








-
In contemporary China, accelerated urbanization has prompted a large number of rural populations to migrate to cities, resulting in a shortage of agricultural labor. This conflict inevitably leads to increased agricultural labor cost. Objectively, it has also accelerated the transition of double-cropping rice to single-cropping rice or mid-season rice production, especially in southeastern China. In addition, the poor quality of early rice, weak market competitiveness and low comparative efficiency result in a reduction in rice planting area, which incurs a threat to regional food security[1]. Therefore, the local governments in southeastern China are adopting a low-stubble mechanized rice ratooning technology in an attempt to deal with this passive situation. The technology for rice ratooning can be used to increase the rice multiple cropping index for coping with insufficient arable land, which is therefore considered to be environmentally friendly, simplified, convenient, labor-saving and highly efficient. Furthermore, ratooning rice can produce a higher grain quality compared with its main crop, and the same is true in the case when it was compared with its counterpart synchronizing in heading time (late season)[2]. Therefore, in recent years, rice ratooning has been rekindled to develop rapidly in southeast China[3, 4], and the low left stubble cultivation technology for rice ratooning has been developed, by which the planting areas of ratoon rice are continuously expanding, and the mean yields of ratooning rice was more than 4,500 kg·hm−2, of which the highest output was 6,750 kg·hm−2, the total grain yield of the main and ratoon rice crops was more than 15 t·hm−2, which slightly corresponds with the total yield of double cropping rice[5]. In recent years, a large number of new rice varieties with high regenerative ability have been developed and released, making the technology scale up quickly[2]. Noteworthy, the fertilization pattern of low cut stubble was different from that of high cut stubble in rice ratooning[1]. In the case of rice ratooning under the low stubble height regime, a proper application rate of fertilizer was used to boost root vigor before harvesting the main crop. However, if a high dose of fertilizer application was used, it would inevitably accelerate the spout and elongation of axillary buds in this time. This in turn results in cut damage to the axillary buds after mechanically harvesting the main crop, especially under low cut stubbles. We also found that inappropriate application of bud-promoting fertilizer could also lead to significantly increased greenhouse gases, especially N2O emission, decreased nitrogen utilization efficiency, and affected rice quality[6]. Therefore, suitable fertilization management, especially at the late growing stage of the main crop, is the key to determining high yield and good quality of the main crop and ratooning rice from mechanically low cut stubble[1,4]. Several scholars have documented that under appropriate total nitrogen supplies, a reasonable nitrogen application method for the main crop not only could increase its output, but also has a good carry-over effect on subsequent ratooning rice[2]. However, the long-run large-scale application of chemical fertilizers often causes soil compaction and acidification, and hence decreases available nutrients and organic matter content, in turn declines nitrogen fertilizer utilization and increases farmland non-point source pollution. Moreover, long-run uses of sole inorganic nitrogen fertilizer not only increase the concern about agricultural environmental problems, but also often boost overgrowth of the main crop in the early growth stage, thus increasing ineffective tillers. Meanwhile, it often results in insufficient nutrient supplies in the later growth period due to the rapid release characteristics of chemical fertilizer, consequently triggering premature senescence and hence declining yield of the main crop, thereby producing negative carry-over effect on the regeneration rate and yield of the regenerated rice. Previous studies mostly focused on farmland productivity, nutrient absorption and soil fertility, but ignored the effects of combined organic-inorganic fertilizer with an appropriate proportion on plant nutrient absorption and utilization efficiency at the grain-filling and ripening stage in the main crop and its carrying-over effect on ratooning rice from low cut stubbles. It therefore has been suggested that properly combined application of organic and inorganic fertilizers is a fertilization system for rationally using natural resources to improve soil fertility, and maintain high and stable crop yields based on the quick-acting properties of chemical fertilizers and the better durability of organic fertilizers in the nitrogen mixture supplies[7−13]. The current research focal point was how to apply this principle as mentioned above to make a reasonable configuration of organic and inorganic fertilizers to improve soil fertility for balancing the physiological demands of the main and its ratooning rice crops in early and late growth stages, and hence to increase nutrient utilization and realize the targets for one planting with two higher harvests in rice ratooning practice. This experiment of combined organic/inorganic fertilizers was conducted using different substitutions of chemical nitrogen with organic nitrogen according to equivalent nitrogen amounts to further study the effects of different combined organic and inorganic fertilizer on the physiological trails and yield performance of the main and ratooning rice crops. This was to clarify the physio-ecological action mode of the organic nitrogen in replacement of inorganic nitrogen, and hence to recommend the most optimal configuration with organic and inorganic nitrogen fertilizers for providing a scientific basis to establish a cost effective fertilization system with best nutrient efficiency, uniform population, stable and high grain yield for rice ratooning in the southeast China.
-
A three-line indica hybrid rice cultivar Yongyou 1540, widely popularized in southeast China, was used as the experimental material. The topsoil (0–20 cm layer) of the experimental field had a clay loam texture with the following properties: 5.22 pH, 25.38 g∙kg−1 organic matter, 1.67 g∙kg−1 total nitrogen, 0.73 g∙kg−1 total phosphorus, 7.37 g∙kg−1 alkalin hydrolysis nitrogen, 167.91 mg∙kg−1 total potassium, 19.83 mg∙kg−1 available phosphorus and 101.64 mg∙kg−1 available potassium, respectively. The nutrient content of commercial organic fertilizer was 53.51 g∙kg−1 organic matter, 8.01 g∙kg−1 total nitrogen, 1.57 g∙kg−1 total phosphorus and 1.94 g∙kg−1 total potassium.
