








-
The genotype-specific patterns of defense activation will critically govern the outcome from interactions between plant and its invading pathogens[1−3]. It is well acknowledged that plant responses to infection from necrotrophic pathogens include multi-phased and multi-layered defense mechanisms[4]. Recently, the molecular defense networks in apple roots activated by infection from a soilborne necrotrophic pathogen Pythium ultimum have been systematically investigated through a series of transcriptome analyses[5−8]. Apple laccase genes were identified as a primary component among the transcriptomic changes during defense activation in apple roots[5,7,8]. The role of laccase directed lignin biosynthesis and cell wall fortification to plant disease resistance has been proposed for several decades in different pathosystems[9,10]. More recently, transcriptomic and proteomic analyses have provided the direct and detailed molecular evidence regarding the functions of laccase genes and lignification processes in relation to genotype-specific resistance in several crops[11−15].
Lignins are heteropolymers covalently associated with polysaccharides in plant cell walls[10,16]. Lignin biosynthesis is the result of oxidative polymerization of three p-hydroxycinnamyl (p-coumaryl, -coniferyl and -sinapyl) alcohols which is mediated by both laccases and peroxidases[17]. The lignification process is crucial for several aspects of plant physiologies including preserving the integrity of the plant cell wall and imparting strength to vascular tissues[17−20]. Additionally, accumulating evidence indicates that lignified cell wall serves as physical barriers against invasions of phytopathogens and other environmental stresses[15,21−23]. As lignification is a non-reversible process, the pathways of monolignol biosynthesis, polymerization and lignin deposition are tightly controlled during development and response to stresses[21,24,25]. For the less investigated pathosystem between apple root and P. ultimum, the contribution of apple laccase encoding genes to root resistance traits remains elusive.
Laccases (EC 1.10.3.2) are glycosylated multi-copper oxidases (MCOs) that serve as electron transfer proteins and catalyze the oxidation of a variety of aromatic and phenolic compounds[26,27]. The presence of cupredoxin-like domains in the sequences of all MCOs, including polyphenol oxidases (PPOs) and ascorbate oxidases, allows oxygen to be reduced to water without producing harmful byproducts[27,28]. Despite their wide taxonomic distribution (bacteria, fungi, plants, and insects) and diversity of substrates, laccases have a common molecular architecture[27,29]. Plant laccase is composed of three cupredoxin domains that include one mononuclear and one trinuclear copper center[24,30]. The small, 10−20 kDa, cupredoxin-like domain possesses relatively simple 3D structures, primarily composed of beta sheets and turns[27]. Plant laccase activity has been implicated in a wide spectrum of biological activities and, in particular, plays a key role in morphogenesis, development and lignin metabolism[20]. Accumulated evidence suggests the roles of laccase-directed lignin deposition for plant resistance[11,13,15,23]. However, the specific genes and their detailed physiological or biochemical roles remain largely unclear, as plants usually express multiple laccase genes in most tissues, and often with overlapping expression profiles and functional redundancy[12].
It has been hypothesized that cell wall lignification at the infection sites, through impeding pathogen penetration, offers time or opportunity for plant cells to mount a more effective defense activation such as biosynthesis of phytoalexin and resistance proteins[10]. The intensity and swiftness of tissue lignification at the early stage of infection could be pivotal in differentiating resistance and susceptibility to infection from a fast-growing necrotrophic pathogen like P. ultimum. Based on our recent transcriptome analyses[5−7], including miRNA profiling and degradome sequencing[8], laccase directed lignification appeared to be an integral part of the defense system in apple root towards P. ultimum infection. The objective of the current study is to advance our knowledge regarding the bioinformatic features of these identified laccase encoding genes, the genotype-specific expressions patterns, as well as lignin deposition patterns in apple root tissues in response to P. ultimum infection. Results from this study will be essential to identify target genes for subsequent functional analysis including transgenic expression manipulation.
-
As part of the effort to elucidate the molecular regulation of apple root resistance to P. ultimum infection, three transcriptome analyses have been performed sequentially[5,7,8]. The laccase encoding genes are among the notable groups in our first transcriptome analysis which was aimed to determine the timeline of transcriptome changes in apple root in response to P. ultimum infection[5,31]. The subsequent comparative transcriptome analysis revealed that several gene models encoding laccases appeared to be differentially induced between a resistant apple rootstock genotype of G.935 and a susceptible genotype of B.9 in response to P. ultimum infection[7]. Among them, four gene models annotated as Arabidopsis laccase 7 homologues exhibited upregulation in response to P. ultimum infection (Fig. 1). Two of them, MDP0000168850 and MDP0000294031 were highly expressed based on the comparison of normalized transcript levels between mock inoculated roots (C for control) and P. ultimum inoculated (denoted as P) at three time points of 24, 48 and 72 h post inoculation (hpi). The expression of MDP0000168850 appeared to be at relatively comparable induction levels between both genotypes. In contrast, the expression of MDP0000294031 demonstrated a genotype-specific pattern, i.e., a higher expression level was specifically observed in the root of a resistant G.935. The consistent upregulation of these laccase genes, particularly the earlier (at 24 hpi), and stronger expression of MDP0000294031 in the resistant G.935, suggested the potential roles of these laccase genes during defense activation in apple root towards P. ultimum infection.
