









HTML
-
Anthocyanins are important pigments with multiple biological functions in plants[1]. By the activity of six aglycones (cyanidin, petunidin, peonidin, pelargonidin, delphinidin, and malvidin) and multiple chemical modifications (glycosylation, acylation, and methylation), anthocyanins exhibit various colors in plants[2−5]. The accumulation of anthocyanins also impacts many other biological processes, such as UV-light protection, insect attraction, biological and other abiotic stress (low-temperature, drought, and salt) defenses, not just in tissue coloration[6−9].
Anthocyanins are ubiquitous in plant seeds, roots, fruits, and flowers and are also known as effective antioxidants[10]. It was reported that anthocyanins were generated in vegetative tissues for scavenging reactive oxygen species under stress conditions[11, 12]. Furthermore, anthocyanins play multiple roles in the human diet and food consumption[13]. Anthocyanins are effective antioxidants that can reduce the incidence of diseases such as cancer, atherosclerosis, and cardiopathy[14−16].
Given the importance of anthocyanins in plant development and the human diet, anthocyanin biosynthesis has been extensively studied in many higher plants, such as carrot, Arabidopsis, and grape[17−19]. In anthocyanin pathways, the early biosynthetic genes (EBGs) including chalcone isomerase (CHI), chalcone synthase (CHS), flavonoid 3’-hydroxylase (F3’H), and flavanone 3-hydroxylase (F3H) are responsible for the production of flavonoid precursors. The late biosynthetic genes (LBGs) of anthocyanin include dihydroflavonol 4-reductase (DFR), leucoanthocyanidin dioxygenase (LDOX), and flavonoid 3-O-glycosyltransferase (UFGT)[20]. The expression levels of structural genes are often positively associated with anthocyanin content in plants[19]. Previous studies have demonstrated that the expression of anthocyanin biosynthetic genes were activated by the R2R3-MYB, basic helix-loop-helix (bHLH), and WD40 repeat protein complex (MBW)[20, 21]. At the transcriptional level, MBW complex regulates anthocyanin production by interacting with the DNA-binding sites[22, 23]. The R2R3-MYB transcription factors in Arabidopsis could be divided into different subgroups and the members in subgroup 6 function in the regulation of anthocyanins[24, 25]. The post-translational mechanisms of MBW complex also play important roles in anthocyanin regulation, such as phosphorylation and cysteine nitrosylation[26−28]. MBW-mediated anthocyanin regulation is also influenced by both developmental and physiological factors in higher plants[29, 30]. Moreover, the R2R3-MYB transcription factors (TFs) are the key regulators of MBW complex determining the tissue coloration of plants. R2R3-MYB (PeMYB2, PeMYB11, and PeMYB12) TFs activate the downstream structural genes to promote the formation of anthocyanin in Phalaenopsis spp[31].
Celery (Apium graveolens L.), an important vegetable crop of the Apiaceae family, is cultivated worldwide[32]. Celery has medicinal value and abundant phytochemical nutrients, such as cellulose, vitamins, and flavonoids[33−35]. Purple celery accumulates anthocyanins in the petioles and its skin appears purple[5]. The functions of AgMYB2 in the regulation of anthocyanin biosynthesis have been investigated in previous research[36]. In this study, another R2R3-MYB TF in S6 subgroup, AgMYB1, was identified from purple celery and its role in controlling anthocyanin biosynthesis was also determined.
-
‘Liuhe huangxin celery’ (Q1) is a landrace with yellow-green leaves from Nanjing, China. ‘Nanxuan liuhe purple celery’ (PQ1) has purple petioles (Fig. 1). Nicotiana benthamiana and Columbia ecotype of Arabidopsis thaliana were used for functional verification. The seeds of celery, N. benthamiana, and A. thaliana were stored in the State Key Laboratory of Crop Genetics and Germplasm Enhancement (Nanjing Agricultural University; 32°04′ N, 118°85′ E). Plant materials were cultivated in an artificial climatic chamber with the conditions as previously described[5].

Figure 1. Phenotypes of different celery varieties. The black line at the right corner represents 5 cm in that pixel. Q1, ‘Liuhe huangxin celery’; PQ1, ‘Nanxuan liuhe purple celery’.
RNA extraction and cDNA synthesis
-
RNA was extracted from celery, Arabidopsis, and tobacco plants using an RNAsimple Total RNA Kit (Tiangen, Beijing, China). cDNA was synthesized using Prime-Script RT reagent Kit (TaKaRa, Dalian, China) according to the product manual. cDNA was stored for further gene cloning and quantitative real-time PCR (RT-qPCR) assays.
Identification of MYB transcription factors in celery
-
The sequences of MYB transcription factors in celery were downloaded from CeleryDB, a genomic database for celery (http://apiaceae.njau.edu.cn/celerydb)[37, 38]. The sequences of 126 Arabidopsis R2R3-MYB transcription factors were obtained from TAIR (http://www.arabidopsis.org)[39]. The amino acid sequences of the above MYB transcription factors were used to construct the phylogenetic tree using MEGA 5.0[40]. The MYB transcription factors in celery were divided into various subgroups based on the phylogenetic relationships with Arabidopsis R2R3-MYB[25].
Clone and analysis of AgMYB1 gene
-
The predicted AgMYB1 gene was amplified with special primers (Forward: 5'-ATGAAGAGTGGCAACGCTTCAAAG-3'; Reverse: 5'-TTAATTATCATCTGCTGGATTTAGA-3') and subsequently cloned into pMD-19 vector (Takara). The positive plasmid was verified for further sequencing (Genscript, Nanjing, China). The sequence alignments of anthocyanin-related R2R3-MYB proteins were achieved using Clustal X[41]. The phylogenetic analysis of different R2R3-MYB TFs was performed using MEGA 5.0 software[40]. The phylogenetic tree was constructed using the neighbor-joining method and the reliability was set to 1000 bootstrap replicates.
RT-qPCR assay
-
The expression levels of various genes in plants were detected by RT-qPCR assay. The RT-qPCR primers (Forward: 5'-AACAGATGGTCACTAATCGGTGGAAG-3'; Reverse: 5'-CAGCAGTAGTTGGAGCAATGTAACG-3') of AgMYB1 gene were designed using Primer Premier 6.0 software. RT-qPCR primers of anthocyanin-related structural genes in Arabidopsis and tobacco followed the methods used in previous studies[42−44]. RT-qPCR assay was performed with SYBR Premix Ex Taq (TaKaRa, Dalian, China) and conducted on a CFX96 Real-Time PCR system (Bio-Rad) following the manufacturer’s instructions. The relative gene expression values were calculated using Schmittgen's method[45]. The AgTUB-B, AtActin2, and NtActin genes were selected as the internal control for celery, Arabidopsis, and tobacco, respectively[46]. Each sample was performed with three biological replicates.
Transient expression assay of AgMYB1
-
In order to verify the activating role of AgMYB1 in anthocyanin production, the transient expression assay was conducted as previously described[47, 48]. The AgMYB1 gene was amplified with specific primers (Forward: 5'-TTTACAATTACCATGGGATCCATGAAGAGTGGCAACGCTTCAAAG-3'; Reverse: 5'-ACCGATGATACGAACGAGCTCTTAATTATCATCTGCTGGATTTAGA-3'). AgMYB1 and previously reported AgMYB2 were cloned into binary vector pCAMBIA-1301 between the Bam HI and Sac I sites. The recombinant pCAMBIA-1301 vector containing entire AgMYB1 or AgMYB2 was transformed into Agrobacterium tumefaciens strain GV3101 (pSoup-p19). The A. tumefaciens strain hosting AgMYB1 and AgMYB2 was infiltrated into the tobacco leaves, respectively. Three independent replicates were performed for each infiltration. After 10-day growth, the samples of tobacco leaf were collected for anthocyanin determination and RNA extraction.
Arabidopsis transformation
-
The genetic transformation assay of AgMYB1 gene in Arabidopsis plants was performed following the floral-dip method[49]. The transgenic Arabidopsis harboring AgMYB1 gene was confirmed using the β-glucuronidase (GUS) assay[50] and polymerase chain reaction (PCR) amplification with special primers (Forward: 5'-ATGAAGAGTGGCAACGCTTCAAAG-3'; Reverse: 5'-AAGCACAACAAATGGTACAAG-3').
