









-
Artocarpus heterophyllus Lam. (jackfruit), a tropical fruit crop of the genus Artocarpus in the Moraceae family, is hailed as the 'Queen of Tropical Fruits'. It boasts significant industrial value and occupies a strategic position in the domains of tropical horticulture and economic development[1]. Notably, red-fleshed jackfruit has attracted increasing attention because of its superior nutritional properties and favorable commercial traits. The academic community generally accepts that A. heterophyllus originated from the Western Ghats in Southern India. Through subsequent artificial selection, introduction, and domestication, its cultivation range has gradually expanded to tropical and subtropical regions of SouthEast Asia, Pacific Island nations, and Central and South America, forming an intercontinental cultivation distribution pattern[2].
In terms of jackfruit genome assembly, the Sahu team first constructed a draft genome of A. heterophyllus using Illumina short-read sequencing technology in 2020. This draft contained 162,440 scaffolds with a total length of 982 Mb, and 35,858 genes were annotated[3], laying a preliminary data foundation for subsequent molecular research. In 2022, Lin et al. utilized PacBio long-read sequencing data combined with Illumina short-reads for sequence correction, and supplemented with a RAD-Seq to construct a high-density genetic linkage map of the F1 generation. Eventually, a chromosome-level genome of A. heterophyllus with a size of 985.63 Mb was obtained, comprising 482 scaffolds with a GC content of 34.86% and repetitive sequences accounting for 54.02%. A total of 41,997 protein-coding genes were annotated in this A. heterophyllus-S10 genome, and comparative genomic analysis has further revealed that the genus Artocarpus underwent a recent whole-genome duplication event[4].
However, the frequent occurrence of extreme climate events such as high temperatures and humidity in tropical regions has caused substantial economic losses to the jackfruit industry. Additionally, anthracnose, a major fungal disease of jackfruit caused by Colletotrichum siamense severely compromises fruit quality and limits its industrial processing potential[5]. Therefore, identifying genes related to stress responses in jackfruit is of great significance for the sustainable development of the jackfruit industry. Plant transcription factors are key proteins that regulate various biological processes in plants. The NAC proteins represent a major plant-exclusive transcription factor family, with essential roles in modulating plant growth, developmental processes, and stress adaptation mechanisms[6].
In previous studies, the NAC gene family has been extensively characterized in various tropical plant species. For example, 79 MaNAC genes were identified in Morus atropurpurea, which belongs to the same family as jackfruit[7]. These MaNAC genes exhibited varying degrees of response to drought stress and sclerotinia disease, among which MaNAC12, MaNAC32, MaNAC44, and MaNAC67 were identified as the most strongly responsive candidate genes. In addition, 66 CpNAC genes were identified in the genome of Carica papaya, of which 25 showed differential expression under drought stress[8]. In the tropical fruit tree Litchi chinensis, 112 LcNAC genes were identified, and promoter analysis revealed that these LcNAC genes contain abundant cis-acting elements associated with phytohormone responses, light responses, stress responses, and plant growth and development, with some members involved in the regulation of pericarp maturation and anthocyanin biosynthesis[9]. A total of 142 AmNAC genes were identified in the Avicennia marina genome, among which AmNAC010/040 were suggested to be subject to relatively relaxed selective constraints during neofunctionalization. Transcriptomic analyses further indicated that AmNAC transcription factors participate in the development of pneumatophores and leaf salt glands, and exhibit responsive expression patterns under salinity, flood, and cadmium stresses[10].
Collectively, these studies demonstrate from multiple perspectives including evolutionary features, cis-regulatory element composition, and stress-responsive expression patterns, that the NAC gene family plays a conserved and critical role in the regulation of plant growth and development, as well as in stress adaptation in tropical plant species.
The functional diversity and evolutionary conservation of the NAC gene family at the molecular level is closely associated with the origin of the NAC domain and its core regulatory functions. The NAC domain was initially defined based on the conserved sequence characteristics of NAM (NO APICAL MERISTEM) from Petunia hybrida and ATAF1, ATAF2, and CUC2 from Arabidopsis thaliana. Mutational analysis of P. hybrida has revealed that disruption of the NAM gene abolishes shoot apical meristem development, suggesting a crucial role of NAM in regulating the positioning of shoot apical meristems and primordia[11]. Correspondingly, alterations in the cup-shaped cotyledon genes (i.e., CUC1 and CUC2) induce defects in the detachment of cotyledons, sepals, and stamens, as well as compromised formation of shoot apical meristems. These findings demonstrate that CUC2 regulates plant embryonic development and flower development[12,13], highlighting the irreplaceable role of the NAC transcription factor family in plant survival and adaptive evolution. Collectively, these findings underscore the conserved and critical roles of the NAC family in development and stress adaptation. However, such systematic characterization of the NAC family is currently lacking in jackfruit, leaving their regulatory potential unexplored.
Despite the availability of chromosome-level reference genomes for jackfruit, existing genomic resources are predominantly derived from yellow-fleshed cultivated types, and systematic studies on red-fleshed jackfruit and its stress response mechanisms remain limited. In particular, a chromosome-level reference genome based on red-fleshed phenotypic material with high sequence contiguity and consensus quality is still not well established.
To address these gaps, we employed third-generation HiFi sequencing technology integrated with transcriptome data to construct a high-quality, chromosome-level reference genome for red-fleshed jackfruit (MDM2). The genome has a total length of 1.03 Gb with a scaffold N50 length of 39.36 Mb. BUSCO assessment indicated a genome completeness of 93.8%, while the assembly achieved a long terminal repeat assembly index (LAI) of 26.88 and a consensus quality value (QV) of 76.11. In total, 45,366 protein-coding genes were predicted. Compared with previous studies, the assembled genome shows significant improvements in contiguity and completeness, demonstrating the high quality and reliability of the MDM2 genome. Furthermore, through enrichment analysis of expanded gene families, we focused on systematic research of the NAC transcription factor family in jackfruit, aiming to identify candidate AhNAC genes for improving stress responses in this species. In summary, this achievement provides high-quality genomic data for gene function and evolutionary studies of jackfruit and strongly facilitates its molecular breeding process.
-
The plant material used in this study was A. heterophyllus (Fig. 1a), cultivar Mengdemi No. 2 (MDM2), bred by Hainan Hengmi Fruit Industry Development Co., Ltd. The fruit are nearly round with relatively smooth, yellowish-green peel, and the average single fruit weight is approximately 10 kg. The flesh is orange-red, with a sweet and fresh flavor.