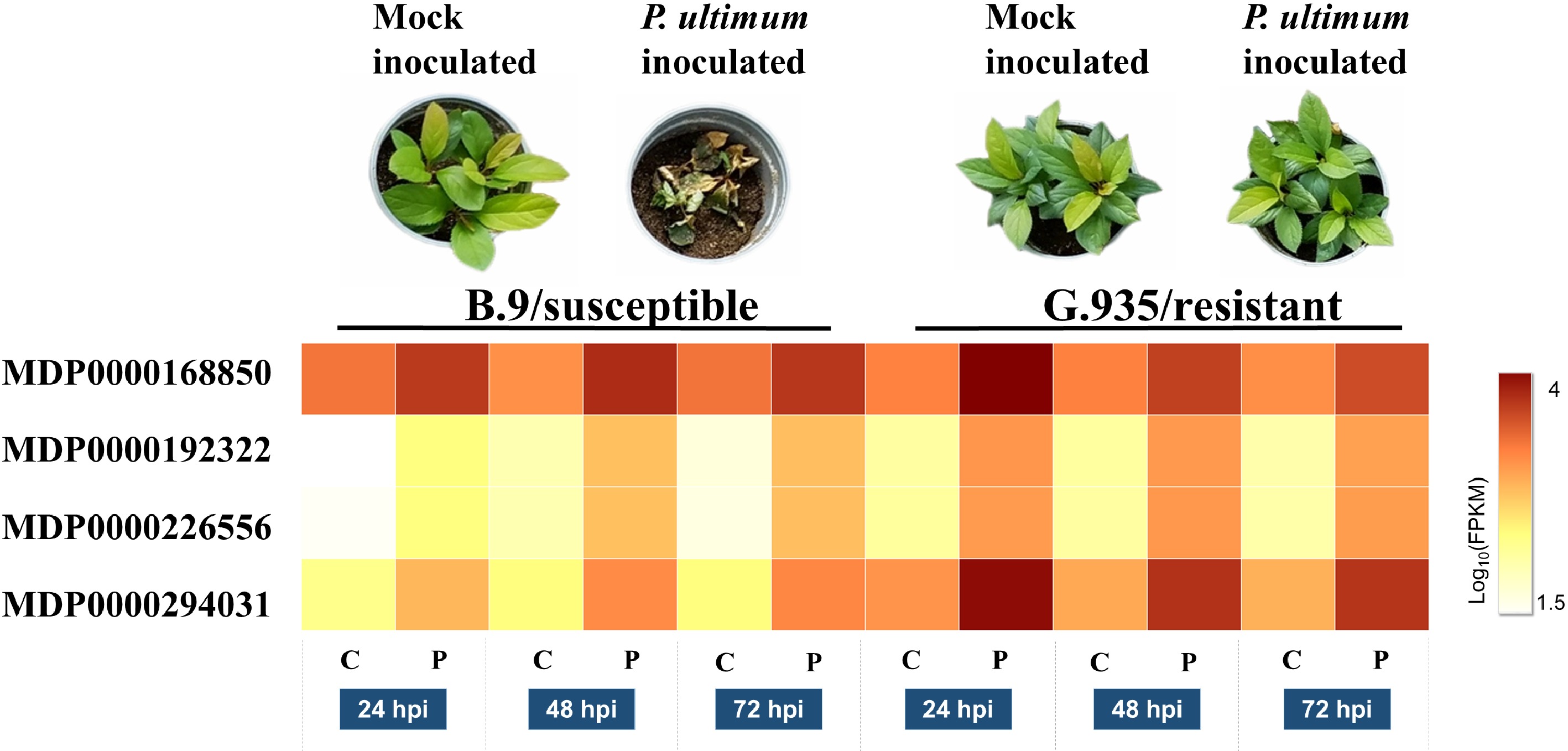
Figure 1. Expression levels of four gene models which encode MdLAC7s in apple root. Images in the top panel shows the resistance phenotypes at 7 dpi for both susceptible B.9 and resistant G.935 apple rootstock genotypes. The heatmap demonstrates the normalized transcript levels between mock inoculated roots (C for control) and P. ultimum inoculated (denoted as P) at three time points of 24, 48 and 72 h post inoculation (hpi) for all four gene models. The levels of expression per genotype/treatment are indicated as the coloration according to legend. The functional annotations of these four genes are according to predicted coding genes of the Malus × domestica Whole Genome v3.0.a1 (www.rosaceae.org/analysis/162).
In a more recent study using microRNA profiling and degradome sequencing, four apple laccase genes were identified as the cleavage targets of a microRNA, i.e., miR397b, between two groups of resistant and susceptible apple rootstock genotypes in response to P. ultimum infection[8]. Firstly, miR397b was shown to be differentially expressed between resistant and susceptible group (three genotypes in each group), with significantly downregulated expression in roots of three resistant genotypes in response to P. ultimum infection. Secondly, the degradome sequencing data indicated that MdLAC-3, -5 and -7 encoding genes are the cleavage targets of miR397b. Thirdly, the cleavage activity of HF27792, or a gene encoding MdLAC7b, exhibited differential cleavage intensity between the resistant and susceptible genotype groups in response to P. ultimum infection (Table 1). The post-transcriptional regulation of laccase gene expression also suggested the potentially critical roles of laccase and root tissue lignification during defense activation to P. ultimum infection.
Table 1. Laccase genes targeted by specific microRNA397b during P. ultimum infection by degradome sequencing and microRNA profiling.
Gene ID Annotation Targeted by Relative cleavage intensity L1 L2 L3 HF40034 MdLAC 3 miR397b 15 N/A N/A HF23917 MdLAC 5 miR397b 2 11 N/A HF26400 MdLAC 7a miR397b 30 N/A N/A HF27792 MdLAC 7b miR397b 45 N/A 82 The IDs of these target gene are based on the Malus x domestica HFTH1 Whole Genome v1.0 of an anther-derived homozygous line HFTH1 (www.rosaceae.org/species/malus_x_domestica_HFTH1/genome_v1.0). Relative cleavage intensity is referred to the detected tag abundance in three degradome libraries, where L1 (or library #1) represents the data from the degradome library constructed from the pooled RNA samples of mock inoculation (of both resistant and susceptible genotypes), L2 (or library #2) represents the data from the degradome library constructed from the pooled RNA samples of P. ultimum inoculated root tissues from three resistant genotypes, L3 (or library #3) represents data from the degradome library constructed from the pooled RNA samples of P. ultimum inoculated root tissues of susceptible genotypes.