Extraction and determination of anthocyanins
-
Total anthocyanins were extracted following previous methods, with some modification[51]. The whole plants of 35-day-old Arabidopsis were collected and used for anthocyanin extraction and determination. Briefly, the plant samples of Arabidopsis and tobacco were ground in liquid nitrogen. The powder samples were transferred to an extraction solution and extracted overnight at room temperature in the dark. The extracting solution was centrifuged at 15,294 g for 10 min at 4 °C to remove sediment. The absorbance of extraction at 530, 620, and 650 nm were determined using a spectrophotometer. The normalized optical density (OD) of anthocyanin was calculated based on the formula:
OD = (OD530−OD620) − 0.1 × (OD650 − OD620)
And the total anthocyanin content was calculated based on the normalized OD and expressed by cyanidin 3-O-galactoside equivalents in 100 g fresh weight (mg/100 g FW)[52]. The extraction and measurements were conducted for three biological replicates.
Measurement of glycosylation capacity of Arabidopsis crude enzyme
-
To determine the glycosylation capacity of wild type (WT) and transgenic Arabidopsis plants, the glycosylation products catalyzed by crude enzymes were detected. Approximately 0.2 g of fresh samples of WT and transgenic Arabidopsis plants were used for the extraction of crude enzymes. The extraction of crude enzymes and determination of glycosylation capacity was carried out following previous methods[53]. The relative glycosylation capacity of Arabidopsis crude enzyme was calculated based on the peak area of the glycosylation product. The measurement was performed with three biological replicates.
Yeast two hybrid assay
-
Yeast two hybrid (Y2H) assay of AgMYB1 and bHLH (AgbHLH2, AtTT8, and AtEGL3) proteins were performed according to the Matchmaker Gold Yeast Two-Hybrid System (Clontech, http://www.takarabio.com). AgMYB1 was fused with the pGADT7 vector between Eco RI and Xho I sites with specific primers (Forward: 5'-GCCATGGAGGCCAGTGAATTCATGAAGAGTGGCAACGCTTCAAAG-3'; Reverse: 5'-ACGATTCATCTGCAGCTCGAGTTAATTATCATCTGCTGGATTTAGA-3'). As previously reported, AgMYB2 and bHLH TFs were cloned into pGADT7 and pGBKT7 vectors, respectively[36]. Using Gietz’s method, the recombinant vector and empty vector were co-transformed into the Y2HGold yeast strain[54]. The transformed yeast cells were photographed following 3 days of incubation.
Antioxidant capacity measurement
-
The ferric reducing/antioxidant power (FRAP) is a typical method used to determine plant antioxidant capacity[55]. The total antioxidant capacity of Arabidopsis plants was measured using Total Antioxidant Capacity Assay Kit with FRAP method (Beyotime Institute of Biotechnology, Nanjing, China) based on the manufacturer’s instructions. Total antioxidant capacity quantities are represented in the FeSO4 concentration equivalents per g fresh weight (mM/g FW). Each sample was conducted for three biological replicates.
Statistical method
-
The Duncan’s multiple-range test (at a 0.05 significance level) was used to compare the differences of gene expressions and anthocyanin content. Statistical analysis was carried out using SPSS software Version 17.0.
Plant materials
-
According to the genomic sequence in CeleryDB, 154 MYB transcription factors were identified from celery[37, 38]. The phylogenetic tree was constructed using the sequences of MYB transcription factors in celery and Arabidopsis (Fig. 2). The celery MYB transcription factors were classified into different subgroups based on the previous results found in Arabidopsis[25]. In Arabidopsis, the functions of different subgroups of R2R3-MYB factors were diverse. The subgroup 6 of Arabidopsis R2R3-MYB includes AtMYB75, AtMYB90, AtMYB113, and AtMYB114, which are involved in the regulation of anthocyanin biosynthesis[24, 56]. Phylogenetic tree analysis showed that there were two celery MYB transcription factors, Agr10145 and Agr41800, in the S6 subgroup, namely AgMYB1 and AgMYB2 respectively. We suggest that these two transcription factors are related to anthocyanin accumulation in celery.
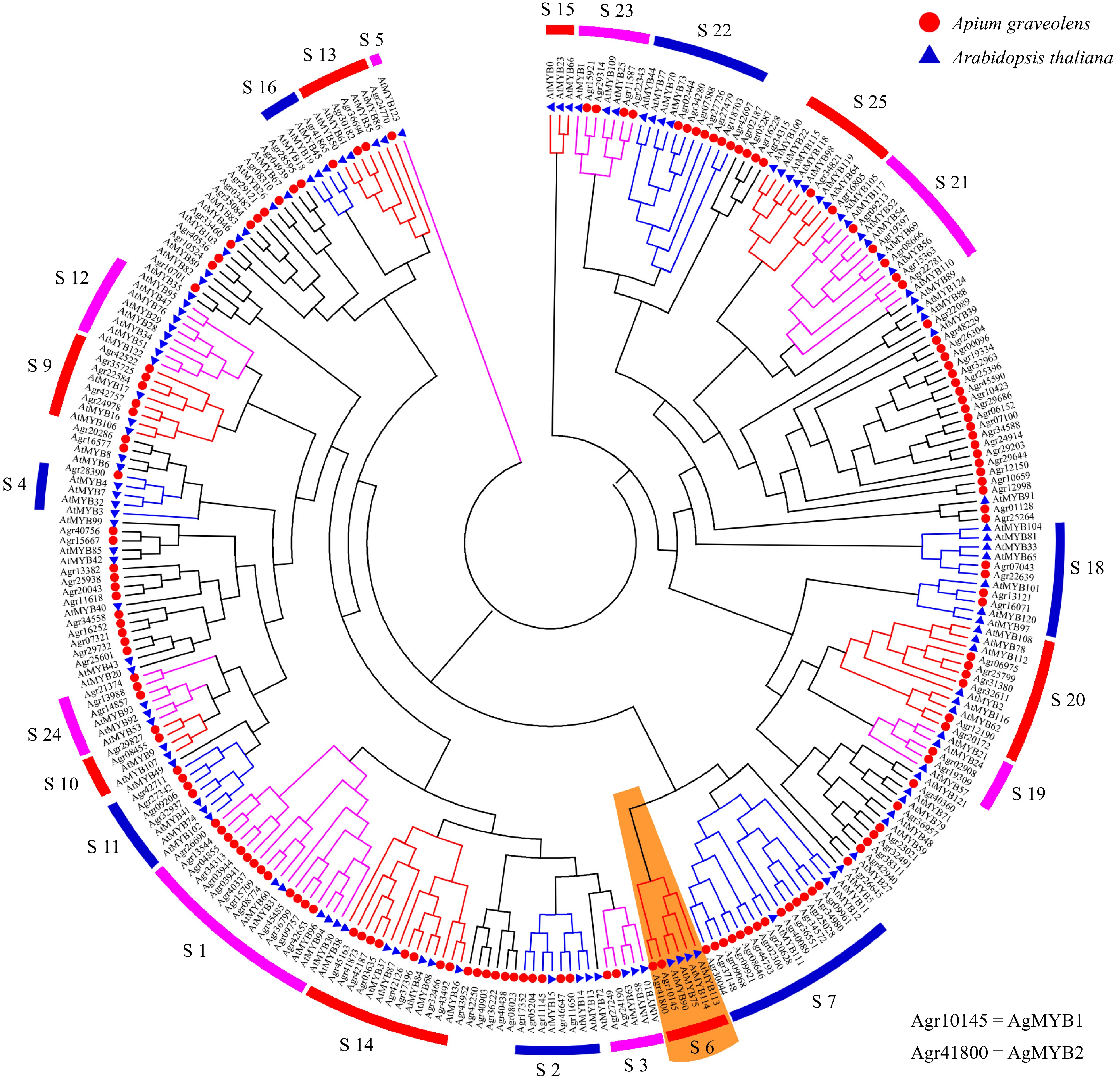
Figure 2. Phylogenetic tree of MYB transcription factors from celery and Arabidopsis. Different subgroups are represented using different colors. The S6 subgroup is shaded orange.