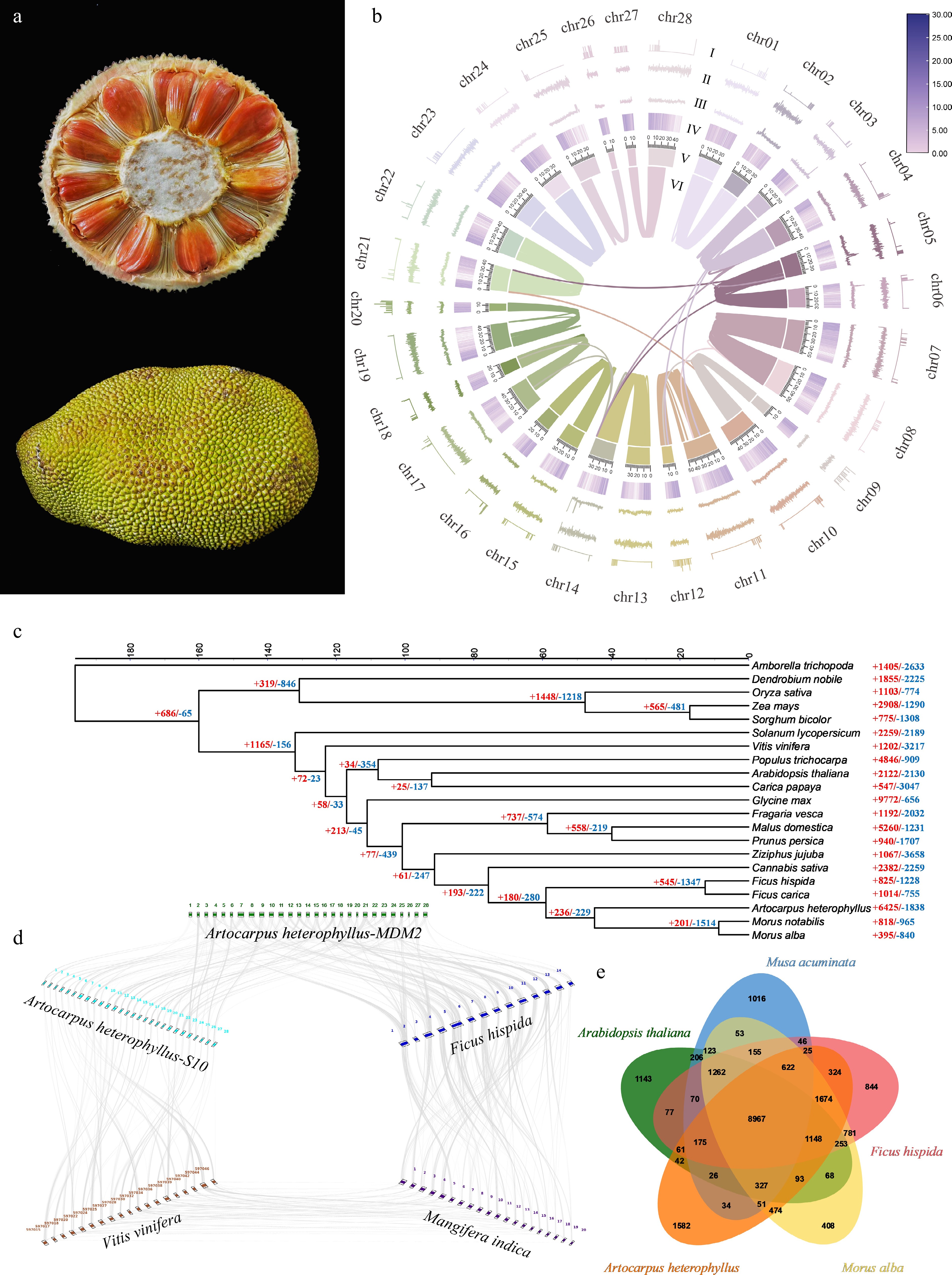
Figure 1.
(a) Morphological features of A. heterophyllus-MDM2. (b) Circos diagram of the A. heterophyllus-MDM2 genome features. (I: N [unknown bases]. II: GC skew. III: GC content. IV: gene density distribution. V: chromosome length. VI: intra-genomic chromosomal synteny among chromosomes of A. heterophyllus-MDM2 genome). (c) Phylogenetic tree and divergence time estimation of A. heterophyllus and its related species. (d) Genome collinearity analysis among A. heterophyllus-MDM2, A. heterophyllus-S10, M. alba, F. hispida, and M. indica; syntenic blocks are highlighted with grey lines connecting different chromosomes, and numbers around the rectangles indicate the chromosome IDs of each genome. (e) Venn diagram showing the shared and unique gene sets among the studied genomes.
Experimental plants were cultivated in the experimental orchard located in Dongcheng Town, Danzhou City, Hainan Province, China (19°42'30" N, 109°26'43" E). Vigorous and disease-free A. heterophyllus individuals were selected for sample collection. Genomic DNA was extracted from young, fully expanded leaves, while total RNA was isolated from six distinct tissues: young leaves, young stems, mature stems, flowers, early-stage infructescences, and fully mature infructescences. The entire sampling procedure was completed within 30 min to minimize RNA degradation. All harvested samples were immediately snap-frozen in liquid nitrogen to preserve nucleic acid integrity and stored at –80 °C until subsequent molecular experiments.
High-quality genomic DNA was isolated via liquid nitrogen homogenization coupled with a CTAB lysis protocol[14], ensuring the recovery of intact high-molecular-weight nucleic acids suitable for long-read sequencing. Sequencing was carried out on the PacBio Revio platform, generating 1,985,271 HiFi reads for the MDM2 with a total data output of 44.30 Gb (43 ×). This dataset afforded robust long-read depth to underpin subsequent high-quality de novo genome assembly.
For transcriptomic profiling, total RNA was extracted from six distinct jackfruit tissues using the Dynabeads mRNA Purification Kit to enrich poly (A)-tailed mRNA and minimize rRNA contamination. Short-read sequencing was performed on the MGISEQ-2000 sequencer, yielding 61.19 Gb of clean sub-read bases. This comprehensive transcriptomic dataset captures tissue-specific gene expression dynamics, providing a solid foundation for downstream gene annotation, functional analysis, and validation of genome assembly quality.
Genome assembly and annotation
-
To assemble the MDM2 reference genome, we first processed 44.30 Gb of high-quality HiFi reads with Fastp v0.23.4[15] for quality control, removing low-quality bases and adapter sequences. The filtered reads were then de novo assembled into a 1.82 Gb primary assembly using Hifiasm v0.25.0-r726[16] with default parameters, yielding 998 contigs with a contig N50 of 16.76 Mb. The initial assembly represented a diploid-aware output produced under default settings, which retained substantial redundant sequences arising from haplotype separation and repeat expansion. Therefore, redundant sequences were removed from the primary assembly using Purge_dups v1.2.5[17], based on combined evidence of sequencing coverage depth and sequence similarity. Coverage thresholds (3, 8, 13, 18, 26, and 40) were determined using the calcuts module, together with whole-genome self-alignment, enabling accurate identification and removal of redundant fragments. This procedure resulted in a non-redundant haploid contig assembly of approximately 1.03 Gb. For chromosome-level scaffolding, we anchored and oriented approximately 96% of the non-redundant contigs onto 28 pseudochromosomes using Ragtag v2.1.0[18] with the A. heterophyllus-S10 genome as a reference.
Synteny analysis was performed using NGenomeSyn v1.43[19], and the chromosomes of the MDM2 assembly and the reference genome showed good homologous correspondence at the large scale, verifying the accuracy of the chromosome-level scaffolding (Supplementary Fig. S1). A multi-dimensional quality assessment was performed to ensure the high confidence of the genome. QUAST v5.3.0[20] was employed to evaluate contiguity, while BUSCO v5.6.0[21] was utilized to determine the representation of conserved orthologous genes. The structural integrity of the genome was further quantified using the LAI, which was derived from LTR retrotransposons predicted by both ltrharvest v1.6.5[22] and LTR_FINDER_parallel v1.3[23]. Finally, Merqury v1.3[24] validated the consensus quality and overall completeness of the assembly.
Repetitive sequences in the MDM2 genome were identified using an integrated approach combining homology-based searching and de novo prediction. Homologous sequences were masked by RepeatMasker v4.1.7-p1[25] against the Dfam 3.3 and RepBase-20181026 databases. To capture lineage-specific repeats, we constructed a custom repeat library with EDTA v2.2.2[26]. Furthermore, we enhanced the classification accuracy of transposable elements (TEs) using DeepTE[27], which enabled refined categorization of TE superfamilies. Regarding non-coding RNA (ncRNA) annotation, tRNA genes were predicted using tRNAscan-SE v2.0.12[28]. Other ncRNA species, including rRNA, miRNA, and snRNA, were identified by searching the genome against the Rfam database using INFERNAL v1.1.5[29]. This multifaceted pipeline ensured a high-confidence landscape of the repetitive and non-coding components of the genome.