Bioinformatic characteristics of these induced laccase genes in apple roots
-
The sequence features for these root-expressed members of the laccase gene family, including their intron/exon numbers, domain compositions, cis-elements in the promoter region, and their genomic locations are summarized in Fig. 2. Except MdLAC7a, these laccase-encoding genes are about 2,300−2,500 bp in length and contain 5−6 exons (Fig. 2a). Each gene was predicted to contain three conserved Cu oxidase domains (Fig. 2b). As an exception, MdLAC7a (HF26400) has an additional long exon, which is predicted to encode a PNGaseA domain (Fig. 2a & b ). At the promoter region (2 Kb region before the starting codon) various binding sites were detected, which suggest the putative regulatory roles of various hormones (ABA, JA, and Auxin) and transcription factors (MYB, MYC and WRKY) on the expression of these laccase genes (Fig. 2c). Each of these laccase encoding genes is located on a different chromosome (Fig. 2d).
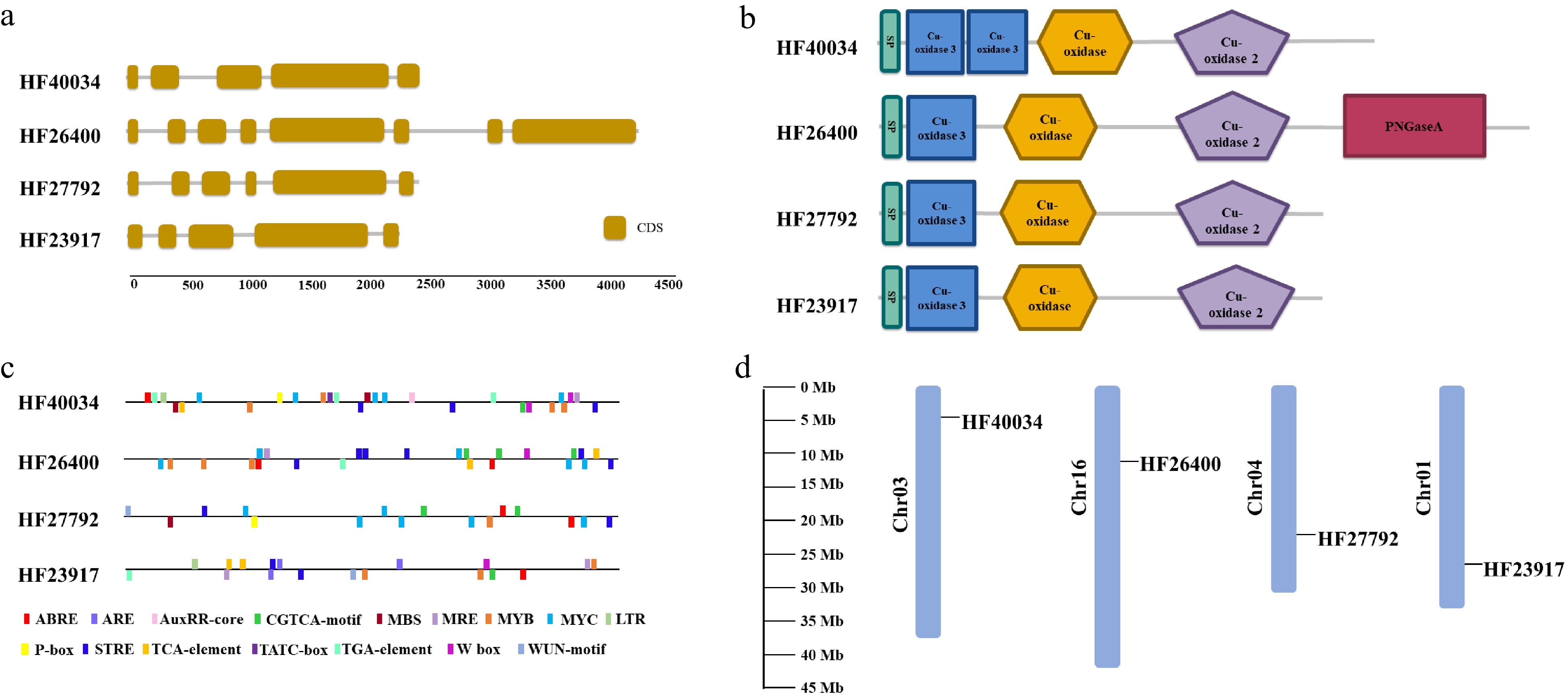
Figure 2. Bioinformatic characteristics of root-expressed apple laccase genes. (a) Numbers and position of introns and exons for each gene. (b) Domain composition contained in their predicted amino acid sequences. (c) Cis elements within 2 Kb of promoter sequences. (d) Location of these laccase encoding genes on apple chromosomes.
The genotype-specific expression patterns during P. ultimum infection
-
The genotype-specific expression patterns of these laccase-encoding genes were examined between a resistant O3R5-#161 and a susceptible O3R5-#132 during P. ultimum infection. Both MdLAC3 and MdLAC5 showed a slight induction and mostly at 48 hpi. The expression of MdLAC7a was upregulated in both genotypes with a relatively similar intensity. Noticeably, MdLAC7b exhibited a differential expression pattern between these two genotypes (Fig. 3). Specifically, a more consistent and stronger expression was observed in the roots of a resistant genotype O3R5-#161, as it was compared with that in the susceptible genotype O3R5-#132. The elevated expression for both MdLAC3 and MdLAC5 in control tissues (C0 and C00), or before exposure to P. ultimum, possibly indicated that these two genes are more responsive to abiotic stress conditions such as transplant processes or mechanical handling during the infection process.