Cloning and sequence analysis of AgMYB1
-
The function of AgMYB2 has been previously studied[36]. The current study focuses on the identification of AgMYB1 in celery. The predicted AgMYB1 gene was amplified from purple celery with specific primers. The sequencing results revealed that the open reading frame (ORF) of AgMYB1 gene was 960 bp and encoded 319 amino acids (Additional file 1: Fig. S1). The constructed phylogenetic tree indicated that R2R3-MYB TFs with similar functions were clustered into the same branch. As shown in Fig. 3a, AgMYB1 TF belongs to the branch of the anthocyanin pathway and it has the closest evolutionary relationship with AgMYB2.
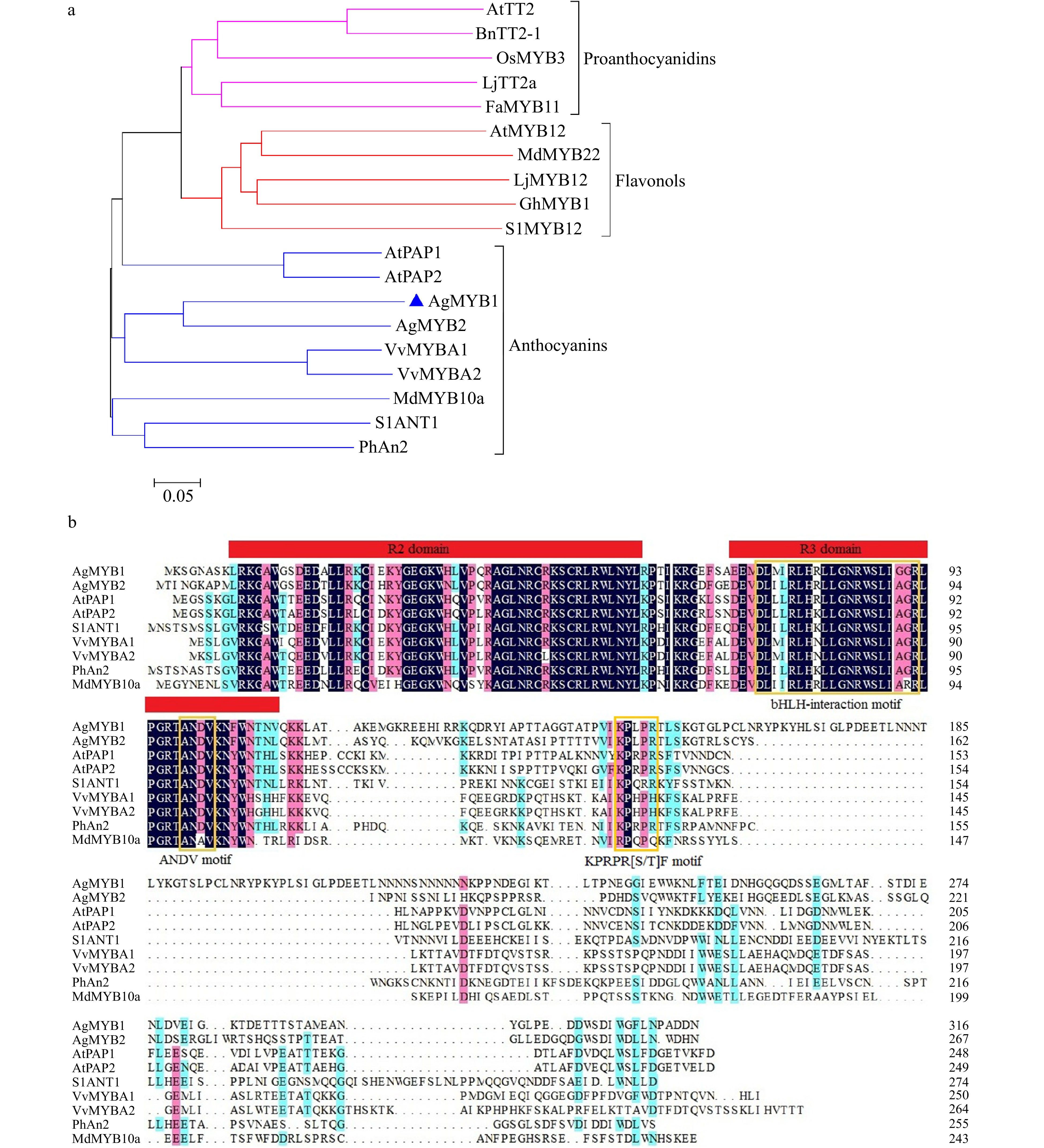
Figure 3. Phylogenetic analysis and sequence alignment of AgMYB1 and other R2R3-MYB proteins. (a) Phylogenetic analysis of AgMYB1 with R2R3-MYB TFs from celery and other plants. (b) Protein sequence alignment of AgMYB1 with other known anthocyanin-related R2R3-MYB TFs. The various motifs are indicated with yellow frames. The R2R3-MYB TFs with similar functions are clustered onto the same branch (anthocyanins, proanthocyanidins, and flavonols). The accession numbers of R2R3-MYB TFs: Arabidopsis thaliana AtPAP1 (AAG42001), AtPAP2 (AAG42002), AtMYB12 (ABB03913), AtTT2 (NP_198405); Solanum lycopersicum SlANT1 (AAQ55181), SlMYB12 (ACB46530); Vitis vinifera VvMYBA1 (BAD18977) and VvMYBA2 (BAD18978); Fragaria×ananassa FaMYB11 (AFL02461.1); Oryza sativa OsMYB3 (BAA23339.1); Gerbera hybrida GhMYB1 (CAD87007.1); Petunia×hybrida PhAn2 (AAF66727); Malus domestica MdMYB10a (ABB84753.1) and MdMYB22 (AAZ20438.1); Lotus japonicus LjMYB12 (BAF74782), LjTT2a (BAG12893); Brassica napus BnTT2-1 (ABI13034).
To further analyze the sequence structure of the AgMYB1 protein, sequence alignment of the AgMYB1 protein with other anthocyanin-related R2R3-MYB TFs was performed, including A. graveolens AgMYB2, A. thaliana AtPAP1 and AtPAP2, Solanum lycopersicum SlANT1, Vitis vinifera VvMYBA1 and VvMYBA2, Petunia × hybrida PhAN2, and Malus × domestica MdMYB10a (Fig. 3b). Sequence alignment indicated that the R2R3 domain in AgMYB1 and other anthocyanin-related R2R3-MYB proteins was highly conserved. AgMYB1 contained the typical bHLH-interaction motif, ANDV motif, and KPRPR[S/T]F motif, which are related to the regulation of anthocyanin biosynthesis in higher plants.
RT-qPCR analysis of AgMYB1 gene in purple and non-purple celery varieties
-
To recognize the relationship between color phenotypes and AgMYB1 transcripts, an RT-qPCR assay was conducted in the petioles of purple and non-purple celery plants (Fig. 4). At three developmental stages, the purple celery showed higher AgMYB1 gene transcripts compared with non-purple celery. The expression of the AgMYB1 gene in purple celery was approximately 17-fold higher than that of non-purple celery at the second development stage. We suggest that the transcripts of the AgMYB1 gene are involved in the phenotype difference in purple and non-purple celery varieties.