Gene structures were predicted through an integrated approach, combining homology-based prediction, RNA-Seq-assisted prediction, and ab initio prediction. For homology-based prediction, protein sequences from Cannabis sativa, Ficus carica, Morus alba, Morus notabilis, Ziziphus jujuba, Broussonetia papyrifera, and Durio zibethinus were selected as reference homologs. To obtain comprehensive transcript sequences, RNA-Seq data were subjected to both de novo assembly and genome-guided assembly using Trinity v2.15.2[30]. The two sets of assembled transcripts were merged, and contaminants were removed by aligning against the UniVec database (
ftp://ftp.ncbi.nlm.nih.gov/pub/UniVec/ ). The decontaminated sequences were then mapped to the genome using PASA v2.5.3[31] to generate integrated transcript sequences. Open Reading Frames (ORFs) were extracted from the PASA v2.5.3 results for gene prediction, and complete gene models combined with homologous protein sequences were used to train Hidden Markov Models (HMMs) for Augustus v3.3.3[32] and SNAP v2017-03-01[33]. For ab initio prediction, GeneMark v3.68[34] was additionally employed. Intron positions were identified from RNA-Seq data via STAR v2.7.11b[35]. These intron annotations, combined with the reference genome sequence, served as input to train HMMs using GeneMark v3.68. The gene structures generated from the aforementioned methods, the repeat-masked genome, and the homologous protein data were integrated for gene structure annotation using Maker v3.01.03[36]. Functional annotation of the genome was performed by alignment against databases, including eggNOG and COG, using eggNOG-mapper v2.1.12[37].Phylogenetic analysis and divergence time estimation
-
To investigate the genomic family characteristics and phylogenetic history of A. heterophyllus, this study selected nine Rosales species, including M. notabilis, C. sativa, Z. jujuba, Prunus persica, Malus domestica, Fragaria vesca, Ficus hispida, F. carica, and M. alba. Additionally, six representative angiosperms, including C. papaya, Vitis vinifera, Solanum lycopersicum, A. thaliana, Populus trichocarpa, Glycine max, and four monocotyledonous plants, Zea mays, Oryza sativa, Sorghum bicolor, and Dendrobium nobile were included, with the basal angiosperm Amborella trichopoda serving as the outgroup to construct a phylogenetic analysis framework. This framework was constructed based on three single-copy orthologous genes, identified by OrthoFinder v3.1.0[38], which cover all 21 studied species, exhibit a broad phylogenetic span, and contain no sequence gaps. Divergence times among the studied species were estimated using R8s v1.81[39] under the approximate likelihood method, with calibration constraints derived from TimeTree[40] (
https://timetree.org/ ), which compiles divergence time estimates from published studies. Two calibration points were applied: the divergence between O. sativa and V. vinifera, constrained to 142.1–163.5 million years ago (Mya), and that between P. trichocarpa and G. max, constrained to 99.0–111.3 Mya. The estimated divergence times were integrated into the phylogenetic tree using the iTOL (Interactive Tree Of Life)[41] visualization tool, providing robust support for in-depth analysis of evolutionary relationships among the studied species.Gene family dynamics and synteny analysis
-
To further investigate the evolutionary relationships between A. heterophyllus and other species, collinearity analysis was performed pairwise among A. heterophyllus-MDM2, A. heterophyllus-S10 reference genome, M. alba, Mangifera indica, and Ficus microcarpa using JCVI v1.5.8[42]. All pairs exhibited significant collinear characteristics, reflecting conserved genomic structural relationships among these species.
Gene family clustering was conducted for M. alba, Musa acuminata, A. thaliana, F. hispida, and A. heterophyllus via the Orthovenn3 (
https://orthovenn3.bioinfotoolkits.net/ )[43]. Genomic data of all species involved in this analysis were downloaded from the NCBI database.Based on the species tree generated by OrthoFinder v3.1.0, CAFE5[44] software was employed to simulate the gain and loss dynamics of gene families across the phylogenetic tree. Special focus was placed on the significant expansion and contraction of gene families in the A. heterophyllus lineage. Functional enrichment analyses were further performed on these significantly changed gene families to reveal the functional adaptive characteristics driven by gene family number variations during the long-term evolutionary process of this species.
Identification and characterization of NAC family genes in A. heterophyllus
-
NAC transcription factors constitute a plant-specific transcription factor family, with well-documented roles in regulating plant growth and development, stress resistance, disease resistance, and hormone signaling in A. thaliana, O. sativa, and other species. Traits unique to jackfruit, such as enlarged fruit and tropical stress tolerance, may also be modulated by NAC transcription factors. However, comprehensive analyses of the NAC gene family in jackfruit remain lacking.
To address this gap, we performed a genome-wide identification of the NAC gene family based on the de novo assembled jackfruit MDM2 genome, aiming to screen key candidate genes associated with stress resistance. A. thaliana NAC protein sequences were retrieved from the TAIR database (
www.arabidopsis.org ) and used as queries for BLASTp[45] searches against the jackfruit protein dataset with an E-value < 1e–30 to identify potential homologous sequences. Additionally, the HMM profile of the NAC core conserved domain (NAM, PF02365) was obtained from the Pfam database[46] (http://pfam.xfam.org/ ), and HMMER[47] was employed for candidate gene prediction. After merging the candidate sequences and removing redundancies, the presence and integrity of NAC-specific conserved domains were validated using the NCBI Conserved Domain Database (CDD) to ensure the identification of authentic AhNAC family members.Phylogenetic analysis was conducted using MEGA12 software[48], with the Neighbor-Joining (NJ) method and 1,000 bootstrap replicates for reliability assessment. The resulting phylogenetic tree of NAC proteins from A. heterophyllus and A. thaliana was visualized and optimized using the online tool iTOL. AhNAC genes were subsequently classified by referencing the gene IDs and established subfamily classifications of AtNAC proteins.
Conserved protein motif and gene structure analysis of NACs
-
Chromosomal distributions and physicochemical properties of the AhNAC genes were analyzed and visualized using TBtools v2.119[49]. Conserved motifs were identified via the MEME suite (
http://meme-suite.org/tools/meme )[50], while the exon-intron architectures and untranslated regions (UTRs) were illustrated using the Gene Structure Display Server within TBtools, integrated with the jackfruit genome annotation. -
PacBio HiFi sequencing generated 44.3 Gb of raw reads with about 43 × coverage. Initial assembly of the HiFi data was performed using Hifiasm v0.25.0-r726, yielding a genome size of approximately 1.82 Gb with a contig N50 of 16.76 Mb. Redundant sequences derived from haplotype separation and repeat expansion were removed using Purge_dups v1.2.5, reducing the genome to a non-redundant haploid assembly of ~1.03 Gb. Contigs were subsequently scaffolded and anchored onto 28 pseudochromosomes using Ragtag v2.1.0 with the S10 genome as a reference, resulting in the chromosome-level assembly of the MDM2 genome (Table 1).
Table 1. Statistics of the A. heterophyllus-MDM2 genome assembly.
Assembly feature MDM2 Total sequence length (bp) 1,027,292,696 Number of chromosomes 28 Number of scaffolds 60 Scaffolds N50 (bp) 39,361,022 Scaffolds L50 12 GC content (%) 34.82 Number of genes 45,366 Total TE length (bp) 605,212,121 LAI 26.88 BUSCO (%) 93.8 QV 76.11 The final genome had a total length of 1,027,292,696 bp, consisting of 60 scaffolds with a scaffold N50 of 39.36 Mb and a scaffold N90 of 18.59 Mb. The GC content was 34.82%, and the BUSCO completeness score reached 93.8%. Assembly quality assessment showed that the genome had an LAI of 26.88, indicating excellent continuity. Additionally, the QV was 76.11, further validating the high accuracy and completeness of the assembled consensus sequence (Fig. 1b).