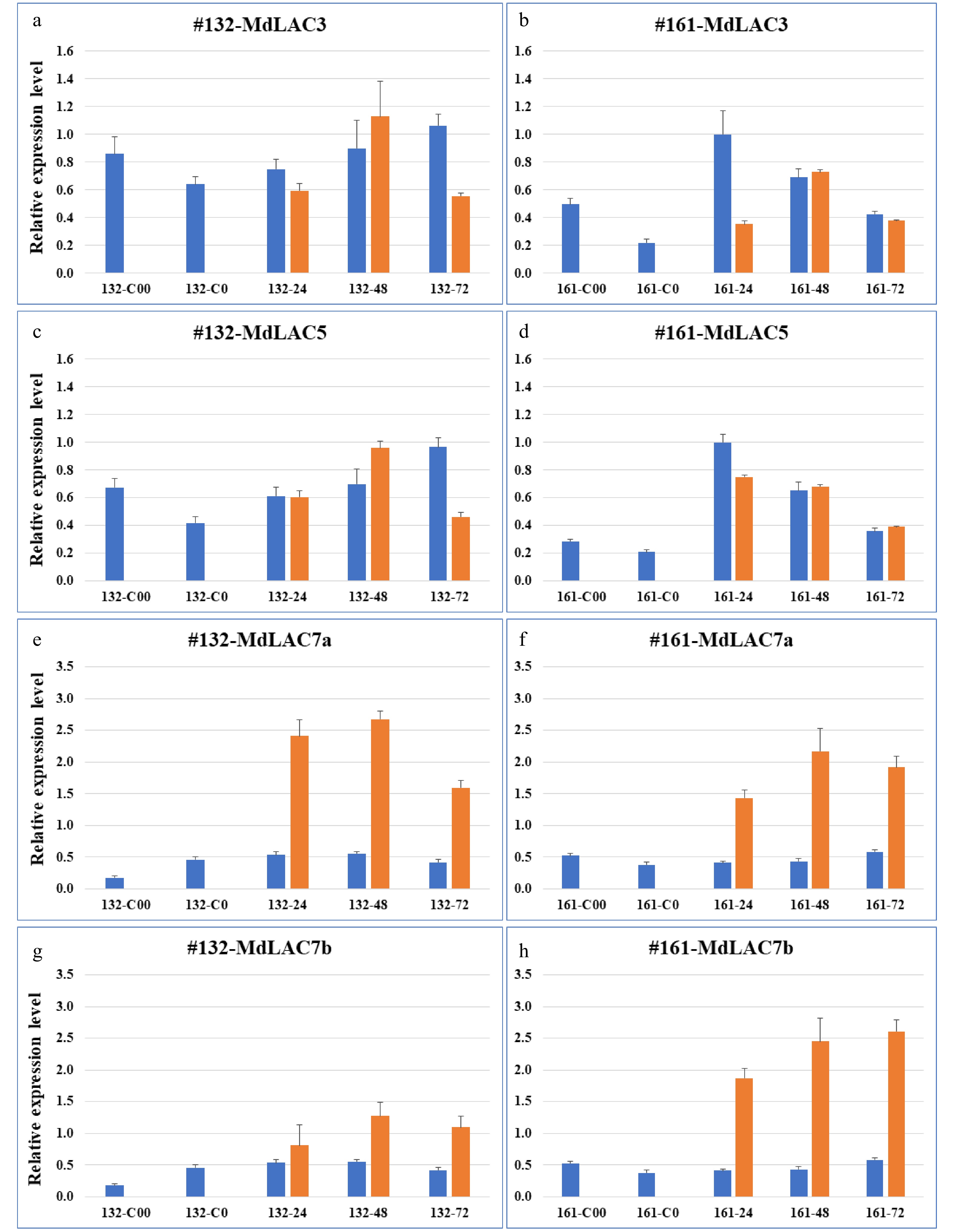
Figure 3. Expression patterns of four apple laccase genes in roots of two apple rootstock genotypes. The left panels, (a, c, e & g) denote the expression of MdLAC3, MdLAC5, MdLAC7a and MdLAC7b, respectively, in root tissue of a susceptible O3R5-#132; The right panels, (b, d, f & h) denote the expression of MdLAC3, MdLAC5, MdLAC7a and MdLAC7b, respectively, in root tissue of a resistant O3R5-#161. Blue bars represent expression level in mock-inoculated control tissues; and orange bars represent expression levels in P. ultimum-infected root tissues. Values on the Y-axis denote the relative expression level, using value of control tissue for MdLAC3 at 161-24 hpi as a calibrator. Labels on the X-axis indicate the tissue collection including five time points for control tissues and three timepoints after P. ultimum inoculation. For each genotype, C00 denotes the root tissues in culture medium (or two weeks before inoculation), and C0 denotes root tissue after one-week in-soil acclimation or one week before mock inoculation. The numbers of 24, 48 and 72 indicate the timepoints of hours post (mock- or P. ultimum) inoculation (hpi). Values represents the averages and sd of three technical repeats of qRT-PCR analyses for each of the two biological replicates.
Sequence features, microRNA cleavage site and three-dimensional structure of MdLAC7b
-
The differential response of MdLAC7b between resistant O3R5-#161 and susceptible O3R5-#132 added evidence for its roles contributing to apple root resistance to P. ultimum infection. The specifics of amino acid sequences, its known post-transcriptional regulation by microRNA397b mediated cleavage, and the predicated three-dimensional folding pattern of its polypeptides as well as copper ion binding sites were further investigated. A signal peptide at the N-terminus was identified indicating MdLAC7b is a secreted protein like many other plant laccases[24,27]. Three cu-oxidase domains (highlighted in blue, yellow and green, respectively) were predicted, as well as 11 asparagines (N, in red) predicted to be the sites for N-glycosylation, which are responsible for copper retention, enzyme stability and activity (Fig. 4a). The site where the miR397b attacks was located at the 5' region of the large exon of MdLAC7b based on the data from a recent degradome sequencing analysis[8] (Fig. 4b). The folding pattern of a three-dimensional structure of MdLAC7b and the predicated ligands of three copper ions was predicted using Discovery studio 4.1 software (Fig. 4c). Cu1 is coordinated with two histidines, one cysteine and one leucine, Cu2 is coordinated by another two histidines and one H2O ligand, while six histidines coordinate the Cu3 pair in a symmetrical manner, with a bridging OH ligand.