Experimental design
-
The experiment was carried out in the ratooning rice base in Chongluo Township, Jianyang District, N 27°35′4.68″, E 118°21′50.88, Nanping City, Fujian Province, Southeast China from March to November in 2018 and 2019, respectively. In the design of the combination treatment with different proportions of inorganic and organic nitrogen, we applied total nitrogen 225 kg∙hm−2, and based on actual nitrogen contents in inorganic and organic fertilizers, eight combinations of the organic and inorganic fertilizers were set, namely: no fertilizer (CK) used in the whole season, chemical fertilizer application only (CF), 85% inorganic nitrogen + 15% organic nitrogen (GM1), 70% inorganic nitrogen + 30% organic nitrogen (GM2), 55% inorganic nitrogen + 45% organic nitrogen (GM3), 40% inorganic nitrogen + 60% organic nitrogen (GM4), 25% inorganic nitrogen + 75% organic nitrogen (GM5), and 100% organic fertilizer (GM6). Moreover, organic nitrogen was applied as basal fertilizer based on the design in each combined nitrogen fertilizer treatment, and the chemical nitrogen fertilizer was applied as topdressing at the ratios of 3:1:1:4 at transplanting, early tillering, middle tillering and panicle formation stages of the main crop. Fertilizer application at tillering stage of the main crop was split into two time points, 7 d after transplanting (early tillering stage) and 15 d after transplanting (middle tillering stage). The same application rate of fertilizers for ratooning rice was applied in the second cropping season. The amounts of total nitrogen applied in chemical fertilizers were 187.5 kg∙hm−2, of which 10% nitrogen fertilizer for root-vigor preserving and bud survival promoting was applied at 25 d after the fully heading stage of the main crop (Yongyou154, hybrid rice), and the rest of the nitrogen fertilizer was used twice for bud and tillering promotion at 3 and 9 d after the main crop was harvested. The application ratio of the two time fertilizer amounts was 1:1. The ratios of N : P5O2 : K2O were kept at 1:0.8:1 in the second season for ratooning rice. The area of each experimental plot was 100 m2, and the plots were arranged in completely randomized block design (RCBD) with three repeats. Each plot was isolated by a ridge covered with black waterproof mulch film. The height of the ridge was 40 cm and the width is 50 cm, and irrigation and drainage of each plot were independent. In 2018, the seedlings of the main season rice crop were raised on March 12, transplanted on April 14, fully headed on July 21, and matured on August 15; the ratooning season rice fully headed on September 23 and matured on October 26. In addition, in 2019, the same experiment and tests were carried out in the same plot as that in the previous year, i.e., the main season rice crop was sown on March 15, transplanted on April 10, fully headed on July 24, and matured on August 21, and its ratooning season rice fully headed on October 1 and matured on November 3, 2019. Rice transplanting and its harvest were conducted by combining the planting density of 30 cm × 17 cm, and 2−4 plants per hill. The cutting height of the main crop was 25 cm.
Determination items and methods
Root activity of rice plants
-
The sap amounts of rice plants were collected at the active tillering stage (AT),booting (BS), fully heading stage (HS), the day close to the stage fertilizing for root-vigor preserving and bud promoting (CS), and ripening stages (RS) of the main season crop, as well as fully heading (RHS) and ripening stages (RRS) of ratooning season rice by using the cotton absorption method described by Huang et al.[5]. Briefly, approximately 15 g absorbent cottons were placed in a ziplock bag, then marked and weighed and recorded as W1. Three representative uniform plants with the same number of tillers or effective panicles were randomly selected and sampled in each treatment, and the number of tillers per hill were recorded as A. The rice plant was then cut off at 10 cm from the ground and the wound stubbles were covered completely with the weighed cotton and wrapped tightly with the corresponding ziplock bag. Sap collection was performed from 6 pm to 6 am the following day. Afterwards, the cotton and the ziplock bags were weighed and recorded as W2. The bleeding sap intensity (SI) was calculated in a single stem based on the following formula:
$ (SI)(mg\cdot h^{-1})= [W_{2}-W_{1}]\cdot A^{-1}\cdot 12^{-1 } $ Nitrogen content and its utilization efficiency in rice plants
-
The sampled plant parts were weighed, ground into powder and passed through a 0.25 mm sieve, then the powder samples were put into the ziplock bag, then marked and saved at room temperature until used. The concentrated sulfuric acid-hydrogen peroxide digestion method was used to prepare the test solution of total nitrogen, phosphorus and potassium of the rice plants. 0.25 g of the rice plant sample powders were added into the cleaned and dried digestion tube, and marked, 5 ml of concentrated sulfuric acid was then added to each tube, the digestion tube was then put into the digestion furnace and the temperature adjusted to 265 °C. It was then heated for 15 min; after preheating, the temperature was adjusted to 365 °C, the digestion tube was then removed and cooled to room temperature after digestion for 45 min, 4 ml of hydrogen peroxide solution was then added for the first time. The digestion tube was then returned to the digestion furnace to digest after 30 min, in turn the tube was cooled, thereafter, 2 ml of hydrogen peroxide solution was added for the second time and left to digest for 30 min, it was then removed again for cooling. 1 ml of hydrogen peroxide solution was then added for the third time, digested for 45−60 min, if the solution is still not clear and transparent, we continued to add 1 ml of hydrogen peroxide solution and digest for another 30 min. This step was repeated until the solution became clear and transparent, then the digestion was stopped. The solution was then cooled to room temperature, distilled water was used to increase the volume to 100 ml, and then it was poured into a clean centrifuge tube, labeled and stored for testing. The total nitrogen, phosphorus, and potassium content of rice plants were measured using a German automatic chemical discontinuous analyzer (Smartchem, 2000).
Nitrogen utilization efficiency was then calculated based on the formula:
Plant nitrogen content (%) = [(Leaf dry weight per unit area × Leaf nitrogen content + Dry weight of stem and sheath per unit area × Nitrogen content in stem and sheath + Dry weight per panicle per unit area × Nitrogen content of panicle) / Aboveground parts of whole plant (Dry weights of stems, sheaths, leaves and panicles) per unit area] × 100
Nitrogen accumulation in leaves (stem, sheath and spikelets) (kg·hm−2) = Dry weight of leaf (stem sheath, spikelets) per unit area at each stage × Nitrogen content in leaves (stem sheath and spikelets)
Nitrogen physiological efficiency (%) = [(Grain yield in nitrogen treatment area − Grain yield in the area without nitrogen treatment) / Total nitrogen uptake by plants in nitrogen treatment area − Total nitrogen uptake by plants in the area without nitrogen treatment)] × 100
Nitrogen agronomic efficiency (kg/kg) = (Grain yield in nitrogen treatment area − Grain yield in the area without nitrogen treatment) / Nitrogen application rate
Nitrogen uptake efficiency (%) = [(Nitrogen uptake by plants in nitrogen treatment area − Nitrogen uptake by plants in the area without nitrogen treatment) / Nitrogen application rate] × 100
Partial productivity of nitrogen fertilizer (kg/kg) = Grain yield of nitrogen treatment area / Nitrogen application rate
Fertilizer contribution rate (%) = (Yield of nitrogen treatment area − Yield in the area without nitrogen treatment) / Yield of nitrogen application area
Dry matter accumulation and allocation in rice
-
Rice plants were randomly sampled from different plots at AT, BS, HS, CS, RS of the main crop and RHS, RRS of the ratooning season rice, respectively. Five representative plants were selected and sampled based on the average of plants at different stages of main and ratooning rice crops. The sampled plants were quickly cleaned with water, and then separated into roots, stems, sheaths, leaves, panicles, and all these different parts of the main crop stubbles were placed into dry-oven at 105 °C to be deactivated for 30 min. Subsequently these were dried at 75 °C to a constant weight, then weighed and cooled at room temperature. Simultaneously, exportation rate of stem and sheath dry matters (ERSSDM), translocation rate of stem and sheath dry matters (TRSSDM) were calculated based on the formula:
$ ERSSDM (\%) = [(A)-(B)/A]\cdot 100 $ $ TRSSDM (\%) = [(A-B)/GW]\cdot 100 $ Where A refers to dry matter weight at the heading stage of rice, B stands for dry matter weight at the mature stage of rice (B), and GW refers to fully filled grain weight in the formula.