Repeat sequence characterization and genome annotation
-
Repetitive sequences totaling 605,212,121 bp were masked in the MDM2 genome using EDTA v2.2.2, accounting for 58.91% of the total genome length. TEs were dominated by long terminal repeat retrotransposons (LTR-RTs), with a total length of 459,633,274 bp, representing 76.12% of all TEs. We subsequently conducted phylogenetic clustering analysis on the identified LTR-RTs (Fig. 2b), which grouped these elements into multiple distinct clades. Among these, Gypsy LTR-RTs accounted for 24.72% and Copia LTR-RTs 29.65%, serving as the primary drivers of genome expansion. DNA transposons were the secondary TE type, contributing 18.05% of the total TE length, and mainly included subfamilies such as hobo-Activator, Tc1-IS630-Pogo, MULE-MuDR, Tourist/Harbinger, and rolling-circle transposons. Non-LTR retrotransposons only accounted for 0.36%, consistent with the typical TE distribution characteristics of higher plant genomes (Table 2). Other repeat sequence types constituted less than 1%, indicating high completeness and accuracy of TE annotation.
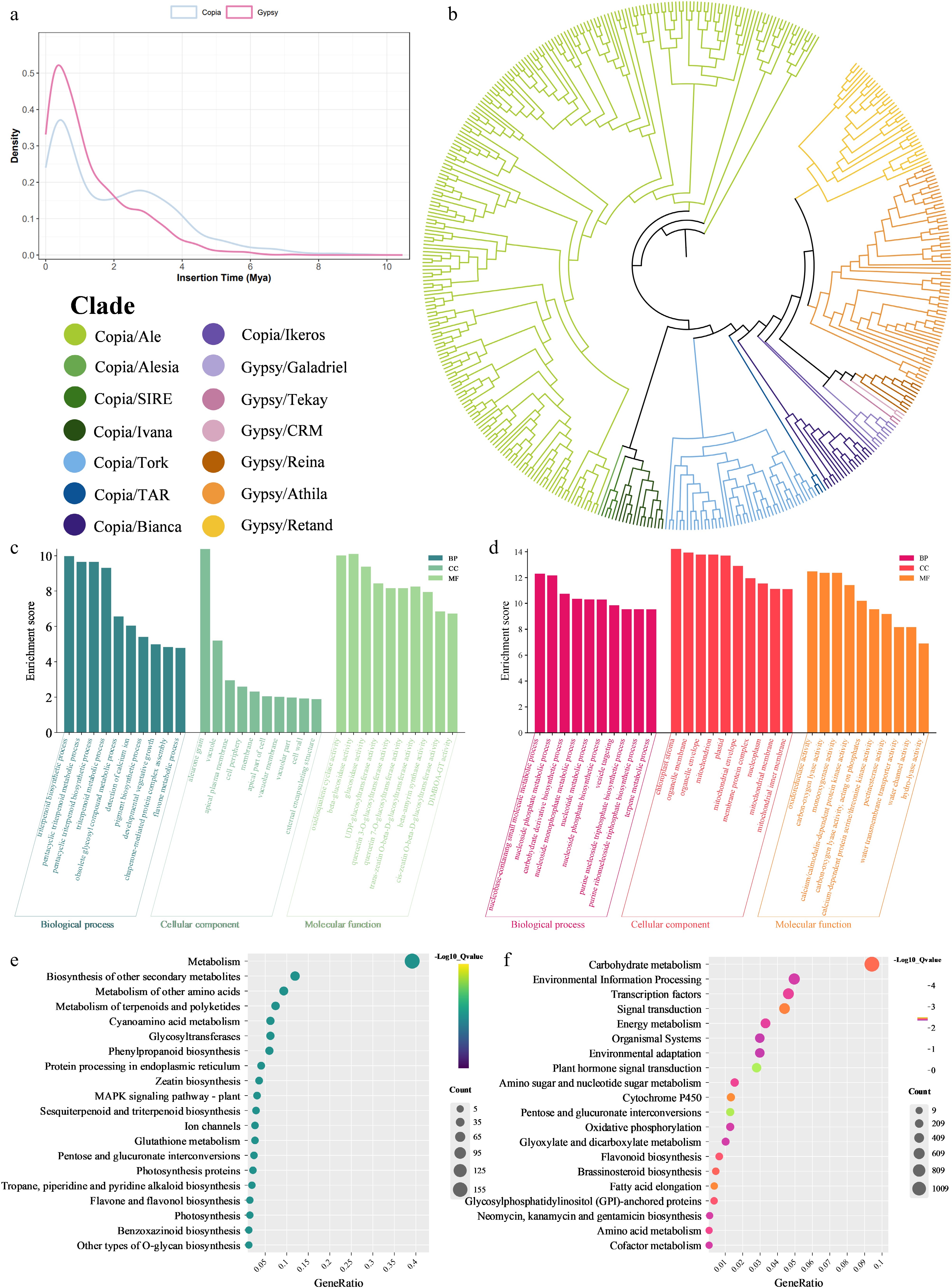
Figure 2.
(a) Density plot of LTR retrotransposon insertion times. (b) Phylogenetic clustering of LTR-RTs. (c) GO enrichment analysis plots of contracted gene families. (d) GO enrichment analysis plots of expanded gene families. (e) KEGG enrichment analysis plots of contracted gene families. (f) KEGG enrichment analysis plots of expanded gene families.
Table 2. Statistics of repeat elements in the A. heterophyllus-MDM2 genome.
Type Number of elements Length (bp) In genome
(%)DNA transposons 514,818 109,019,163 10.61 Long interspersed nuclear elements (LINEs) 8,203 2,163,258 0.21 Rolling-circles 1,646 1,315,557 0.13 Long terminal repeats (LTR) 627,910 459,633,274 44.74 Simple repeats 90 5,989 < 0.01 Total − 605,212,121 58.91 To investigate the evolutionary dynamics of LTR retrotransposons, insertion time estimation was performed on high-quality and intact Gypsy and Copia LTR-RTs in the MDM2 genome. Both types of LTR retrotransposons exhibited characteristics of recent burst insertion (Fig. 2a). The Gypsy superfamily showed an insertion peak at approximately 0.38 Mya, with an average insertion time of 1.31 Mya and a median of 0.89 Mya, displaying a typical feature of 'high-intensity recent activity'. The Copia superfamily had an insertion peak at 0.44 Mya, slightly later than that of the Gypsy superfamily, with an average insertion time of approximately 2 Mya and a median of 1.64 Mya, reflecting a relatively wider time span of insertion events compared to the Gypsy superfamily. When further combined with the TE classification proportion results, the total copy number of Copia was slightly higher than that of Gypsy. However, Gypsy, relying on a higher recent insertion concentration, became the core factor driving the short-term rapid expansion of the MDM2 genome, while the insertion pattern of Copia was relatively scattered, playing a role in the long-term evolutionary process of genome structure.
NcRNAs in the genome were comprehensively annotated using tRNAscan-SE v2.0.12 and INFERNAL v1.1.5 to characterize their distribution patterns. Results showed that a total of 4,633 ncRNA molecules were identified, with a combined length of 816,229 bp. Ribosomal RNAs (rRNAs) were the dominant type, accounting for 2,210 copies with an average length of 281.8 bp. This high abundance is consistent with their functional role as core components of the protein synthesis machinery, which requires numerous copies to maintain translational efficiency. The rRNAs included four subtypes (5S, 18S, 28S, and 5.8S), among which, 5S rRNA existed as tandem repeat clusters and dominated with 1,735 copies. A total of 991 transfer RNAs (tRNAs) were identified, with an average length of 73.6 bp. Small nuclear RNAs (snRNAs) numbered 782, with an average length of 107.18 bp, and were further classified into CD-box, HACA-box, and splicing subtypes. Among regulatory ncRNAs, 265 microRNA (miRNA) copies were detected, with an average length of 123.8 bp.