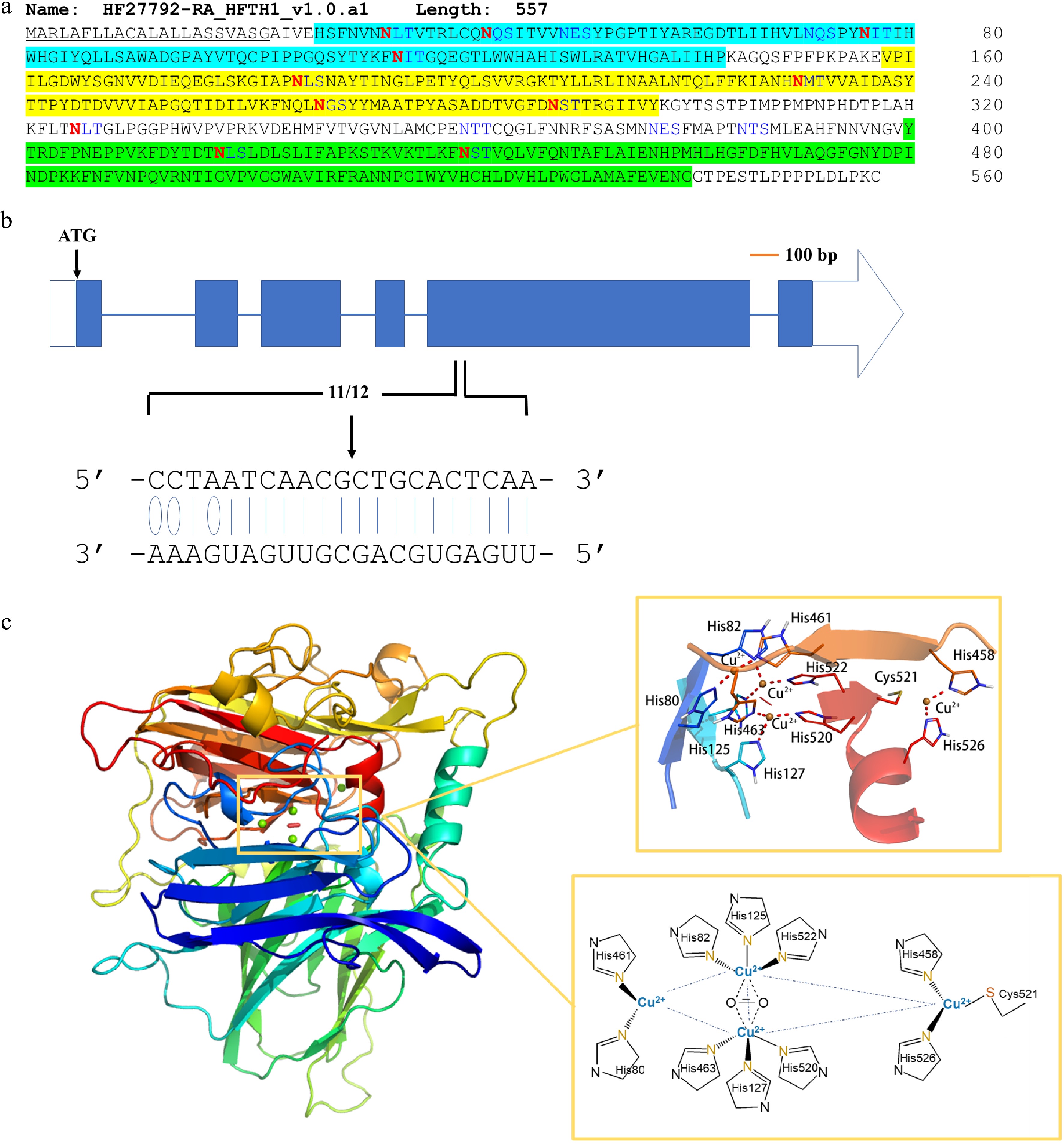
Figure 4. Sequence features and structure modeling of MdLAC7b. (a) The amino acid sequence of MdLAC7b. The protein consists of 557 amino acids. It has a signal peptide (underlined) at the N-terminal predicted by SignalP 4.1 Server, and three conserved Cu-oxidase domains were identified (highlighted in blue, yellow and green, respectively) according to Pfam. Twelve asparagines predicted to be N-glycosylated by NetNGlyc 1.0 Server were indicated in red. (b) The specificity of MiR397b mediated cleavage of MdLAC7b transcript was revealed by degradome sequencing in a previous study[8]. The blue boxes represent exons, and the horizontal lines represent introns. The white box represents 5′-UTR, while the white arrow represents 3′-UTR. Solid lines indicate the Watson–Crick pairing and the oval indicates G:U wobble pairing between MdLAC7b target sequence and the complementary miRNA397b sequence. (c) Left panel, three-dimensional structure of MdLAC7b predicted using Discovery studio 4.1 software. Upper right panel, the view of ligands at the copper center of MdLAC7b. Lower right panel, Cu1 is coordinated with two histidines, one cysteine and one leucine, Cu2 is coordinated by another two histidines and one H2O ligand, while six histidines coordinate the Cu3 pair in a symmetrical manner, with a bridging OH ligand.
Lignin deposition patterns between resistant and susceptible genotypes and in response to infection
-
The patterns of lignin deposition in apple root tissues, before and after pathogen infection, were examined using Wiesner's staining on hand-sectioned root tissues. Two apple rootstock genotypes of O3R5-#161 and O3R5-#132, which are known to be resistant or susceptible to P. ultimum infection, respectively[32]. Both genotypes demonstrated the easily detectable lignin staining in vascular tissues even before pathogen exposure (Fig. 5a & c). Under pathogenic pressure, the enhanced lignification in infected root tissues was observed for both genotypes (Fig. 5b & d). Furthermore, several spots can be easily identified at the outer layer of the root cortex tissue of the resistant genotype #161, which appeared to be enhanced tissue lignification (Fig. 5d, arrows). Specific root sections at a similar position or the distance from root tips, were selected for sectioning for better comparability across genotypes and treatments.