Determination of rice yield and its components
-
The effective panicles of more than 50 rice plants were randomly sampled and recorded in each plot on the day that the main and its ratoon rice crops were harvested. Then, the averages of effective panicles were calculated. According to the average of effective panicles, six representative rice plants were selected from each plot to investigate the number of grains per panicle, empty grains per panicle and filled grains per panicle, seed setting percentage, 1000 grain weight and theoretical yield. In addition, in order to calculate plant height and harvest index of the main crop (HI) and ratooning rice (RHI), the rice was cut from the stem base close to the field surface at the mature stage of the main crop and ratooning rice (including old rice stubbles). Where HI or RHI = Grain yield (g) / Total dry matter weight including grain yield and residual stubble per plant (g).
Data processing and statistical analysis
-
Office 2016 software for data processing was used for making the tables and figure drawings. DPS 7.05 statistical software was used to analyze the experiment results.
-
Figure 1 displays that the bleeding sap intensity of all fertilization treatments was greater than that of the zero applied fertilizer regime in the whole growth period of the main and ratoon rice crops. The main and ratoon rice crops in all treatments performed the highest intensity of the root bleeding sap at both heading stages of the main crop and its ratoon rice (Fig. 1). At BS stage, the bleeding sap intensity of the single stem in the main crop was the highest under the treatment with chemical fertilizer (CF), followed by that under GM2 regime, but both were not significantly different. Furtherthermore, the obviously decreasing tendency in the bleeding sap intensity was determined under the other combined rates of organic and inorganic fertilizers. However, there was no significant difference in bleeding sap intensity under GM1, GM3 and GM4 treatments, it was significantly higher than that under GM5, GM6 and CK regimes. At the heading stage of the main crop, insignificant difference in the bleeding sap intensity was detected under GM1 and GM2 treatments, and the same was true in the cases under GM4, GM5 and GM6 treatments, but all fertilization treatments were significantly higher than CK without nitrogen treatment in terms of the sap intensity. At the CS stage of the main crop, except for GM6, the other treatments displayed insignificant difference in the sap intensity. After the fertilization treatment for preserving vigor roots and promoting bud sprout, the sap intensity in the GM2 regime increased to the maximum at the ripening stage of the main crop. Although there was no significant difference in the sap intensity in GM1 and GM2 treatments, they were significantly higher than those in other treatments. At the same time, the sap intensity in CF treatment decreased to significantly lower than that in the treatments with GM1 and GM2, but the difference was not obvious compared with those under GM3, GM4, GM5 and GM6 regimes. In the period of ratooning season, the sap intensity increased to the peak at the heading stage of ratooning season rice under fertilization treatments with the same amounts of chemical nitrogen for tillering-promotion applied on day 5 after the main crop was harvested, and the largest value of the sap intensity was found in the CF regime. Further, the second place was found in GM1 and GM2 treatments, but they were not significantly different in three fertilization treatments. Insignificant difference was also detected in GM4, GM5 and GM6 treatments except for that in CK. At RRS stage, the sap intensity of the ratoon plants was much higher in GM2 treatment, but insignificantly different in all fertilization treatments (except for that in CK). Therefore, an appropriate organic nitrogen proportion in the fertilizer mixture (such as GM2 treatment) could improve the bleeding sap intensity, thereby increasing root-vigor at earlier and late growth stages of the main and ratoon rice crops, which lays an important physiological foundation of nutrient absorption and utilization as well as high-yielding formation in rice ratooning.
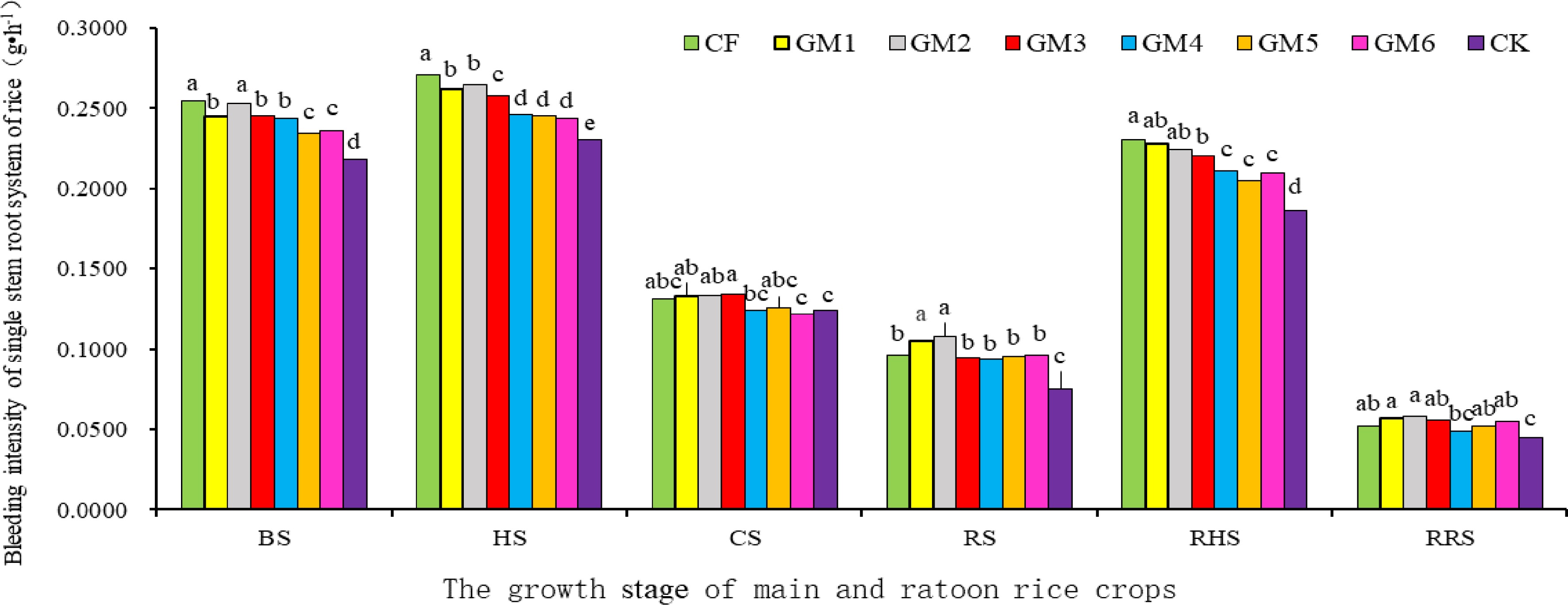
Figure 1.
