago. D. zibethinus cv. Chuongbo exhibits substantial gene family expansion and a high abundance of species-specific genes, reflecting key genomic innovations underlying its unique biological traits. Additionally, comparative analysis of the TERT gene family across 27 Malvaceae species uncovered strong evolutionary constraints that maintain a predominant single-copy configuration, with two copies identified in several Gossypium taxa. This high-quality, gap-free genome provides a foundational resource for elucidating genome architecture, gene evolution, and the molecular basis of unique traits in durian and related Malvaceae species.
-
Key words:
- Durio zibethinus cv. Chuongbo /
- Gap-free genome /
- Evolution /
- TERT /
- Malvaceae