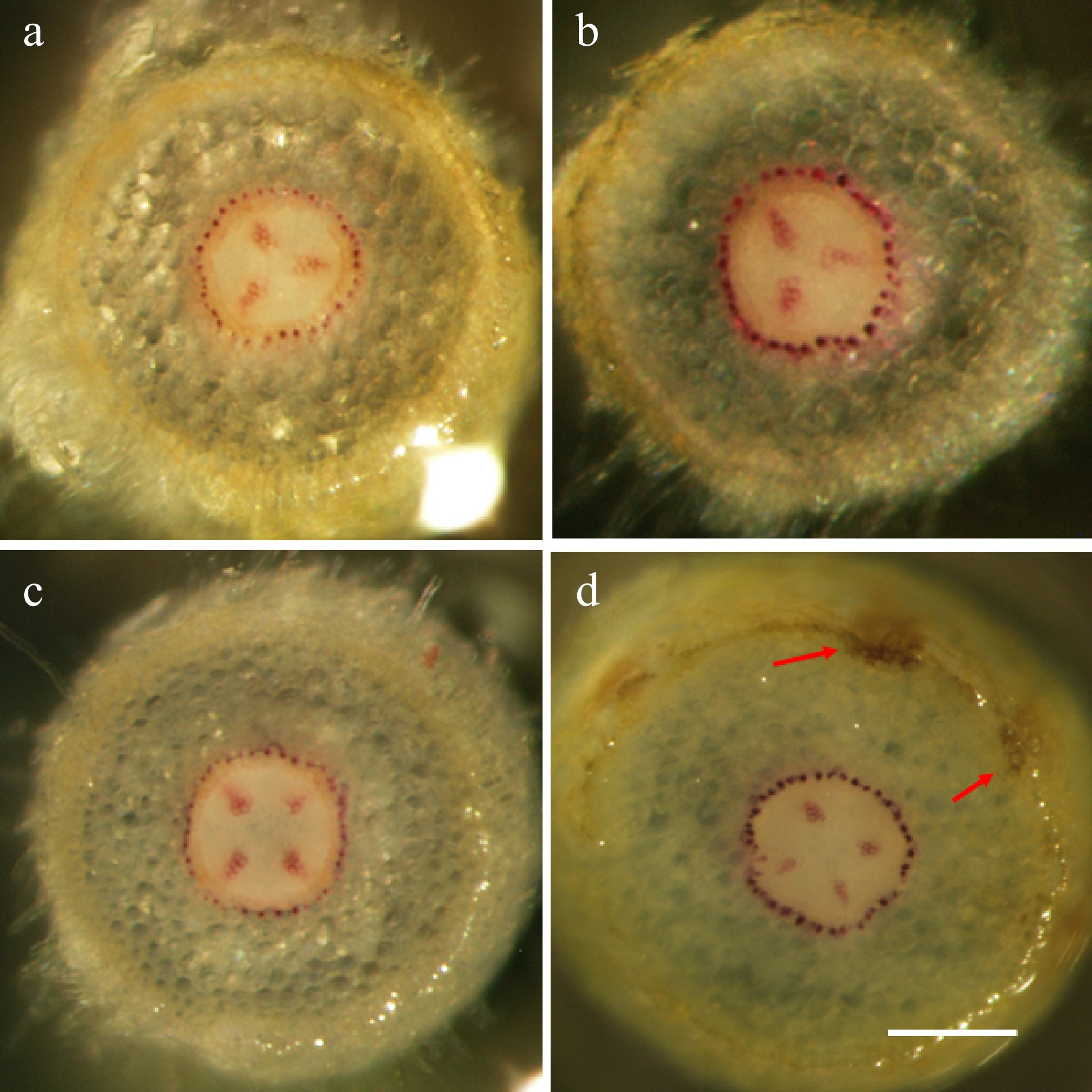
Figure 5. The genotype-specific patterns of lignification in apple root tissues. Wiesner staining of hand-sectioned root tissue was carried out at 72 hpi on selected root branches from both treatments (mock-inoculation and P. ultimum infection) and for both genotypes. (a) The representative image from a susceptible cultivar O3R5-#132 with mock-inoculation; (b) The representative image from a susceptible cultivar O3R5-#132 after P. ultimum inoculation; (c) The representative image from a resistant cultivar O3R5-#161 with mock-inoculation; (d) The representative image from a resistant cultivar O3R5-#161 after P. ultimum inoculation. Root segments at a similar position, or equal distance from the root tip, were selected for sectioning for a better comparison between treatments and genotypes. The bar at the lower part of panel (d) represents a length of 100 µm.
-
Plants are known to be equipped with an array of defense mechanisms to protect themselves from infection[4,33−35], however, a unique combination of these mechanisms are activated in a specific organ or tissue type to the infection even from the same pathogen. Among various biochemical pathways and cellular processes within the defense networks in apple root, which were revealed by three transcriptome analyses, expression regulation of laccase genes and perhaps the lignification of root tissues appeared to be a focal point. Their differential induction between resistant and susceptible apple rootstock genotypes likely play a key role of defense activation at the early stages of P. ultimum infection. Large-scale and high-throughput genomic approaches, such as transcriptome analyses, offer the advantage of generating a global view over the molecular defense responses in apple root[5,7,8]. However, the data from such approaches are potentially error-prone in terms of gene identity and its verifiable expression pattern. This is particularly necessary for those species with more complex genomes such as apple[36,37].