Effects of treatment with different ratios of organic and inorganic fertilizers on the root sap intensity of main and ratoon rice crop plants in single stem level. Note: Different lower case letters indicate significant differences in p < 0.05 level between different treatments in the same part and the same period. BS: booting; HS: fully heading stage, CS: the day close to the stage fertilizing for root-vigor preserving and bud promoting, RS: ripening stages of the main season crop, RHS: fully heading of ratooning season rice, RRS: ripening stages of ratooning season rice.
Effects of different configurations with organic and inorganic fertilizers on dry matter accumulation and grain yield in rice
-
The results revealed that the nitrogen content in stem and sheath was the highest at heading stage of the main crop and the lowest at RRS stage of ratoon rice. In terms of the changing trend of different treatments in the main and ratoon rice crops, the nitrogen content in stem and sheath of conventional fertilization treatment (CF) was the largest at HS stage of the main crop, and decreased with the increase of organic fertilizer proportion in the organic/inorganic mixture at the same stage. In the CS stage as shown in Fig 2, the nitrogen content in stem and sheath of the main and ratoon rice crops decreased rapidly in all treatments, and the largest declining rate was observed in CF treatment. During this period, the nitrogen content of each fertilization treatment was significantly higher than that of CK (without fertilization treatment). The nitrogen content of sole chemical fertilizer (CF) treatment showed the highest in stem and sheath among all fertilization treatments, and insignificant difference was observed among other treatments of organic/inorganic fertilizer mixtures. Furthermore, the nitrogen content of all fertilization treatments increased from CS to RS stage of the main crop, the reverse was true in the case of CK treatment. In terms of ratooning season rice, the nitrogen content in stem and sheath of CF was higher at the RHS stage of ratoon rice. However, at the RRS stage of ratooning season rice, the nitrogen content in the stem and sheath of all treatments decreased rapidly in a way that it ended up with no significant difference in nitrogen content among fertilization treatments except for CK treatment, suggesting that compared with other combined application of organic and inorganic nitrogen, sole chemical nitrogen fertilizer treatment (CF) increased the amount of allocated nitrogen in the stem sheath of the main crop rice, but there was no significant difference in the RRS stage of ratooning season rice.
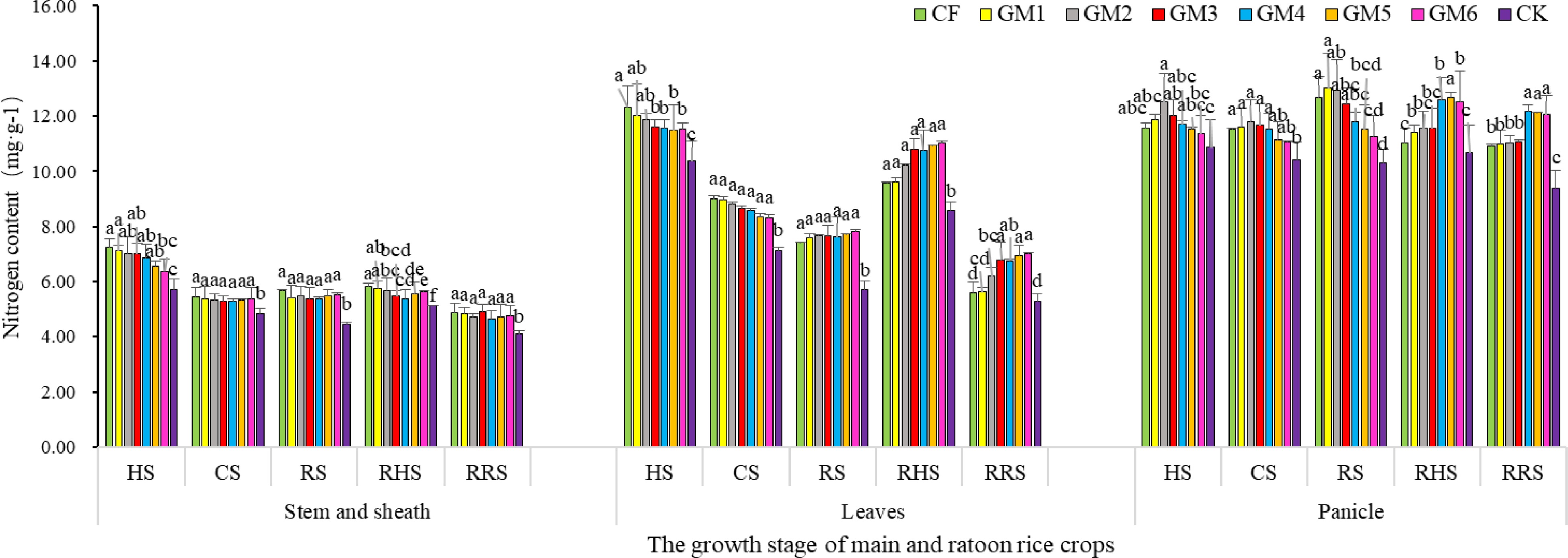
Figure 2.
Effect of treatment with different ratios of organic and inorganic fertilizers on nitrogen content in various parts in different periods of main and ratoon rice crops at various phases of grain-filling. Note: Different lower case letters indicate significant differences in p < 0.05 level between different treatments in the same part and the same period. HS: fully heading stage, CS: the day close to the stage fertilizing for root-vigor preserving and bud promoting, RS: ripening stages of the main season crop, RHS: fully heading of ratooning season rice, RRS: ripening stages of ratooning season rice.