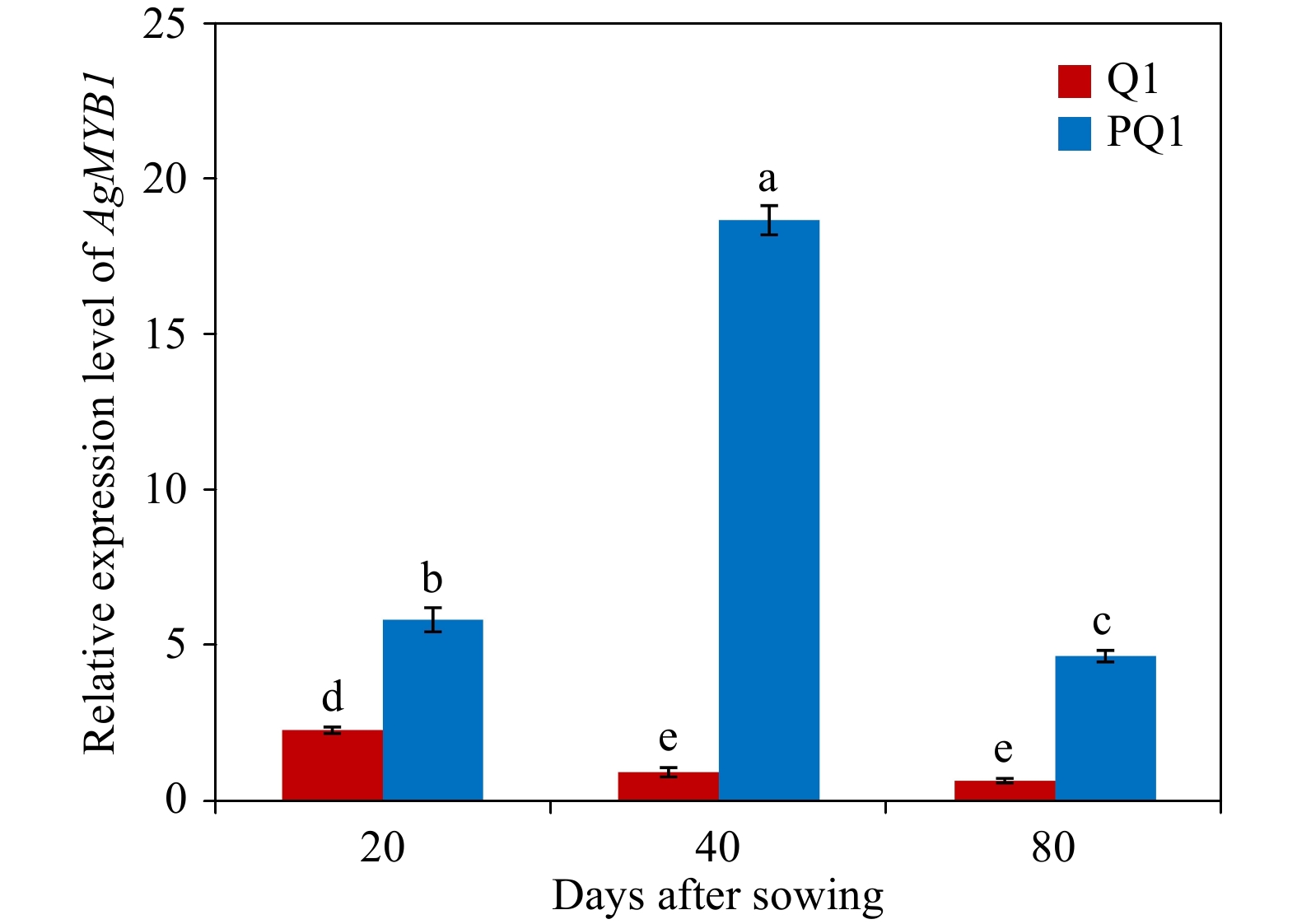
Figure 4. Relative transcript levels in various developmental stages and varieties. Q1, ‘Liuhe huangxin celery’; PQ1, ‘Nanxuan liuhe purple celery’. The values represent the mean of three independent experiments ± SD. The lowercase letters over the columns represent the significant differences at P < 0.05.
Transient expression of AgMYB1 in tobacco leaves
-
AgMYB1 and AgMYB2 were transiently expressed in tobacco to investigate the anthocyanin-promoting function. As shown in Fig. 5, the tobacco leaves expressing AgMYB1 and AgMYB2 appeared with a red pigmentation and contained significantly higher total anthocyanin content compared to control tobacco leaves. To understand the relationship between expression levels of structural genes and anthocyanin production in tobacco, RT-qPCR assay of NtCHI, NtCHS, NtF3H, NtF3’H, NtDFR, NtANS was conducted. As for the two R2R3-MYBs, the total anthocyanin contents and structural gene expressions in tobacco hosting AgMYB2 were significantly higher than those in tobacco hosting AgMYB1.
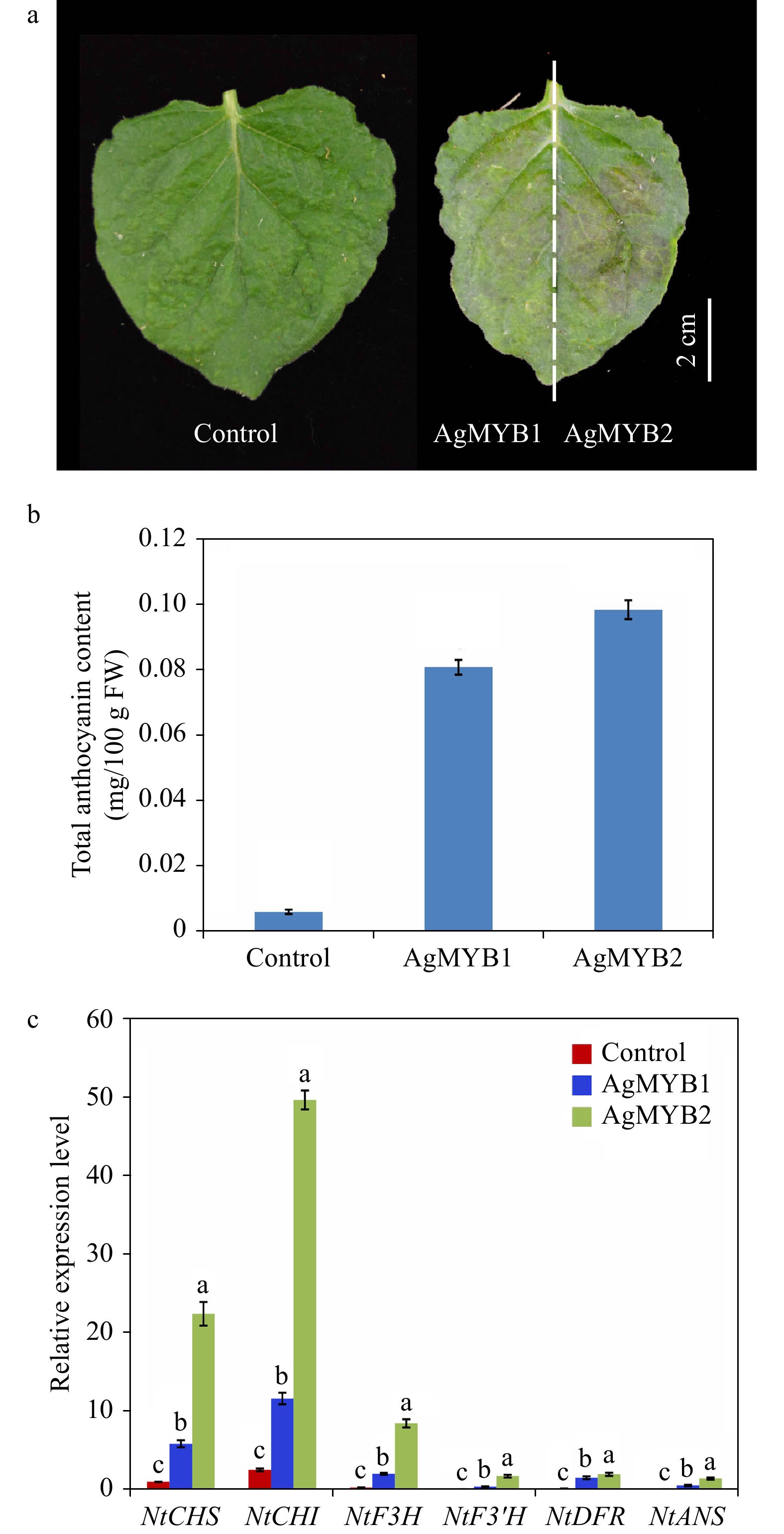
Figure 5. Transient expression of AgMYB1 and AgMYB2 in tobacco leaves. (a) Tobacco leaves infiltrated with Agrobacterium strain harboring AgMYB1 or AgMYB2. (b) Total anthocyanin content of control tobacco and tobacco transient expressing AgMYB1 or AgMYB2. (c) The relative transcript levels of anthocyanin biosynthetic genes in tobacco leaves. The AgMYB1 and AgMYB2 genes were transiently expressed in the left and right halves of tobacco leaves, respectively. The values represent the mean of three independent experiments ± SD. The lowercase letters over the columns represent the significant differences at P < 0.05.