The results from the current study demonstrated that these laccase genes contain the conserved cu-oxidase domains as well as the typical laccase gene structure, and they are located on different chromosomes. Their distinct expression patterns may be attributed to the various cis elements at the promoter regions and/or post-transcriptional regulation schemes such as selective cleavage by microRNAs. Genotype-specific expression patterns suggested that MdLAC7b likely play a critical role contributing to the resistance trait. The sequence features and three-dimensional modeling of MdLAC7b, based on its predicted protein sequences, added details related to its proposed functions such as its trafficking to the apoplast, Cu ion binding sites and cleavage site by miR397b. The genotype-specific lignin deposition patterns added additional experimental evidence potentially connecting the laccase directed lignification process with apple root resistance traits to P. ultimum infection.
It can be argued that a quicker and more efficient lignin deposition at the early stage of infection is crucial to impede or slow-down the initial progression of the fast-growing P. ultimum. Such a passive barrier may physically block or delay the disruptive effect of toxins, enzymes and/or effector from this pathogen. Even with such a basal defense strategy, a more robust cellular defense activation can be fully developed for ultimately defeating the pathogen invasion. On the other hand, laccase directed lignin deposition likely only functions as one of the many components in a comprehensive and complex defense system. The effective resistance or tolerance to infection from a necrotrophic pathogen like P. ultimum relies on several factors functioning sequentially, additively, or synergistically to achieve the optimized defense output[4,5,7,38,39]. For example, the metabolites from the phenylpropanoid pathway possesses at least two major roles in host resistance to necrotrophic pathogens, i.e., lignin deposition and phytoalexin accumulation. The biosynthesis of monolignols, the precursor for lignin formation, is part of the phenylpropanoid pathway with a direct impact on the lignification process[20,40]. As observed in many other pathosystems, activation of the phenylpropanoid pathway is one of the most notable transcriptomic changes upon P. ultimum infection in apple roots[5,7,8]. In our previous transcriptome datasets, genes encoding biosynthesis enzymes in several key steps of the phenylpropanoid pathway, such as PAL, CHS and CAD, were almost uniformly upregulated in apple roots upon P. ultimum infection[5,7]. Additionally, several families of TFs (transcription factors) such as MYBs NACs and MYCs, as well as transporter-encoding genes such as those for ABC transporter or MATE family members, showed a quicker, stronger and more consistent upregulation in the root of a resistant apple rootstock genotype as compared with that in susceptible genotypes[7,8]. Therefore, the variation at the biosynthesis and transport of monolignols may also contribute to the lignification process, in addition to the elevated upregulation of laccase gene expression per se.
The preliminary data from the current study is consistent with the notion that apple laccase genes and the lignin biosynthesis are an important part of the defense network in apple root towards P. ultimum infection. Further studies are needed to substantiate the identity of MdLAC7b as a primary candidate which may contribute to the variable resistance responses between apple rootstock genotypes. Many questions need to be answered related to the detailed functional roles of MdLAC7b to apple root defense activation. What is the critical timeline of lignin deposition in infected root tissues? Is MdLAC7b acting alone or is it a limiting factor to form a functional enzyme complex? How do other regulatory points in the phenylpropanoid pathway such as TFs (MYB, MYC, TCP…) affect the lignification process during P. ultimum infection? Does the relationship between the observed resistance response and lignin deposition apply to other apple rootstock genotypes (beyond O3R5 genetic background)? What is the potential connection between the observed intensity at the lignified vascular tissue and the over-flowed diffusible phenolic compounds or phytoalexins? Maybe the more relevant question is: how does lignin deposition at the initial phase of infection facilitate the effective defeat of the pathogenic arsenal? Further experimental evidence from carefully designed experiments is required for a better understanding on the roles of laccase directed lignification during defense activation to P. ultimum infection. Among them, transgenic manipulation of MdLAC7b expression and associated assays to measure the changes at biochemical and enzymatic levels should generate more definitive answers to these questions.
-
Tissue culture based micro-propagation procedures were used to obtain individual apple plants for infection assays and tissue collection as described previously[31]. Both B.9 and G.935 are widely used commercial apple rootstock genotypes, while O3R5-#161 and O3R5-#132 apple rootstock genotypes are the progenies from a rootstock cross population between 'Ottawa 3' and 'Robusta 5' (O3R5). A synchronized micro-propagation process was carried out to generate apple plants with non-contaminated root tissues and equivalent developmental stages for both genotypes. Four weeks after root induction in tissue culture medium was followed by 'in-soil' acclimation for one week in a growth chamber allowing further root tissues differentiation before P. ultimum inoculation. To minimize transplanting shock from tissue culture medium to soil conditions, a transparent 7'' Vented Humidity Dome (Greenhouse Megastore, Danville, IL) was placed on top of a 10 × 20-inch flat tray holding the pots for retaining humidity. An identical watering schedule of every other day was applied to both plant genotypes and treatments, i.e., mock inoculation and P. ultimum inoculation.
Inoculum preparation, infection procedure and root tissue collection
-
The P. ultimum isolate used in this study originates from the roots of 'Gala'/M26 apple grown in Moxee, WA, USA[41]. The procedures of inoculum preparation, quantification and root-dip inoculation were as described previously[31]. Mock-inoculated control plants and P. ultimum inoculated plants were transplanted in individual 4" diameter pot and placed in separate trays. Root tissues from mock inoculated and P. ultimum inoculated plants for both genotypes were sampled at designated timepoints according to experimental design. For hand-sectioning, apple roots were carefully excavated from soil, rinsed under running tap water, and floated in tap water until root branches were selected for sectioning. For gene expression analysis, root tissues were separated from aboveground tissues and flash frozen using liquid nitrogen and stored at −80 °C until RNA isolation. Pooled root tissues were collected from at least three individual plants per genotypes and/or treatments.
Total RNA isolation and high-throughput mRNA sequencing
-
Total RNA isolation was carried out following the lithium-chlorite method previously described by Zhu et al.[31]. Root tissues of both resistant O3R5-#161 and susceptible O3R5-#132 were represented by two biological replicates, and each replicate included the pooled root tissues from three plants. RNA quantity was determined using a Nanodrop spectrophotometer (ND-1000; Thermo Fisher Scientific) and RNA integrity was confirmed by RNA gel.