The results displayed a similar trend change of nitrogen content in leaves as those in the stem, and sheath of the main and ratoon rice crop in all the treatments as shown in stem and sheath, which indicated a decreasing trend at first then increased and decreased again. The leaf nitrogen content reached the maximum at the heading stage, and then decreased rapidly until maturity of the main crop in all treatments. From the HS to CS stage of the main crop, the nitrogen content of leaves in the CF treatment was higher than that in other treatments. Meanwhile, a reduction of nitrogen content in leaves was also accompanied by the increasing proportion of organic fertilizer in the fertilizer mixture treatment, and at maturity stage of the main crop the more organic nitrogen in the fertilizer applied, the greater the leaf nitrogen content of the corresponding treatment. In fully heading and mature stages of ratoon rice, the nitrogen content of leaves was GM6 > GM5 > GM3 > GM4 > GM2 > GM1 > CF > CK in order. It implies that the excessive proportion of organic fertilizer in the mixture fertilizer will lead to the retention of nitrogen in rice leaves at the late stage and eventually lead to the undesirable consequences of stay-green.
Figure 2 also reveals that the changing trend of nitrogen content in the panicle was different from that in stems, sheaths and leaves under different treatments from the HS stage of the main crop to the RRS stage of ratoon rice. At the BS stage of the main crop, the nitrogen content in the panicle was the highest in the GM2 regime, but the variation trend was irregular among different treatments. Compared with that at the HS stage, the nitrogen content in the panicle decreased to a certain extent until the CS stage of the main crop. The nitrogen content in panicles of the main crop was GM1 > GM2 > CF > GM3 > GM4 > GM5 > GM6 > CK. At the RHS stage of ratooning rice, the changes of nitrogen content in the panicles were different under different fertilization treatments. Furthermore, compared with that at the HS stage of the main crop, the nitrogen content decreased in the panicles of ratoon rice under CF, GM1, GM2 and GM3 treatments, while the reverse was true in the case of the other four treatments GM4, GM5, GM6 and CK. At the RRS stage, the panicle nitrogen content of ratoon rice under the treatment with more than 70% organic fertilizer in the replacement was significantly higher than that in the other treatments. Therefore, it is suggested that the higher proportions of chemical nitrogen in the combined fertilizer had the earlier and faster physiological effect, the opposite was true in the case under treatment with the higher proportions of organic nitrogen in the combined fertilizer. It is therefore assumed that a reasonable ratio of organic and inorganic nitrogen (such as GM2) was able to coordinate the contradiction between supply and demand of nutrients, hence making the rice plant absorb nitrogen and allocate it reasonably in various rice plant parts, and ultimately improve the nutrient utilization efficiency and economic returns.
The characteristics of dry matter accumulation and distribution were further analyzed as shown in Fig. 3. At the booting stage of the main crop, the dry matter accumulation in stem, sheath and leaves was higher in GM2 treatment, but it was lowest in leaves under CF treatment compared with that in the other treatments, while the lowest dry matter weight of stem and sheath was detected in the GM6 treatment. At the fully heading stage of the main crop, the dry matter accumulation in different plant parts was increased under all treatments, of which GM2 showed the highest dry matter accumulation in stem and sheath, followed by the GM1 regime. The results further displayed a decreased dry matter accumulation in the stem and sheath under the treatments with the increases of the combined application proportions of organic nitrogen in the fertilizer mixture. In terms of the leaf dry matter weights, all treatments of combined application with different proportions of organic fertilizer were significantly lower than that of the chemical fertilizer treatment (CF), but the GM2 treatment effect on dry matter weight of panicles was enhanced, indicating the highest value of dry matter accumulation in panicles, and significantly higher than that in the other treatments. In the pre stage of fertilizer application for root-vigor and bud preserving, GM1 and GM2 showed significantly higher dry matter weight of stem and sheath than the other treatments, but both of them indicated insignificant difference. In terms of dry matter weights of leaves, insignificant was found in CF, GM1 and GM2 treatments, the same was true in the case of GM3, GM4 and GM5 treatments, but all of them showed higher values than that in CK (without fertilizer). All treatments displayed significantly different dry matter weight of panicles, indicating the following order: GM2 > GM1 > CF > GM3 > GM4 > GM5 > GM6 > CK. At the maturity stage of the main crop, both GM1 and GM2 treatments showed higher dry matter weights of panicle, stem and sheath compared with the other treatments. While in terms of leaf dry matter weight, it was lower in GM2 treatment than that in the CF regime. At the fully heading stage of ratooning rice, the dry matter weights of stem, sheath, leaves and panicles as well as old stubbles were higher in GM1, GM2 and GM3 regimes than that in CF treatment. Meanwhile, at the maturity stage of ratooning rice, the dry matter weights of stem, sheath and leaves, as well as the old stubbles of the main crop, were significantly decreased, the reverse was true in the case of the panicle dry weight. Additionally, a positive performance in terms of panicle weight was noted in GM2, GM1, GM3, and GM4 treatments compared to other treatments. These results further confirmed that suitable combination of organic and inorganic fertilizer was beneficial to increase dry matter production and accumulation in late stage of main crop.
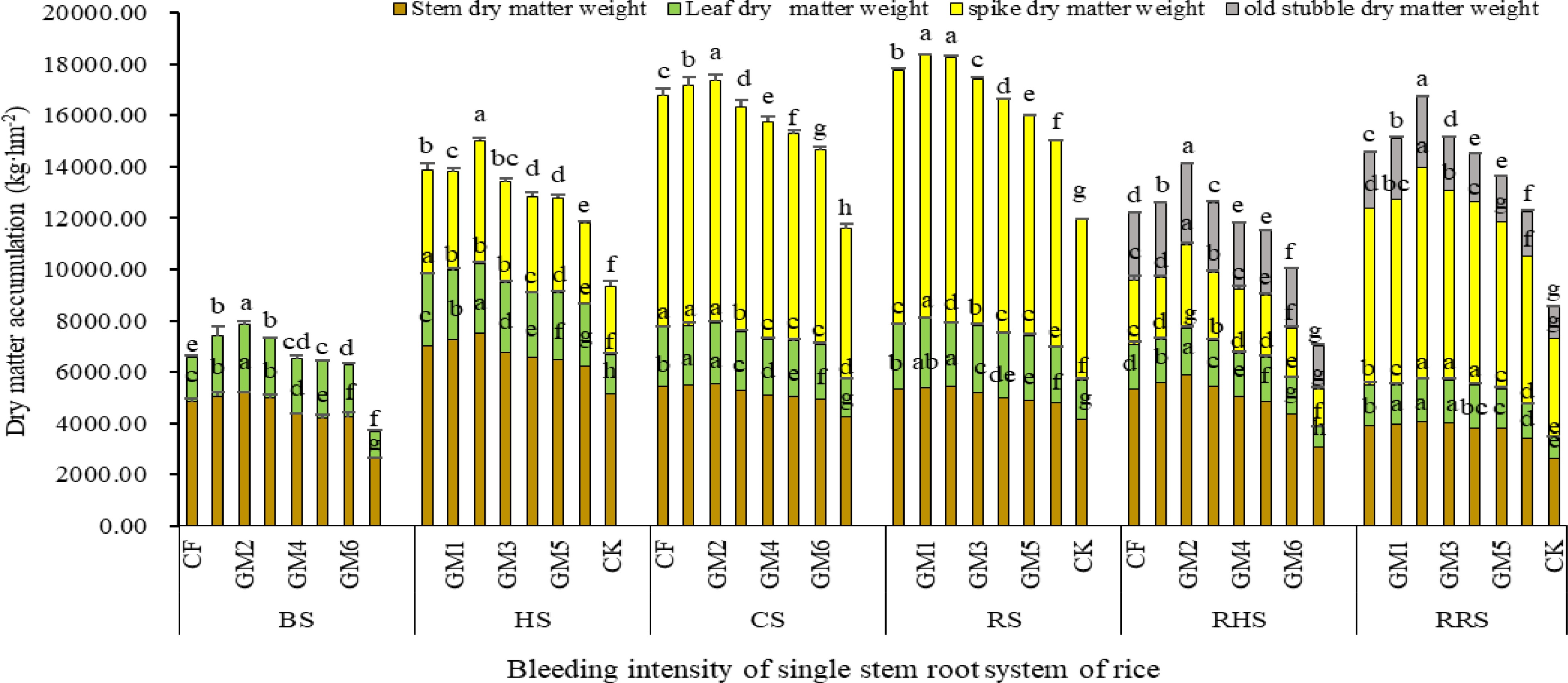
Figure 3.