In addition, 45,366 protein-coding genes were annotated in the MDM2 genome, with a gene density of 44.16 genes per Mb, indicating a relatively dense overall gene distribution. The average gene length was 3,985.64 bp, with an average of 6.82 exons per gene and a mean length of individual exons of 1,983.49 bp. These features are consistent with the typical gene structure characteristics of plant genomes (Table 3).
Table 3. Gene structural characteristics of the A. heterophyllus-MDM2 genome.
Type MDM2 Gene density (gene/Mb) 44.16 Gene number 45,366 Average gene length (bp) 3,985.64 Average CDS length (bp) 1,983.49 Average exon per gene 6.82 Average exon length (bp) 293.65 Average intron length (bp) 456.40 Phylogenetic position and divergence time of A. heterophyllus
-
To investigate the evolutionary position of A. heterophyllus, we performed a phylogenetic analysis based on concatenated alignments of single-copy orthologous genes (Fig. 1c). The results confirmed that the sister group of Artocarpus is Morus, which is consistent with previously reconstructed phylogenetic relationships of Moraceae[51]. Using this established phylogenetic framework, we estimated that the divergence time between A. heterophyllus and its sister genus Morus was approximately 44.85 Mya. The divergence time of this clade from Ficus was further traced back to 59.00 Mya, with the two clades together forming the core evolutionary branch of Moraceae.
The close phylogenetic relationship between Artocarpus and Morus is consistent with their shared morphological and physiological traits, such as rapid growth and high adaptability to hot and humid environments. Genomic blocks between jackfruit MDM2 and S10 showed a highly conserved homologous correspondence pattern, reflecting sequence conservation among different accessions of the same species. Identifiable collinear regions were detected between jackfruit and F. carica, which are more closely related phylogenetically. In contrast, the distribution of genomic homologous syntenic blocks between jackfruit and V. vinifera or M. indica was scattered, indicating a high degree of genomic structural divergence among distantly related species (Fig. 1d). Next, we compared the complexity of gene families among M. alba, M. acuminata, A. thaliana, F. hispida, and A. heterophyllus. A maximum of 8,967 gene families were shared among all five species, while 1,582 gene families were specific to A. heterophyllus (Fig. 1e).
Gene family contraction and expansion in evolutionary context
-
Among Moraceae species, homologous gene family clustering analysis revealed that A. heterophyllus has undergone significant gene family expansion, with 6,425 expanded gene families and 1,838 contracted ones. The expanded gene families of jackfruit are mainly concentrated in core functionally-related families such as transcription factors, signal transduction, and environmental adaptation. Notably, the enrichment levels of transcription factor families, signal transduction pathways, signal and cellular process-related protein families, and protein kinase families all reached extremely significant levels (Fig. 2d). The expansion of these regulatory genes suggests a potential genomic framework for perceiving and integrating environmental and developmental signals. This characteristic might be associated with the prolonged life cycle of woody plants and their adaptation to variable tropical environments, potentially reflecting a trend of reinforcement in the regulatory networks of jackfruit.
Notably, the expanded gene families are significantly enriched in pathways associated with plant-pathogen interactions, flavonoid biosynthesis, and environmental adaptation (Fig. 2f). This points toward a possible genetic basis for defensive capacities and broad-spectrum adaptability. Furthermore, the expansion of specific genes involved in flavonoid and amino acid metabolism provides a potential metabolic foundation for the biosynthesis of defensive metabolites, which may contribute to plant stress resistance and fruit quality formation.
The contracted gene families in jackfruit were mainly enriched in functions related to the biosynthesis and modification of secondary metabolites, specifically triterpenoids, flavones, and phenylpropanoids (Fig. 2c). They were also associated with cellular component localization, including vacuoles and cell walls, and pathways related to core enzyme activities such as glycosyltransferases and key triterpenoid synthesis enzymes. The contraction of these families may reflect an evolutionary adjustment in resource allocation between growth, development, and secondary metabolism, potentially influencing the intracellular transport and storage of specific metabolites (Fig. 2e).
In summary, the significant expansion of jackfruit's gene families not only ensures stable metabolism in complex rainforest ecosystems, but also copes with diverse biotic stresses through abundant functional genes. These genomic characteristics represent candidate genetic factors that might contribute to the longevity and high-yield traits observed in jackfruit.
Identification and phylogenetic analysis of the NAC gene family
-
To further explain the significant expansion of the transcription factor family in jackfruit and fill the research gap regarding the functional NAC gene family in this species, we identified and analyzed the plant-specific NAC gene family, which plays a core role in growth, development, and stress resistance. A genome-wide search of the jackfruit genome was performed using the HMM, and genes containing the NAM domain were screened, resulting in the identification of 110 AhNAC genes. This number is fewer than that of NAC genes identified in A. thaliana.
Physicochemical property analysis was conducted on the identified AhNAC genes, including amino acid length (aa), relative molecular weight (MW), theoretical isoelectric point (pI), instability index (II), aliphatic index (AI), and grand average of hydropathicity (GRAVY) (Supplementary Table S1). The results showed significant variations in the predicted protein sequence lengths of AhNAC genes, ranging from 198 to 959 aa. The MW of the proteins spanned 22,659.44 to 105,840.30 Da. The pI ranged from 4.65 to 9.86, with most encoded proteins having a pI below 7, indicating an acidic property. The II varied between 30.95 and 70.11, with most members falling within the range of 30 to 60, suggesting differential stability among some proteins. The AI ranged from 50.96 to 81.45, indicating a certain degree of differentiation in protein hydrophobicity. All GRAVY values were negative (−0.966 to −0.293), demonstrating that all AhNAC proteins are hydrophilic.
Chromosomal localization mapping of AhNAC genes revealed that these genes are distributed across 25 chromosomes (Fig. 3a), with an uneven distribution among chromosomes. Chromosome 21 contained 8 NAC genes, the highest number among all chromosomes, while no NAC genes were detected on chromosomes 06, 15, and 16.