Identification of candidate laccase genes and their bioinformatic analyses
-
The candidate apple laccase (MdLAC) genes were selected based on the analyses of previous transcriptome datasets. The raw RNA-Seq data were deposited in the public domain with the accession numbers SRP117760 and SRP295189. The genomic sequences, the coding region, and the predicted amino acid sequences of these four laccase genes were downloaded from GDR (www.rosaceae.org), which hosts the apple genome sequences. SMART (http://smart.embl-heidelberg.de) was used to identify the conserved domains for selected apple laccase genes[42]. Gene structures were extracted using TBtools[43]. The protein sequence of HF27792 was used as a query to blast against NCBI, and the Zea mays Laccase 3 (PDB ID: 6KLG) was used as a template, as it shared the highest sequence identity (46.08%) with HF27792. Sequence alignment was performed between HF27792 and 6KLG through Align Sequence to Templates module of Discovery Studio 4.1 software to obtain three-dimensional spatial structure of HF27792 encoded protein. Since the target protein and the template protein were quite conserved in the central Cu2+ binding region, the Cu2+ and oxygen molecules in the template were placed into the corresponding positions of the target structure. Two methods, Ramachandran Plot and Profiles-3D, from Discovery studio 4.1 software were used to evaluate the reliability of the model.
Genotype-specific expression patterns of selected apple laccase genes by RT-qPCR
-
The total RNA was treated with DNase I (Qiagen, Valencia, CA) and then purified with RNeasy cleanup columns (Qiagen, Valencia, CA). Two micrograms of DNase-treated RNA was used to synthesize first-strand cDNA using SuperScriptTM II reverse transcriptase (Invitrogen, Grand Island, NY) and poly dT (Operon, Huntsville, AL) as the primer. The RT-qPCR procedure was performed as previously reported[7]. The target gene expression was normalized to that of a previously validated internal reference gene (MD02G1221400) specific for gene expression analysis in apple roots[44] using the 2−∆∆Cᴛ method (the comparative Ct method)[45]. Primer sequences for laccase genes and internal reference gene for qRT-PCR analysis are listed in Table 2.
Table 2. Primer sequences of laccase and reference genes for qRT-PCR analysis.
Gene IDs Gene description Primers F/R [5'-3'] HF40034 Laccase-3 F-5' CAACCCCAGAACAGATCCAG 3'
R-5' AAACCCAGGAAGAGATGTGC 3'HF23917 Laccase-5 F-5' TGGGCAGTCATTCGATTTGT 3'
R-5' AACAAAGAGGCAGATCCACC 3'HF26400 Laccase-7a F-5' TAATCCGCAAGTACGCAACA 3'
R-5' CAGATCAAGTGGTGGTGGAG 3'HF27792 Laccase-7b 5'- TCCTACACGACTCCTTATGAT -3'
5'- GAGATTGGTGAGGAACTTATGG -3'MD02G1221400 Reference gene F-5' ATGGAGAGATGGAATGGCAAAG 3'
R-5' GTGAGCATCGGATCCCATTTAG 3'Lignin deposition patterns by Wiesner staining
-
The Wiesner staining method[46] on hand-sectioned apple root tissues was used to detect the lignin deposition patterns between genotypes, and before and after the exposure to P. ultimum. At least three plants were used per genotype and treatment. For comparability between genotypes and treatments, the root segments at a similar position (or distance from the root tips), preassembly at the equivalent developmental stages were selected for tissue sectioning.
- This work was supported by the USDA-ARS base fund. The authors would like to thank Soon-Li Teh and Amanda Roelant for language editing and reviewing the manuscript.
- The authors declare that they have no conflict of interest.
- Copyright: © 2021 by the author(s). Exclusive Licensee Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Zhu Y, Zhou Z. 2021. The genotype-specific laccase gene expression and lignin deposition patterns in apple root during Pythium ultimum infection. Fruit Research 1: 12 doi: 10.48130/FruRes-2021-0012

The genotype-specific laccase gene expression and lignin deposition patterns in apple root during Pythium ultimum infection
- Received: 01 October 2021
- Accepted: 10 November 2021
- Published online: 23 November 2021
Abstract: Plant resistance responses against invading pathogens require a coordinated set of cellular processes to optimize the effective defense output. Previous transcriptome analyses have identified a multi-phase and multi-layered defense strategy in apple root towards infection from a necrotrophic oomycete pathogen Pythium ultimum. Among the identified apple genes, members of the laccase gene family represent an actively regulated group at both transcriptional and posttranscriptional levels. In this study, several apple laccase genes were selected for further analyses on their sequence features and potential roles during defense activation in apple roots. Their bioinformatic specifics, genotype-specific induction, and lignin deposition patterns during pathogen infection were examined between two apple rootstock genotypes, a resistant O3R5-#161 and a susceptible O3R5-#132. The sequences these laccase genes contain the conserved cu-oxidase domains and the characteristic gene structures with MdLAC7a as an exception. While MdLAC3 and MdLAC5 showed a partial induction to P. ultimum infection, both MdLAC7a and MdLAC7b genes demonstrated consistent and high-level inducibility. Moreover, MdLAC7b exhibited a differential expression pattern, with a higher expression in the resistant O3R5-#161. Lignin deposition appeared to be stronger in the infected root of the resistant genotype compared to that of the susceptible one. The efficient lignin biosynthesis and deposition at the initial stage of infection is crucial for impeding the progression of this fast-growing necrotrophic pathogen. Future study regarding the role of MdLAC7b, including the transgenic manipulation and biochemical analysis, should provide more definitive evidence for its contribution to resistance to P. ultimum infection.
-
Key words:
- Apple root /
- Defense response /
- Laccase genes /
- Tissue lignification /
- Replant disease /
- Pythium ultimum