Effect of combined application treatment with different ratios of organic and inorganic fertilizers on dry matter accumulation of ground rice plants. Note: Different lower case letters indicate significant differences in p < 0.05 level between different treatments in the same part and the same period. BS: booting, HS: fully heading stage, CS: the day close to the stage fertilizing for root-vigor preserving and bud promoting, RS: ripening stages of the main season crop, RHS: fully heading of ratooning season rice, RRS: ripening stages of ratooning season rice.
Furthermore, GM2 treatment exhibited the best effect on exportation and translocation rates of the stem and sheath dry matter, displaying the highest promoting effect on the two fertilizer rates compared with that in the other treatments, followed by those in GM1 and CF, whereas all other treatments showed lower comparative grades with increasing proportion of organic nitrogen in the mixed fertilizer treatments (Table 1). Therefore, this result further confirmed that good soil fertilizer supply guarantees an efficient plant growth as well as dry matter production and allocation in late growth stage, which greatly contributes to the higher harvest index and grain yield of main and ratooning rice crops.
Table 1. Effects of different ratios of organic fertilizers and chemical fertilizers on dry matter accumulation and transport during filling stage.
Stage Treatment SSWF (kg·hm−2) SSWM (kg·hm−2) PWM (kg·hm−2) DMAF (kg·hm−2) EPMSS (%) TPMSS (%) Maincrop CF 7025.43c 5342.27b 9903.15b 4026.62b 23.96c 16.99d GM1 7258.68b 5397.21ab 10250.41ab 3844.83c 25.64b 18.16b GM2 7518.68a 5436.57a 10380.56a 4778.16a 27.69a 20.05a GM3 6791.52d 5190.93c 9634.42c 3945.44b 23.57d 16.61d GM4 6575.77e 4997.32d 9108.53d 3772.75d 24.00c 17.33cd GM5 6471.57e 4915.14d 8583.96e 3740.11d 24.05c 18.13b GM6 6244.54f 4821.60e 8059.98f 3137.99e 22.78e 17.65c CK 5137.59g 4144.89f 6261.79g 2660.41f 19.32f 15.85e Ratoonrice CF 5340.86cd 3890.75c 6149.61d 2483.25cd 27.15bc 23.58b GM1 5567.44b 3971.67b 6544.57bc 2408.53e 28.66b 24.38ab GM2 5889.77a 4051.99a 7348.64a 3219.63a 31.20a 25.01a GM3 5440.62c 4009.24ab 6604.73b 2626.66b 26.3cd 21.67c GM4 5022.85d 3827.70d 6293.14c 2539.09c 23.79d 18.99d GM5 4861.61e 3789.66d 5783.45e 2435.55de 22.05e 18.53d GM6 4329.81f 3417.23e 5169.39f 1916.51f 21.07f 17.65de CK 3047.31g 2649.84f 3539.34g 1441.24g 13.04g 11.23f SSWH: Stem-sheath dry matter weight in full-heading stage; SSWM: Stem-sheath dry matter weight in mature stage; PWM: Panicle dry matter weight in mature stage; DMAH: Dry-matter accumulation after full-heading stage; DMAH: Dry-matter accumulation after full-heading stage; EPMSS: exportation rate of stem-sheath dry matter; TPMSS: Translocation rate of stem-sheath dry matter. Based on the statistical analysis results, an insignificant difference was found in two years for grain yield and its components of the main crop and ratooning rice under different combined application treatments with different proportions of organic and inorganic fertilizers in the main crop season. The reverse was true in fertilization treatments and the interaction between years and fertilization treatments (Table 1). Therefore, we here use the average of the two year results for the comparison analysis. The results showed that the gain yield of main and ratooning rice crops under GM2 treatment was significantly higher than that in other treatments, which resulted from the higher productive tillers and harvest index, especially in the case of ratooning rice under the optimal fertilizer configuration (GM2). The total yield of the main and ratoon rice crops under GM1, GM2 and GM3 showed an increased effect compared with the conventional fertilization treatment (CF), among which GM2 had the largest increase effect, with an average increase of 10.36% in two years. The yield under GM1 and GM3 treatments increased by 3.37% and 1.9%, respectively, however, a more obvious decrease in rice yield and grain yield was recorded with the increase in the proportion of organic nitrogen in the combined application treatments, in which GM5 and GM6 decreased by 5.83% and 10.31% on average in two years, respectively, which might be attributed to lower number of effective panicles and poor harvest index (Table 1).