Heterologous expression of AgMYB1 in Arabidopsis
-
To further confirm the activation roles of AgMYB1 in anthocyanin production, the gene was overexpressed in Arabidopsis. Transgenic Arabidopsis hosting AgMYB1 gene was screened from 1/2 MS agar plates (hygromycin B resistance). The OE-1 and OE-3 lines of Arabidopsis were selected for further analysis. The transgenic Arabidopsis (T2 generation) was further confirmed by GUS-staining and PCR amplification assay (Additional file 2: Fig. S2). The transgenic lines of Arabidopsis exhibited GUS activity. PCR amplification assay showed that the AgMYB1 gene was amplified from the cDNA of transgenic Arabidopsis but not from the cDNA of WT Arabidopsis. These results indicated that the AgMYB1 gene was stably expressed in transgenic Arabidopsis plants.
The phenotype comparison revealed that the transgenic Arabidopsis overexpressing AgMYB1 exhibited distinctly dark-purple seeds and leaves, compared with WT Arabidopsis (Fig. 6). Anthocyanin production was significantly promoted in transgenic Arabidopsis. Total anthocyanin content in OE-1 and OE-3 lines were 0.548 and 0.249 mg/ 100g FW, respectively (Fig. 6d). In addition, an enzyme activity assay was performed to verify the anthocyanin activating role of AgMYB1 at the protein level. The glycosylation products catalyzed by crude enzymes were detected (Fig. 7). Glycosylation products catalyzed by crude enzyme extracted from OE-1 Arabidopsis were significantly more than those catalyzed by crude enzymes extracted from WT Arabidopsis. The relative glycosylation capacity was calculated based on the peak area of the glycosylation product. As shown in Table 1, the relative glycosylation capacity of OE-1 Arabidopsis was 100%, while that of WT Arabidopsis was 3.59 ± 1.61%.
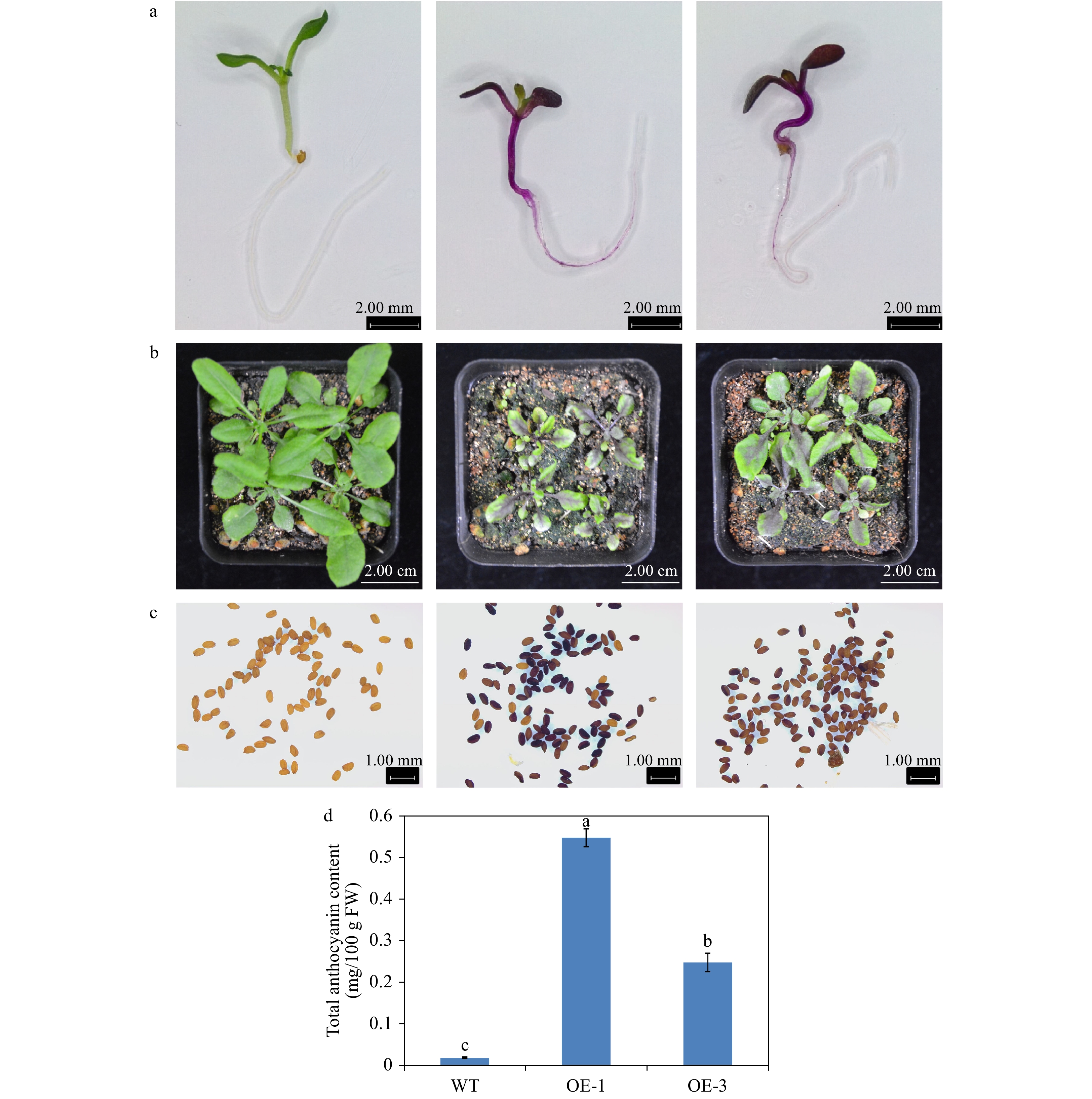
Figure 6. Overexpression of AgMYB1 in Arabidopsis. (a) Seedlings of WT and transgenic Arabidopsis grown on medium plate. (b) Seedlings of WT and transgenic Arabidopsis grown in soil. (c) Seeds of WT and transgenic Arabidopsis plants. (d) Total anthocyanin content of whole plants of WT and transgenic Arabidopsis. The values represent the mean of three independent experiments ± SD. The lowercase letters over the columns represent the significant differences at P < 0.05.