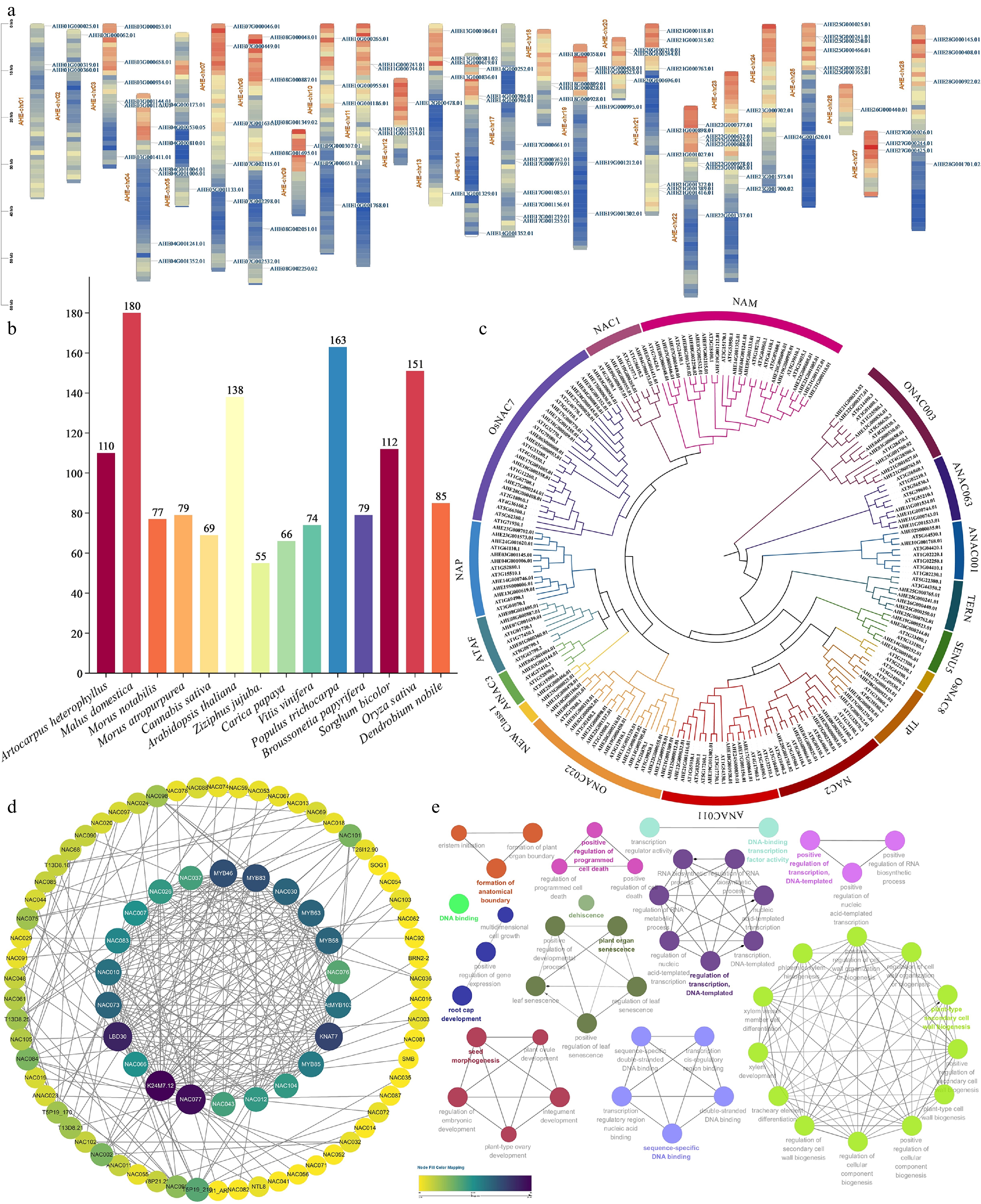
Figure 3.
(a) Schematic presentations for the distribution of AhNAC genes in A. heterophyllus chromosomes. (b) Comparison diagram of NAC gene families of various species. (c) Phylogenetic analysis of NAC proteins from A. heterophyllus and A. thaliana; different subfamilies are highlighted with specific colors. (d) Protein-protein interaction (PPI) network of AtNAC proteins. Nodes represent AtNAC proteins. (e) ClueGO functional enrichment analysis bubble plot.
Phylogenetic classification and functional characteristics of AhNAC genes
-
In an effort to further characterize the conservation relationships of AhNACs, we first counted the number of NAC genes across different species (Fig. 3b). This interspecies variation provides a species-level framework for subsequent conservation analyses. Insights into the conservation relationships of AhNAC genes were gained through multiple sequence alignment of protein sequences encoded by 110 AhNAC genes and 138 AtNAC genes using MEGA12 software. A phylogenetic tree was subsequently constructed via the NJ method (Fig. 3c).
Phylogenetic analysis revealed that the 110 AhNAC encoded protein sequences were clustered into 17 subfamilies, including one novel class. Among these, the NAM subfamily was the largest with 17 AhNAC genes, followed by the OsNAC7 and ONAC022 subfamilies, which contained 15 and 14 AhNAC genes, respectively. The smallest subfamily, designated NEW CLASS I, formed an independent cluster with only three AhNAC genes. No homologous counterparts of these three genes were detected in A. thaliana, indicating they may be unique to the NAC family in A. heterophyllus and potentially associated with species-specific traits.
All 110 candidate NAC genes of A. heterophyllus showed specific matches to pfam02365, with E-values ≤ 1e-37 (Supplementary Fig. S2). They possess the core conserved domain of the NAC family, confirming their membership in the AhNAC gene family with extremely high significance and reliability of domain matching.
The NAM domain of most genes is located in the N-terminal region of the protein, with amino acid positions mainly concentrated in the range of 10–188. This conforms to the typical architecture of NAC proteins, featuring a conserved N-terminal domain paired with a variable C-terminal regulatory region. The positional shift of the NAM domain in a few genes is speculated to be a species-specific characteristic of gene sequence assembly.
Two genes from the ONAC003 subfamily, AHE21G000315.02 and AHE22G000377.01, contain both the core NAM domain of the NAC family and the STKc_MAP3K-like kinase domain, with the matching significance of both domains exceeding the threshold. As the upstream core kinase of the mitogen-activated protein kinase (MAPK) cascade pathway, MAP3K is a key component of plant stress response, and growth and development signal transduction. The NAM domain, as a functional element for NAC transcription factors to bind DNA, indicates that such AhNAC proteins may possess dual functions as signal pathway kinases and transcription factors. These represent a special type of functional differentiation in the A. heterophyllus NAC family, providing novel candidate genes for deciphering the molecular mechanisms underlying its stress resistance or developmental regulation.
Analysis of the conserved motifs in AhNAC family proteins revealed that most contain seven highly conserved motifs, namely motif7, motif1, motif5, motif4, motif2, motif6, and motif3, with substantially consistent distribution positions and quantities. Notably, motif10 is exclusively present in genes of the OsNAC7 subfamily, suggesting it may play a role in the functional specialization of this subfamily. The ONAC003 subfamily includes 6 members with a minimum of 5 motifs, accounting for 5.40% of the total NAC family genes, indicating no significant difference in the number of motifs among individual members within this subfamily.
Construction of AtNAC protein-protein interaction network and prediction of AhNAC homologous interactions
-
In higher plants, all cells form a primary cell wall, while specialized cells can further deposit a secondary cell wall (SCW) inside the primary cell wall. The SCW is mainly composed of cellulose, hemicellulose, and lignin, providing mechanical support for plant upright growth and constructing channels for the long-distance transport of water, nutrients, and photosynthetic products[52]. The formation of SCW is precisely spatiotemporally regulated by a multi-layered network involving transcriptional regulation and post-translational modifications, and disruption of this regulatory network directly leads to abnormal plant growth and development[53]. Previous studies have confirmed that gibberellin (GA) signaling can act as an upstream regulatory cue to activate the downstream MYB-CESA pathway by relieving the inhibitory effect of DELLA proteins on NAC family transcription factors. This mechanism is conserved in cellulose synthesis of SCW in terrestrial plants[54], further improving the multi-layered regulatory network of SCW formation.
Through orthologous sequence alignment between jackfruit NAC genes and Arabidopsis NAC family genes, several jackfruit NAC family genes associated with SCW regulation were identified in this study: AHE18G000809.01 is orthologous to SND1 (AT1G32770), the master switch gene for SCW transcriptional regulation in A. thaliana; AHE17S000030.01 is orthologous to NST1 (AT2G46770), a core NAC gene regulating SCW formation in Arabidopsis; and AHE28G000408.01 is orthologous to VND6 (AT5G62380), a NAC gene specifically regulating SCW deposition in Arabidopsis vessel cells. To intuitively dissect the functional associations among AtNAC family genes, we constructed a protein-protein interaction (PPI) network for the AtNAC gene family (Fig. 3d) and performed functional enrichment analysis (Fig. 3e). The results clarified the core biological processes involved in this family of proteins, including plant-type SCW biogenesis, regulation of transcription (DNA-templated), seed morphogenesis, positive regulation of programmed cell death, and sequence-specific DNA binding. In Arabidopsis, SND1 and NST1 function as top-level master switches that cooperatively activate the transcription of the secondary core regulatory factor MYB46[55,56]. MYB46 and its homologous protein MYB83 are functionally redundant; they drive the transcriptional activation of KNOTTED ARABIDOPSIS THALIANA 7 (KNAT7) by recognizing MYB-responsive elements in the promoters of downstream genes[57−59]. Notably, MYB46 has been demonstrated to directly bind to the promoters of multiple xylan biosynthesis genes, including IRX7/FRA8, IRX8, IRX9, and IRX14, and positively regulate their expression in Arabidopsis inflorescence stems.