Effects of different ratios with organic and inorganic fertilizers on nitrogen use efficiency
-
Table 2 illustrates that the combined application with different proportions of organic and inorganic fertilizers in the main crop have dissimilar effects on nitrogen use efficiency of the main and ratoon rice crops. In terms of nitrogen partial productivity, the performance values of fertilization treatment were significantly higher than CK treatment; notably, with the increase of organic fertilizer proportion, nitrogen partial productivity of the main and ratoon rice crops increased at first and then decreased gradually. Among the fertilization treatments, GM2 had the highest partial nitrogen productivity (48.97 kg/kg), which was significantly higher than other fertilization treatments with 75 kg/kg higher than that of conventional fertilization. Likewise, for nitrogen absorption and utilization efficiency, the nitrogen absorption and utilization efficiency of conventional fertilization treatment was only 33.71%. The nitrogen use efficiency of GM2 was the highest (44.37%), while those of GM1 and GM3 were more than 40%. Compared with conventional fertilization treatment (CF), GM2, GM1 and GM3 increased by 10.66%, 8.12% and 7.86% in nitrogen use efficiency, respectively. Moreover, nitrogen agronomic use efficiency for GM2 reached to 23.82 kg/kg, which was significantly higher than that of other treatments, and increased by 30.55% compared with that in conventional fertilization regime. GM1 and GM3 regimes also showed higher nitrogen agronomic utilization efficiency than the CF treatment, but the other fertilization treatments performed poorly in nitrogen agronomic utilization efficiency than the CF regime. Hence the difference in grain yield of the main and ratooning rice crops results from the different treatments with different configurations, with different proportions of inorganic/organic nitrogen in fertilization treatments (Table 2).
Table 2. Comparison on the grain yield components and related physiological traits of the main and ratoon rice crops under combined application treatments with different proportions of organic and inorganic fertilizers.
Treatments The main crop The ratooning rice MTY
(kg·hm−2)RTY
(kg·hm−2)TTY
(kg·hm−2)MPH
(cm)RPH
(cm)MHI RHI MGD
(days)RGD
(days)NFPP (kg/kg) NAUE
(%)PENU
(kg/kg)NAUE
(kg/kg)NCRF
(%)EP
(104·hm−2)GN TGW
(g)SSP
(%)EP
(104·hm−2)GN TGW
(g)SSP
(%)CF 264.16b 205.24a 22.63a 87.57b 330.60bc 108.55c 21.61a 89.18ab 10750.68bc 6901.61cd 17528.21bc 135a 85a 0.46a 0.51a 142.00 70.00 43.22c 33.71d 41.94c 18.07c 41.80c GM1 267.49b 203.49ab 22.71ab 88.23ab 338.70bc 110.50b 21.65a 90.06ab 10914.32b 7287.35bc 18118.53b 135a 95b 0.49b 0.55b 142.00 72.00 44.82b 41.83b 44.00b 19.67b 43.89b GM2 282.64a 201.69bc 22.69ab 89.52a 356.10a 115.08a 21.47ab 90.66a 11600.46a 7970.31a 19342.97a 140b 95b 0.55c 0.61c 142.00 75.00 48.97a 44.37a 48.79a 23.82a 48.64a GM4 258.34c 194.59d 22.55ab 89.82a 328.66c 110.71b 21.66a 90.32ab 10145.36d 7103.01bc 17243.15c 135a 90c 0.43c 0.50a 148.00 78.00 42.57d 37.22c 40.44d 17.42d 40.92d GM5 247.61d 195.43d 22.63ab 89.56a 308.70d 111.71b 21.45a 90.64ab 9797.27e 6681.84d 16507.49d 135a 85a 0.41c 0.48d 150.00 78.00 40.64e 34.83d 35.98e 15.94e 38.12e GM6 240.58d 194.89cd 22.56ab 90.04a 295.53e 107.62c 21.51ab 90.93a 9486.38e 6234.12e 15722.10e 135a 85a 0.41c 0.45d 150.00 78.00 38.37f 32.17e 30.29f 13.22f 24.46f CK 186.48e 182.83e 22.38b 86.83ab 215.02f 99.52d 21.03b 88.12b 6688.71f 3751.59f 10715.73f − − − − − − − − − − − Year(A) NS NS NS NS NS NS NS NS NS NS NS − − − − − − − − − − − Fertilization
(B)32.43** 12.25** NS 2.34* 33.25** 11.51** NS NS 19.98** 76.13** 99.30** − − − − − − − − − − − A×B NS NS NS NS NS NS NS NS NS NS 163.80** − − − − − − − − − − − The comparison of differences is conducted to the average value of single factor effect and the interaction of double factor in the same column in the average of two year since no difference was found in two year results. The same letter after each data means that the difference is not significant, otherwise the difference is significant. Lower case letters represent significant level at < 5% and upper case letters represent significant level at < 1% , EP: effective panicles; GN: grain number per panicle; TGW: 1000-grain weight; SSP: seed setting percentage; MTY: theoretical yields of the main crop; RTY: theoretical yields of the ratoon rice; TTY: total theoretical yield; MPH: Plant height of the main crop; RPH: Plant height of ratooning rice; MHI: Harvest index of the main crop; RHI: Harvest index of ratooning rice; MGD: growth duration of the main crop; RGD: growth duration of ratooning rice; NFPP: Nitrogen fertilizer partial productivity; NAUE: nitrogen absorption and utilization efficiency; PENU: Physiological efficiency of nitrogen use; NAUE: Nitrogen agronomic use efficiency; NCRF: Nitrogen contribution rate of fertilizer. -
The results indicated that the nitrogen uptake and accumulation in stems, sheaths and leaves of the main and ratoon rice crops were the highest at the heading stage of the main crop, and gradually decreased with proceeding rice plant growth under the fertilizer configurations with different proportions of inorganic and organic nitrogen, simply because during the grain-filling and ripening stage of the main and ratoon rice crops, a large proportion of dry matter was rapidly transferred from the stem, sheath and leaf parts to the grain pools of the main and ratooning rice crops[14]. The result also revealed that the GM2 treatment (70% chemical nitrogen + 30% organic nitrogen) performed the best output compared with the other treatments, which is due to the fact that the rice plants in the early stage need much more nutrients to accumulate biomass, an appropriate proportion of inorganic nitrogen supply can meet the nutrient demands for the growth and development of rice in the early growth stage. However, the carry-over effect of the chemical nitrogen is too short to be favorable for the growth of rice at the late stage, which often leads to decreased root activity, thereby accelerated premature senescence. Therefore, a proper amount of organic nitrogen fertilizer in the combined application treatment could improve the situation since the proper amounts of organic nitrogen in the combined application have better durability of fertility, and the organic nitrogen could be effectively decomposed by rhizosphere microbes to release a large number of small molecules into rhizosphere for direct absorption by rice plants at the late growth stage to support the rice plant physiological demands, thereby preventing root premature senescence and root-vigor decreases, thereby improving the survival status of the axillary buds in the remaining stubbles, consequently increasing grain yield of the main and ratooning rice crops[15−17].