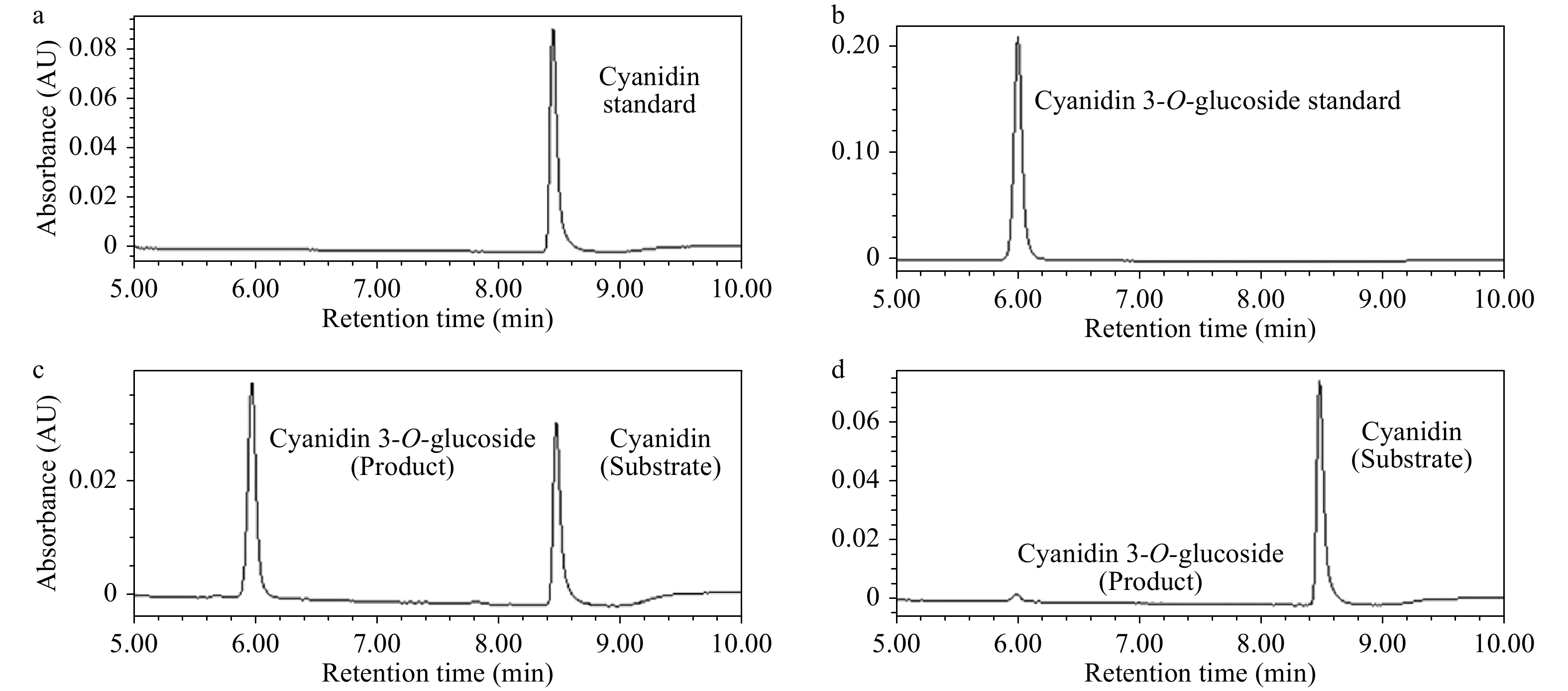
Figure 7. UPLC chromatograms of enzyme activity reactions. (a) Cyanidin standard sample. (b) Cyanidin 3-O-glucoside standard sample. (c) Enzyme activity reaction of crude enzyme extracted from OE-1 Arabidopsis. (d) Enzyme activity reaction of crude enzyme extracted WT Arabidopsis.
Table 1. Relative glycosylation abilities of WT and transgenic Arabidopsis.The glycosylation ability of OE-1 Line of Arabidopsis was set as 100%. The values represent the mean of three independent experiments ± SD.
Arabidopsis category Relative glycosylation ability (%) WT Arabidopsis 3.59 ± 1.61 OE-1 Arabidopsis 100 ± 3.99 The total anthocyanin content in plants were found to correlate with the expression of structural genes in the anthocyanin pathway. RT-qPCR assay was conducted to identify the role of AgMYB1 in activating the transcripts of structural genes in transgenic Arabidopsis plants. As shown in Fig. 8, the relative expression levels of anthocyanin-related structural genes in transgenic Arabidopsis over-expressing AgMYB1, were significantly higher than those in WT Arabidopsis.
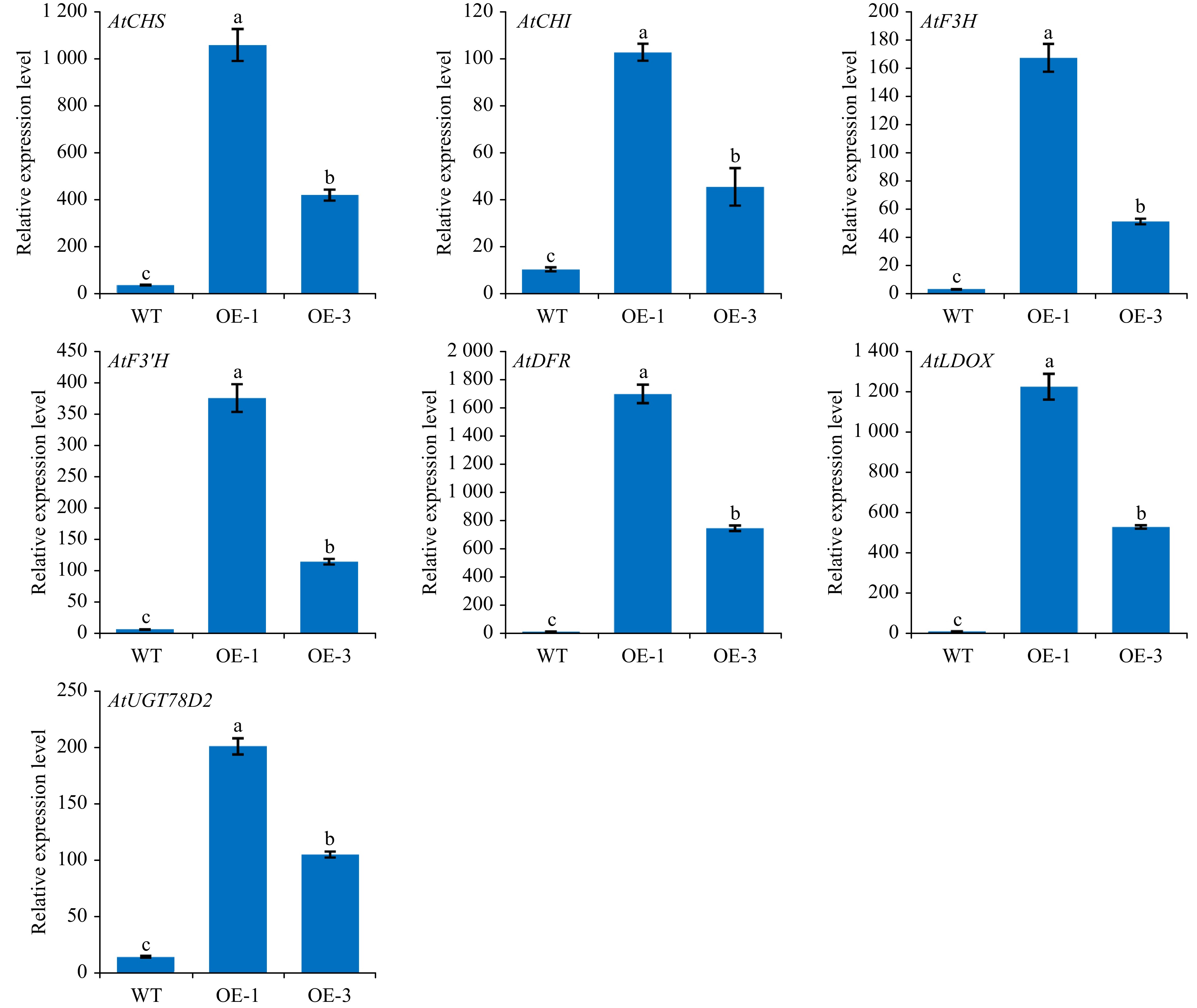
Figure 8. The relative transcripts of structural genes involved in anthocyanin biosynthesis in WT and transgenic Arabidopsis. The values represent the mean of three independent experiments ± SD. The lowercase letters over the columns represent the significant differences at P < 0.05.
AgMYB1 interacts with bHLH proteins
-
R2R3-MYB TF could interact with the bHLH protein to modulate the biosynthesis and accumulation of anthocyanin in many plants. In consideration of the bHLH-interaction motif in the AgMYB1 protein, we performed Y2H assay to verify the interaction of AgMYB1 with bHLH proteins. For interaction analysis, AtEGL3 and AtTT8 from Arabidopsis and AgbHLH2 from celery were selected as the bHLH regulatory proteins in the anthocyanin pathway. The results in Fig. 9a indicate that co-transformed yeast cells harboring AgMYB1-AD and AgbHLH2-BD, or AtEGL3-BD, or AtTT8-BD combinations survived in DDO and QDO selection plates, and the above co-transformed yeast cells also exhibited α-galactosidase activity. In contrast, the co-transformed yeast cells containing empty vectors and AgMYB1-AD, or AgbHLH2-BD, or AtEGL3-BD, or AtTT8-BD did not survive on the QDO selection plates and did not show α-galactosidase activity. Previous study showed that AgMYB2 also interacted with AgbHLH2 protein in yeast[35]. The comparison of Y2H indicated that the yeasts harboring AgMYB2-AD + AgbHLH2-BD grew faster than those harboring AgMYB1-AD + AgbHLH2-BD vectors (Fig. 9b). These results indicated that AgMYB1 interacted with bHLH proteins and the AgbHLH2-binding activity of AgMYB2 was stronger than AgMYB1 in yeasts.