KNAT7 serves as a direct target of MYB46 and is transcriptionally activated by it. Previous studies have indicated that KNAT7 acts downstream of MYB46 and functions as a third-level transcriptional regulator during SCW formation. As a tertiary transcriptional regulator, KNAT7 positively regulates the expression of xylan biosynthesis genes such as IRX9 and IRX14 by specifically binding to the KN1 binding sites (KBS) in their promoters, thereby participating in the regulation of xylan biosynthesis in the SCW of inflorescence stems[60]. Currently, only a limited number of transcription factors, including MYB46 and its downstream target KNAT7, have been shown to be directly involved in regulating xylan biosynthesis during SCW formation in Arabidopsis inflorescence stems[61]. Based on the functional conservation of homologous genes, it is speculated that AHE18G000809.01 and AHE17S000030.01 in jackfruit may act as top-level regulators to activate the expression of MYB46-homologous genes in jackfruit, thereby driving the transcription of downstream SCW-related genes. This conserved hierarchical transcriptional regulatory pattern is presumed to mediate xylan biosynthesis in the SCW of jackfruit inflorescence stems. In contrast, AHE28G000408.01 (orthologous to VND6) may specifically participate in the regulation of SCW deposition in jackfruit vessel cells, forming a functional complement to AhNAC proteins.
Furthermore, the small ubiquitin-like modifier conjugation (SUMOylation) is an important post-translational regulatory mechanism for SCW formation. This study found that the jackfruit gene AHE04G001006.01 is orthologous to the Arabidopsis SUMO E3 ligase gene SIZ1 (AT1G52880). In Arabidopsis, SIZ1 can interact with the C-terminal domain of LBD30 to mediate the SUMOylation of lysine 226 (K226) of LBD30. LBD30 sumoylation plays a crucial role in plant development and SCW biosynthesis. Thus, the SUMO modification of LBD30 regulates SCW formation through the SND1/NST1-directed transcriptional network. This modification is a necessary prerequisite for LBD30 to activate the SND1/NST1 transcriptional network, ensuring the normal assembly of fiber cell SCW. Conversely, mutations at the K226 site of LBD30 or loss-of-function mutations of SIZ1 lead to defects in SCW formation, characterized by the thinning of fiber cell walls and a significant reduction in xylan content. The finding that LBD30 sumoylation acts as an additional regulatory layer to facilitate precise control of SCW formation provides further insights into this key process, which is essential for plant upright growth and the long-distance transport of water and solutes. It also has implications for cell wall modification through the regulation of LBD30 sumoylation in crop improvement.
Combined with the orthologous relationship between AHE04G001006.01 and SIZ1 in jackfruit, it is speculated that AHE04G001006.01 may regulate the activity of NAC family transcription factors such as AHE18G000809.01 (orthologous to SND1) and AHE17S000030.01 (orthologous to NST1) through a conserved SUMOylation mechanism, thereby participating in the post-translational regulation of SCW biosynthesis. This conserved mechanism provides a theoretical reference for analyzing the post-translational modification patterns of homologous proteins in jackfruit and exploring the synergistic functions of AhNAC proteins. Meanwhile, the key SCW regulatory genes identified in this study can serve as targets for molecular breeding of jackfruit to improve agronomic traits such as stem lodging resistance, demonstrating significant theoretical value and application prospects.
-
Jackfruit is renowned as a tropical fruit tree with a unique flavor and nutritional value, yet the genetic mechanism governing its genetic diversity and the formation of important economic traits remain poorly understood. This study aims to further explore the genomic characteristics of jackfruit, enrich genomic research on Moraceae plants, and provide valuable references for constructing a more comprehensive evolutionary framework of jackfruit and its relatives. We obtained a chromosome-level genome assembly of A. heterophyllus-MDM2 with a total length of approximately 1.03 Gb, consisting of 60 scaffolds, and key assembly metrics demonstrated exceptional quality: the scaffold N50 length reached 39.36 Mb, GC content was 34.82%, BUSCO completeness score was 93.8%, LAI was 26.88, and QV was 76.11. Compared with previous studies, this assembly significantly improved genome continuity and completeness, as verified by these metrics to be highly continuous, accurate, and nearly complete—suitable for subsequent functional analysis and gene mining. A total of 605.21 Mb of repetitive sequences were identified, accounting for 58.91% of the genome, with LTR-RTs dominating at 44.74%, followed by DNA transposons at 10.61% and long interspersed nuclear elements at 0.21%; these repetitive elements are speculated to have shaped the genomic structure and evolutionary trajectory of jackfruit. Genome structural annotation predicted 45,366 protein-coding genes, and this high-quality chromosome-level genome assembly fills the gap in genomic resources for jackfruit, providing crucial support for the breeding of superior varieties, conservation of germplasm resources, and improvement of key economic traits.
Comparative genomics analysis revealed the evolutionary relationships of jackfruit: it is most closely related to Morus species, diverging approximately 44.85 Mya, followed by a divergence from Ficus species around 59.00 Mya, with the two clades together forming the core evolutionary branch of the Moraceae family, which is consistent with previously reconstructed phylogenetic relationships of Moraceae. Compared to other moraceous members, jackfruit exhibits a significant expansion of gene families. This involves the amplification of numerous genes related to signal transduction, specifically transcription factors and protein kinases. This expansion is likely associated with jackfruit's adaptive needs to cope with long-term and variable biotic stresses in tropical rainforest environments, reflecting the directional enhancement of its regulatory networks and environmental adaptability. Given that NAC (NAM/ATAF1/2/CUC2) transcription factors are key regulators of plant development and stress responses, research on NAC genes in jackfruit is lacking. This study represents the first identification of 110 high-confidence AhNAC family genes in the jackfruit genome. The number of AhNAC genes is lower than those in A. thaliana, which may be related to the expansion patterns of gene families during evolution, and identifying genes associated with plant growth, development, and stress responses open avenues for enhancing stress resistance in breeding programs. Phylogenetic analysis divided the 110 AhNAC genes into 17 subfamilies, with the NAM subfamily being the largest, while the NEW CLASS I subfamily is unique to jackfruit, with no homologous genes in Arabidopsis—its emergence may be linked to the evolution of species-specific traits such as tropical stress resistance, fruit development, and xylem formation, consistent with the functional specialization of lineage-specific gene subfamilies observed in comparative genomics studies of other plants. Conserved motif analysis of AhNAC family proteins demonstrated that most members contain seven highly conserved motifs (motif7, motif1, motif5, motif4, motif2, motif6, and motif3) with largely consistent distribution patterns and quantities, which is crucial for preserving core biological functions. Notably, motif10 is uniquely present in genes belonging to the OsNAC7 subfamily, indicating its potential involvement in the functional specialization of this subfamily. Within the ONAC003 subfamily, 6 members possess a minimum of 5 motifs, accounting for 5.40% of the total NAC family genes, suggesting high structural conservation among members of this subfamily.