However, the present results revealed that the excessive proportion of organic fertilizer in the mixture fertilizer led to the retention of nitrogen in rice leaves at the late stage and eventually led to the undesirable consequences of stay-green, which is responsible for the good carry-over effect of the organic fertilizer in terms of its fertility. Therefore, too much organic nitrogen in the combined application regime has a stay-green effect on rice growth, consequently resulting in yield reduction in the mixture treatment containing more than 45% organic nitrogen (such as GM3, GM4 etc.). In this experiment, as overused organic fertilizer inevitably results in nitrogen retention and reduces the nitrogen release efficiency, thereby decreasing the nitrogen utilization efficiency. Our present studies suggested that the proper combined application of organic and inorganic fertilizers such as GM2 treatment could make the soil nitrogen supply timely and effective as indicated by the higher root activity and the better nitrogen accumulation and allocation in the main crop under the optimal configuration regime (see Figs 1, 2 & 3 and Tables 1 & 2), which is consistent with the previous studies on conventional rice cultivation[1,8]. Zhang et al. also documented that under an equal nitrogen usage, the substitution of organic fertilizers for chemical fertilizers improved the stability and sustainability of rice yields, reduced soil mineral nitrogen losses, and hence improved the utilization of nitrogen fertilizers[18]. Zhang also suggested that the combined application of organic and inorganic fertilizers could increase the nitrogen uptake of the plants in the later stage and increase the nitrogen use efficiency[19]. Liu et al. reported that under the condition of equal nitrogen replacement, organic and inorganic fertilizers were applied in different mixture combinations, and the soil available nitrogen content was higher in the treatment with a high proportion of chemical fertilizer application in the early stage of rice growth, the reverse was true in the case of the treatment with a high replacement ratio of organic fertilizer at the late growth stage of rice[20].
Previous studies also suggested that with the increase in the proportion of organic fertilizers, the nitrogen fixed by microorganisms increases, and this in turn causes insufficient nutrients to the crops, especially in early plant growth stage[11,21]. Moreover, the microbial biomass carbon, nitrogen and mineral nitrogen in soil under the combined application treatment with organic and inorganic fertilizers were lower than those under sole chemical fertilizer regimes before the rice tillering stage, however, the reverse was true in the case of the heading stage to the grain filling stage. The findings attested that nitrogen supply dynamics should be in accordance with the law of nitrogen absorption and utilization by rice to the highest degree, which is conducive to preventing the delay-green and premature senescence of root and shoot in the late growth stage, in turn improving the status of rice growth and development, and hence leading to increased grain yield of rice[22−27]. Therefore it is suggested that a suitable combined application of organic and inorganic fertilizers could greatly increase rice root activity and nutrient uptake capacity, improve dry matter accumulation and its allocation pattern, enhance nitrogen use efficiency, nitrogen absorption efficiency, agronomic efficiency and nitrogen partial productivity, harvest index and hence increased yield of the main and ratoon rice crops.
-
In summary, the results suggested that under the premise of the same amount of nitrogen fertilizer per hectare (225 kg·hm−2), the organic nitrogen is replaced with 30% equivalent nitrogen to inorganic nitrogen (GM2), the yield performance of the main and ratoon rice crops was the best in the two-year studies. Relative to the CF control, the total yield of the main and ratooning rice crops increased by 10.36%, of which the ratooning rice yield increased by 15.48% under GM2 treatment. The yield performance of the main and ratooning rice crops in two years was higher under GM2 > GM1 > GM3 > CF > GM4 > GM5 > GM6 > CK, and that of ratooning rice was better at GM2 > GM3 > GM1 > GM4 > CF > GM5 > GM6 > CK in a descending order. The finding suggested that the treatment with more than 30% proportion of organic nitrogen in the mixture fertilizer had a significant impact on the yield of the main crop, but the regime with less than 45% organic nitrogen in the mixture treatment had a negative carry-over effect on the yield of the ratooning rice. The optimal combination treatment (GM2) could achieve 44.37% nitrogen absorption and utilization efficiencies, this in turn led to an improvement in the agronomic utilization efficiency and the partial productivity, and hence increased economic and ecological returns.
This research was supported by the national research grants (2016yfd00300508; 2017YFD0301602; 2018yfd0301105), Fujian Taiwan planting resources creation and green cultivation coordination Innovation Center (Fujian 2011 project, no. 2015-75) and the Science and technology development funds of Fujian agricultural and Forestry University (kf2015043).
-
Wenxiong Lin is the Editorial Board member of the journal Technology in Agronomy. He was blinded from reviewing or making decisions on the manuscript. The article was subject to the journal's standard procedures, with peer-review handled independently of the Editorial Board member and his research group.
- Copyright: © 2023 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Weng P, Yang S, Pang Z, Huang J, Shen L, et al. 2023. Effects of the configurations with different organic and inorganic fertilizers on grain yield and its related physiological traits of the main and its ratoon rice crops. Technology in Agronomy 3:2 doi: 10.48130/TIA-2023-0002 

Effects of the configurations with different organic and inorganic fertilizers on grain yield and its related physiological traits of the main and its ratoon rice crops
- Received: 14 September 2022
- Accepted: 30 January 2023
- Published online: 10 February 2023
Abstract: The determination for properly combined application rate of organic and inorganic fertilizers is the key to coordinating soil nutrient supplies and rice growth demands, thereby obtaining improved economic and ecological returns in rice ratooning. A complete randomized block design (RCBD) trial with different configurations of organic and inorganic nitrogen fertilizers was conducted to determine the optimal substitution rate of organic nitrogen for inorganic nitrogen and its effect on the related physiological attributes and yield performance of the main and ratoon rice crops in 2018−2019. The results showed that 30% organic and 70% inorganic nitrogen in the mixture fertilizer (GM2) could produce the best effect on root activity, nitrogen nutrient uptake and its utilization, as well as the dry matter accumulation and its partitioning. This in turn resulted in improved productive panicles and harvest index, and hence increased grain yield by 10.36% and 15.48% in GM2 regime relative to that in CF treatment. Moreover, the uptake and utilization efficiency, agronomic utilization efficiency and partial productivity of nitrogen fertilizer were 44.37%, 23.82 kg/kg and 48.97 kg/kg, respectively under the optimal GM2 treatment, which were much higher than those in CF treatment by 12.66%, 5.05% and 13.4%, respectively. The results suggested that properly combined application of organic and chemical nitrogen fertilizer could improve soil nutrient supply to guarantee an efficient plant growth and rational dry matter allocation, thereby increasing the harvest index and grain yield in rice ratooning.
-
Key words:
- Organic nitrogen replacement /
- Matter allocation /
- Nitrogen utilization /
- Rice ratooning