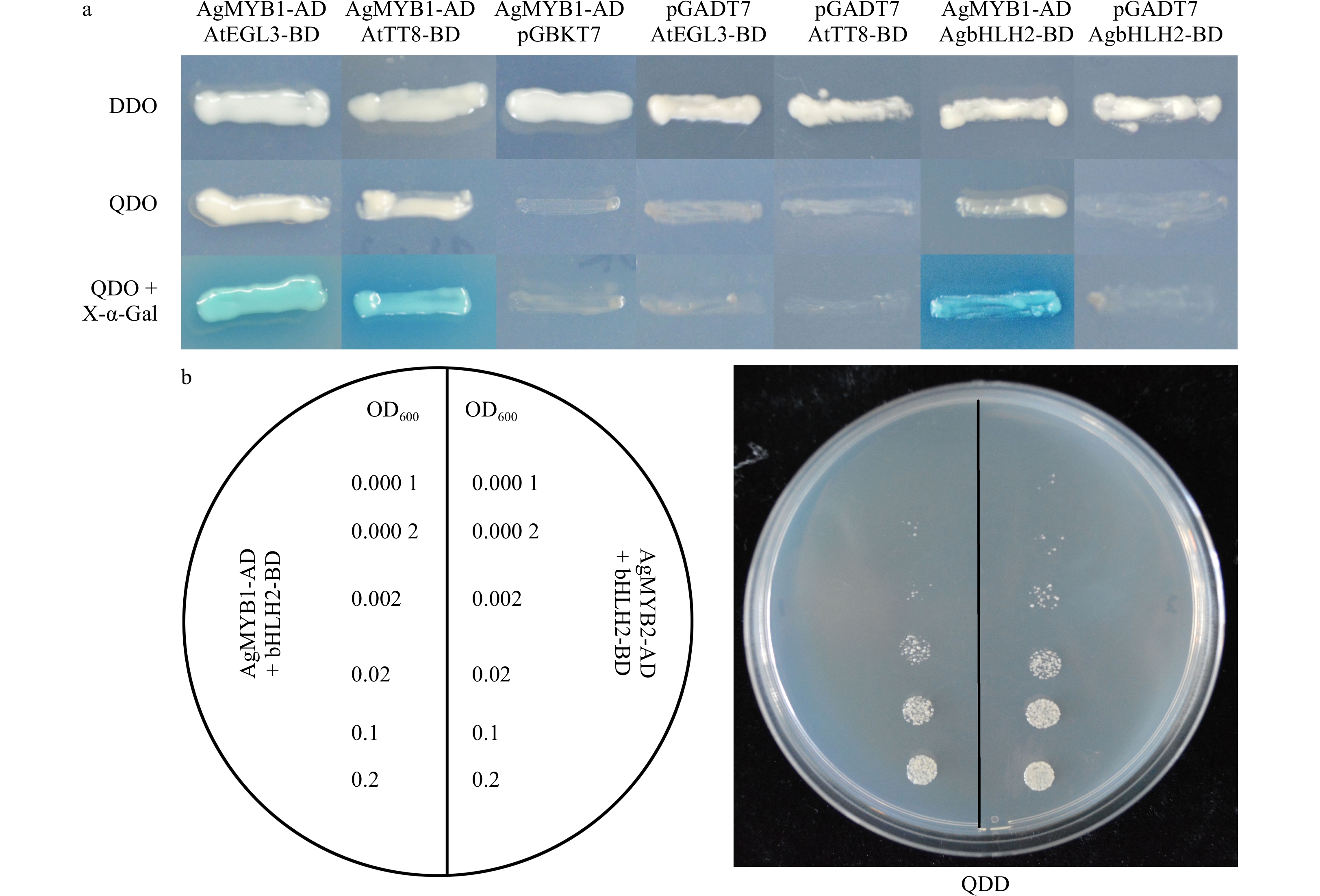
Figure 9. Yeast two-hybrid of celery R2R3-MYB and bHLH TFs. (a) Yeast two-hybrid of AgMYB1 with AtEGL3, AtTT8, and AgbHLH2 proteins. (b) Growth status of yeast cells harboring AgMYB1 and AgMYB2 with different OD600 values. DDO: SD/-Trp/-Leu; QDO: SD/-Leu/-Trp/-His/-Ade; QDO+X-α-Gal: SD/-Leu/-Trp/-His/-Ade/+X-α-Gal.
Analysis of antioxidant capacity in transgenic Arabidopsis
-
To confirm the effects of anthocyanin content in promoting antioxidant capacity, we determined the antioxidant capacity of WT and transgenic Arabidopsis plants. The antioxidant capacity of OE-1 and OE-3 Arabidopsis plants was 10.46 and 7.43 mM/g FW respectively, but the antioxidant capacity of WT Arabidopsis was only 5.52 mM/g FW (Fig. 10). This result suggests that the increase in anthocyanin content enhances the antioxidant capacity in transgenic Arabidopsis overexpressing AgMYB1.
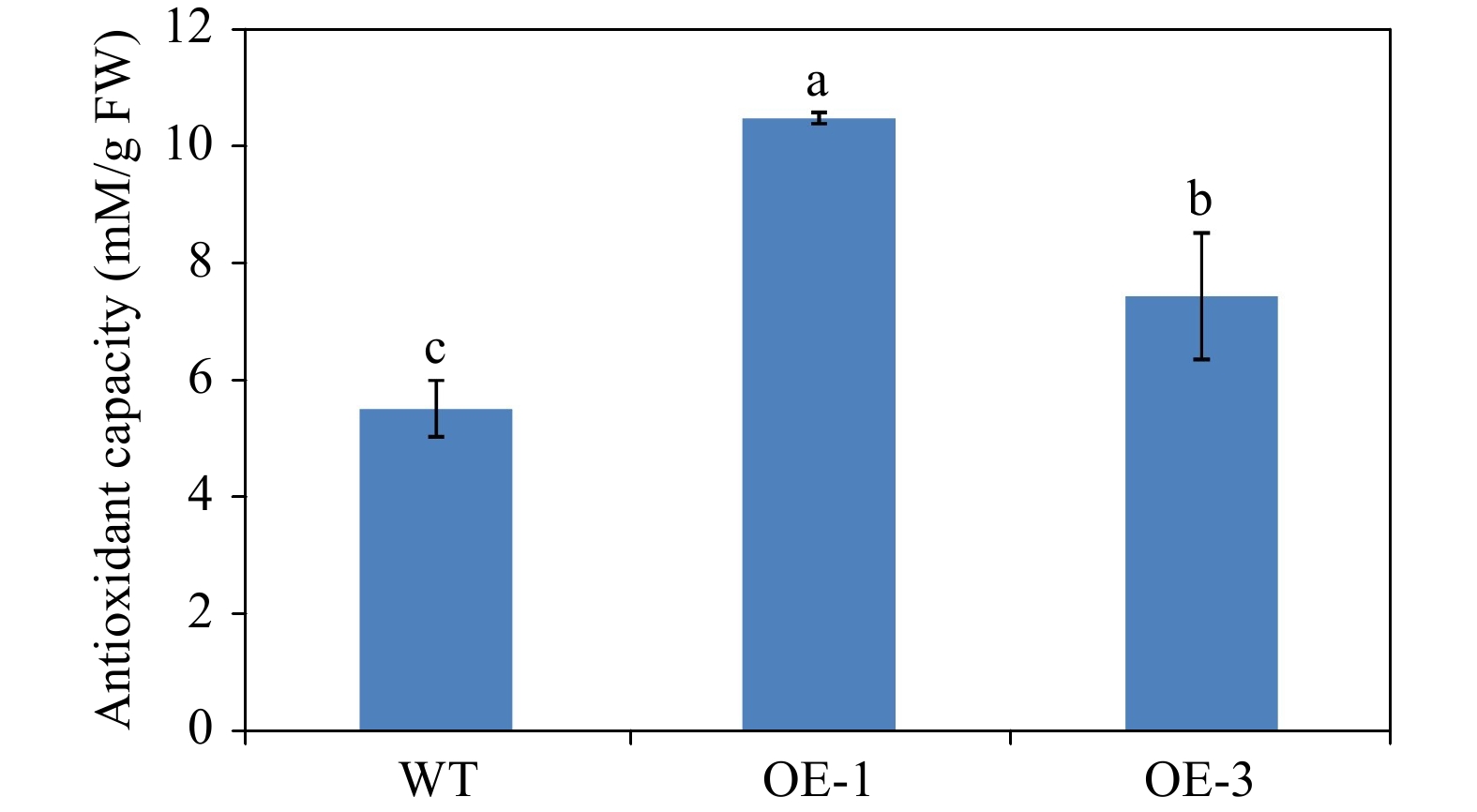
Figure 10. Antioxidant activity measurement of WT and transgenic Arabidopsis plants using FRAP method. The values represent the mean of three independent experiments ± SD. The lowercase letters over the columns represent the significant differences at P < 0.05.