All identified genes carry the NAM domain, which confirms the reliability of the initial identification of the AhNAC family. A small subset of genes encodes additional domains, such as the STKc_MAP3K-like kinase domain in AHE21G000315.02 and AHE22G000377.01 from the ONAC003 subfamily, implying potential functional diversification. Such dual-function proteins may integrate transcriptional regulatory activity with signal transduction capabilities, offering novel candidate genes for investigating the molecular mechanisms underlying A. heterophyllus stress resistance and developmental regulation. Via orthologous sequence alignment, this study identified homologous genes of core SCW regulatory genes and the SUMO E3 ligase gene SIZ1 in A. heterophyllus. This result suggests that the hierarchical transcriptional regulatory pathway of SCW and the SIZ1-mediated SUMOylation post-translational modification mechanism, initially characterized in A. thaliana, is likely evolutionarily conserved in A. heterophyllus. It provides novel empirical evidence for deciphering the conserved evolutionary strategies underlying plant cell wall biosynthesis. The screened core regulatory genes can serve as key targets for molecular breeding; enhancing their functions through biotechnological approaches such as gene editing is expected to significantly improve the mechanical strength of A. heterophyllus stems, optimize xylem structure, and consequently enhance fruit yield and quality. Furthermore, the reinforcement of SCW structure may also potentiate plant resistance against pathogens such as C. siamense, offering a novel technical avenue to address biotic and abiotic stress challenges confronting the jackfruit industry.
Systematic research on the NAC transcription factor family originated from the model plant A. thaliana. Recent evidence has indicated that NAC genes in tropical fruit trees share certain commonalities in composition, evolution, and function. Unlike annual model plants, tropical fruit trees grow long-term in environments characterized by high temperature, high humidity, and frequent pathogenic stress, and the roles of their NAC gene families in stress response and development can more directly reflect adaptive evolution to tropical-specific habitats.
In this study, a NEW CLASS I subgroup containing three members was identified in the jackfruit NAC gene family, and no Arabidopsis homologs were found in this subgroup. Notably, M. atropurpurea, another member of the Moraceae family with a similar ecological niche, also exhibits similar evolutionary characteristics in its NAC family, including two new classes and two unclassified subgroups[7]. Both species have long adapted to the high-temperature and high-humidity environments in tropical and subtropical regions, and their NAC gene families both show species-specific new subgroup characteristics. This may suggest that extreme tropical environmental stress is one of the key selective pressures driving the functional divergence of the NAC family in Moraceae plants. Such environmental stress may promote the evolution of species-specific functions by inducing the NAC gene family to generate unique subgroups or new classes, so as to cope with multiple challenges in tropical environments, such as high-temperature stress tolerance, pathogen defense induced by high humidity, and water metabolism regulation.
NAC transcription factors play an important role in plant defense against various pathogen invasions. Previous studies have confirmed that 20 MaNAC genes in mulberry respond to infection by Ciboria shiraiana, among which MaNAC32, belonging to the ONAC022 subgroup, shows an approximately 200-fold upregulation and is an ortholog of JUB1 (JUNGBRUNNEN 1). In tomato, JUB1 (SlJUB1), as a nuclear-localized NAC transcription factor, can be induced by abiotic stresses such as drought and high salinity. It positively regulates tomato drought tolerance by directly binding to, and activating the promoters of target genes, including SlDREB1, SlDREB2, and SlDELLA, balancing hormone signals and ROS homeostasis, enhancing antioxidant enzyme activity, and reducing oxidative damage and water loss[62]. In M. acuminata, MaNAC5, a transcriptional activator of the NAC family, can be induced by Colletotrichum musae, salicylic acid (SA), and methyl jasmonate (MeJA). It mediates banana resistance to anthracnose by interacting with MaWRKY1 and MaWRKY2 to form a complex, which coordinately activates the transcriptional activity of pathogenesis-related genes such as MaPR1-1 and MaPR2[63].
Based on the functional conservation of NAC genes in Moraceae plants and other tropical fruit trees, it is speculated that the 14 genes in the ONAC022 subgroup of jackfruit may play a certain role in resisting pathogen invasion through similar regulatory pathways. In the future, experiments such as homologous gene cloning, expression profile analysis after pathogen inoculation, and transgenic functional verification could be performed to further clarify the specific disease resistance mechanism of JUB1 homologs in jackfruit, thereby providing potential candidate gene resources for jackfruit disease-resistant breeding.
In summary, the chromosome-level genome assembly of A. heterophyllus established in this study addresses a critical gap in genomic resources for tropical fruit plants within the Moraceae family. It provides a foundational framework for future genetic research on this species and serves as a key resource for exploring genome evolution and the genetic basis of agronomic traits in A. heterophyllus. The identification and classification of the NAC gene family yield candidate genes for enhancing stress response in A. heterophyllus, with key findings including dual-functional NAC proteins and lineage-specific subfamilies that reflect functional diversification, while retaining core functions. These results advance our understanding of the molecular mechanisms governing environmental adaptation and trait formation in A. heterophyllus, and provide valuable genetic resources for its molecular breeding and industrial application.
-
The authors confirm contributions to the paper as follows: study conception and design, project leading: Yu X, Xia Z; plant materials collection: Chen Y, Wang L; data analyses: Chen Y, Xia C, Liu Z. All authors reviewed the results and approved the final version of the manuscript.
-
Raw High-Fidelity sequencing data have been deposited in the Genome Sequence Archive (GSA, NGDC) under accession number CRA037874 (publicly accessible at https://ngdc.cncb.ac.cn/gsa). The whole-genome sequence data of this study has been deposited in the Genome Warehouse (National Genomics Data Center, NGDC) under accession number GWHHNGU00000000.1 (publicly accessible at https://ngdc.cncb.ac.cn/gwh)[64,65].
-
This work was supported by the startup funds for Tropical High-efficiency Agricultural Industry Technology System of Hainan University (THAITS-7). We thank the editor and anonymous reviewers for their insightful comments and suggestions.
-
The authors declare that they have no conflict of interest.
-
accompanies this paper online at: https://doi.org/10.48130/tp-0026-0004.
-
Received 6 January 2026; Accepted 14 February 2026; Published online 18 March 2026
- Supplementary Table S1 Physicochemical properties of proteins encoded by AhNAC gene family.
- Supplementary Fig. S1 Collinearity alignment between the de novo assembled MDM2 sequence and the reference S10 sequence.
- Supplementary Fig. S2 Gene structure, conserved motif distribution, and expression profile of the 110 AhNAC genes.
- Copyright: © 2026 by the author(s). Published by Maximum Academic Press on behalf of Hainan University. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Chen Y, Xia C, Liu Z, Wang L, Xia Z, et al. 2026. Chromosome-level genome assembly and genome-wide analysis of the NAC gene family of Artocarpus heterophyllus. Tropical Plants 5: e006 doi: 10.48130/tp-0026-0004 

Chromosome-level genome assembly and genome-wide analysis of the NAC gene family of Artocarpus heterophyllus
- Received: 06 January 2026
- Revised: 02 February 2026
- Accepted: 14 February 2026
- Published online: 18 March 2026
Abstract: Artocarpus heterophyllus Lam. (jackfruit) is a unique tropical economic plant renowned for its massive fruit as well as substantial nutritional and medicinal values. Notably, red-fleshed jackfruit has garnered significant research interest due to its distinctive nutrient profile and specialized fruit development. However, its genetic diversity and genetic mechanisms remain to be further explored. Herein, we generated a chromosome-level genome assembly of red-fleshed jackfruit, which was anchored to 28 pseudochromosomes, with a total size of 1.03 Gb. The assembly was constructed from 60 scaffolds with a scaffold N50 length of 39.36 Mb, and 45,366 protein-coding genes were predicted. Comparative genomic analysis revealed that jackfruit diverged from the common ancestor of the genus Morus approximately 44.85 million years ago, and that multiple gene families underwent significant expansion during this period. Notably, we identified 110 NAC family genes from the A. heterophyllus genome. As a well characterized family of transcription factors involved in plant development and stress responses, these NAC genes represent important candidate genes potentially associated with fruit development and stress tolerance in jackfruit. This study substantially enhances our understanding of the evolution and genetics of jackfruit and its gene families, and provides key candidate genes for deciphering the molecular mechanisms underlying important traits such as fruit development and stress tolerance in jackfruit.