Identification and classification of MYB transcription factors in celery
-
Plant tissue coloration is determined by various plant metabolites (carotenoids, anthocyanins, and betalains)[19, 57]. The accumulation of various anthocyanins results in the formation of purple, blue, red, or black organs in different species[48, 58, 59]. A previous study reported that the transcripts of anthocyanin structural genes were related to the anthocyanin content and skin colors in different celery plants[60]. The anthocyanin pathway is mainly controlled by MBW complex protein where MYB TFs always play a central role in plants[20, 24]. The S6 subgroup of R2R3-MYB factors plays an important role in anthocyanin biosynthesis in Arabidopsis[25]. In this study, two R2R3-MYB transcription factors (AgMYB1 and AgMYB2) in S6 subgroup were identified from celery, based on the genome-wide analysis of MYB family factors. The expression level of AgMYB2 was higher in purple celery than that in non-purple celery and the anthocyanin-regulatory role of AgMYB2 has been investigated to activate anthocyanin biosynthesis in our previous research[36]. In the current study, we isolated another R2R3-MYB regulator from purple celery, AgMYB1, and the results indicated that it is a transcriptional activator in the anthocyanin biosynthetic pathway. However, the bHLH-binding and anthocyanin-activating activities of AgMYB2 were stronger than AgMYB1.
Phylogenetic analysis showed that AgMYB1 TF was clustered with anthocyanin regulatory MYB TFs from other species, such as Arabidopsis AtPAP1 and AtPAP2[61], Solanum lycopersicum SlANT1[62], Malus domestica MdMYB10a[48]. The biological function of plant TFs usually depends on the protein sequence structure[63]. Sequence alignment of AgMYB1 and other anthocyanin-related R2R3-MYBs revealed that the R2R3 domain was highly conserved in these TFs. The anthocyanin-related bHLH-interaction motif, ANDV motif, and KPRPR[S/T]F motif were recognized in AgMYB1[63−65]. The RT-qPCR analysis indicated that the transcripts of AgMYB1 in purple celery were significantly higher than that in non-purple celery. The difference of AgMYB1 expression levels may be due to the differences of promoter sequence in different varieties, or the regulation of upstream regulatory genes. These results suggest that AgMYB1 TF is involved in the regulation of anthocyanin biosynthesis in purple celery.
Transient expression of AgMYB1 and AgMYB2 in tobacco promote anthocyanin production and result in red pigmentation in tobacco leaves. The tobacco leaf hosting AgMYB2 gene harbored darker pigments than that of leaves hosting the AgMYB1 gene. The total anthocyanin content and expression levels of anthocyanin biosynthetic gene in tobacco leaf overexpressing AgMYB2 were also higher than those in leaves overexpressing AgMYB2. In addition, AgMYB1 TF was heterologous expressed in Arabidopsis to further confirm its regulatory role in activating anthocyanin production. The transgenic Arabidopsis overexpressing AgMYB1 exhibited dark-purple organs at different developmental stages. The phenotypic differences between WT and transgenic Arabidopsis can be attributed to the increase of total anthocyanin content. Previous studies revealed that overexpression of R2R3-MYBs promoted pigmentation and enhanced the anthocyanin production in many plants, e.g. overexpression of RsMYB1 and OjMYB1 in tobacco and Arabidopsis plants, respectively[43, 53]. The transcripts of anthocyanin biosynthetic genes were activated by R2R3-MYB in plants[42]. The expression of anthocyanin biosynthetic genes were up-regulated in transgenic Arabidopsis hosting AgMYB1. The glycosylation process of anthocyanin enhances its stability and water-solubility in plants[66]. Anthocyanin glycosylation was enhanced in transgenic Arabidopsis overexpressing AgMYB1. Previous studies reported that R2R3-MYB and bHLH TFs interacted and co-regulated the anthocyanin pathway in many species[24, 67]. Considering the bHLH-interaction motif in the R2R3 domain, the combination of AgMYB1 and bHLH proteins was also investigated in this study. The growth assay of yeast with various OD values indicated that the bHLH-binding activity of AgMYB2 was stronger than that of AgMYB1. TFs regulate the transcript of target genes via interacting with its upstream promoters. McMYB10 directly binds with the promoter of McF3’H and activates its expression, thereby promoting anthocyanin biosynthesis in crabapple[42]. We also performed a yeast one hybrid assay with AgMYB1 with the promoter of AgF3’H, whereas the promoter showed strong self-activation and affected the verification of interaction. The strong self-activation may be due to the activation of transcription factors inside the yeast. Foods rich in anthocyanins have health-promoting values by improving antioxidant activities in the human diet[68]. The expression of specific TF improved the antioxidant capacities of plants and it is considered beneficial for human health[69]. Our results showed that the overexpression of single R2R3-MYB, AgMYB1, without co-expression of other TFs, can improve anthocyanin production and antioxidant capacity in transgenic plants.
The regulatory mechanism of anthocyanin biosynthesis in higher plants is complicated. Many regulatory genes involved in anthocyanin biosynthesis were identified and genetically mapped in carrot[70, 71]. A previous study reported that DcMYB7, rather than DcMYB6, was responsible for anthocyanin accumulation in purple carrot taproots[72]. So far, AgMYB1 and AgMYB2 were identified from S6 subgroup of R2R3-MYB family, and we have made comparison and analysis in this study. The regulatory mechanism of anthocyanin pathway controlled by AgMYB1 and AgMYB2 in purple celery is still unclear. In future, we will focus on the interaction and regulatory mechanism between AgMYB1 and AgMYB2 and how they co-regulate anthocyanin biosynthesis in purple celery.
-
In this study, we mainly focused on the identification of AgMYB1, an R2R3-MYB TF related to anthocyanin biosynthesis in purple celery. The results indicate that AgMYB1 can promote anthocyanin production and antioxidant activity in transgenic plants. The present study presents useful information for the research of anthocyanin regulation and provides a strategy for cultivating plants with high levels of anthocyanins.
-
This study was financially supported by the Jiangsu Agricultural Science and Technology Innovation Fund [CX(2018)2007], Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD); Postgraduate Research & Practice Innovation Program of Jiangsu Province (KYCX18_0692).
- The authors declare that they have no conflict of interest.
- Additional file 1: Fig. S1 Nucleotide acid and deduced amino acid sequence of AgMYB1 from Apium graveolens.
- Additional file 2: Fig. S2 GUS staining (a) and PCR assay (b) of AgMYB1 with specific primers from cDNA of WT and transgenic Arabidopsis.
- Copyright: © 2021 by the author(s). Exclusive Licensee Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.


|