









-
Nipponbare is a japonica rice cultivar that has been widely used as the standard reference genotype for rice[1]. The rice (Nipponbare) genome was one of the first crop genomes to be sequenced more than 20 years ago[2]. The 1st sequence of the rice genome was completed in 2002 and was a major milestone in the field of plant genomics by the International Rice Genome Sequencing Project, 2005[3]. These international collaborative efforts provided the first genome of a crop plant. The Nipponbare genome assembly contained gaps, primarily due to repetitive DNA sequences. In 2005, these gaps were estimated to be approximately 18.1 Mb in total, with the majority originating from centromeres and telomere regions. Sequencing technological advancements and ongoing research efforts, have improved the rice genome sequence over time[4,5]. Thorough efforts were made to improve the quality of the Nipponbare reference genome assembly in 2013, resulting in greatly enhanced accuracy of cDNA sequences and gene annotation, while it remained incomplete[5]. In the human genome, recent significant strides have been made in assembling and characterization the previously unexplored 8% of the human genome, especially including telomere sequences[6].
Reference genome assemblies often contain gaps, especially regions with repetitive sequences, termed the 'dark side' of the genome[7,8]. New sequence technology allows improved assembly quality, with less gaps, leading to a more complete and accurate representation of the genome. The achievement of a higher quality and more complete reference genome will provide new insights into genomics and breeding, supporting pan-genome studies and genome wide association studies[9]. Recently many other Oryza genomes have been sequenced and assembled, including indica and wild rice species[9−11] Most recently the Nipponbare genome sequence gaps and telomere sequence were addressed[12,13]. Despite these advancements, a fully haplotype resolved assembly has not been reported. In this study, we have used PacBio HiFi reads to produce a more accurate genome sequence assembly. The novel genome assembly is not only almost 11.3 Mb longer than the IRGSP-1.0 reference but also exhibits improvements in all chromosomes (Fig. 1), including the addition of telomeric regions in all chromosomes (T2T), with the addition of fully resolved haplotypes (haplotype 1 and haplotype 2, telomere-to-telomere-T2T) (Tables 1, 2 & Supplemental Tables S1−S10).
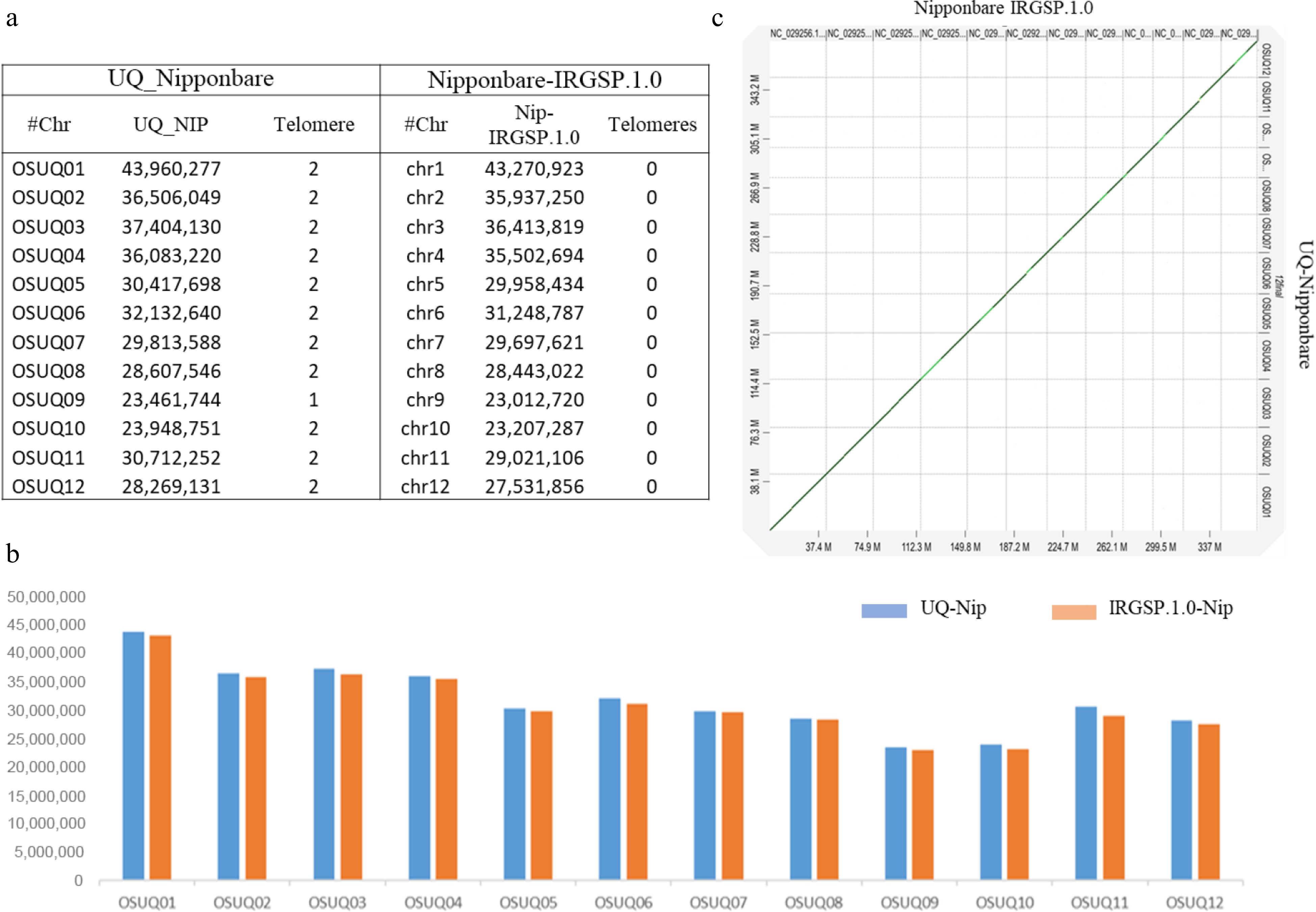
Figure 1.
Comparison of the UQ Nipponbare genome with previously published reference genome assembly IRGSP.1.0 Nipponbare. (a), (b) For UQ Nipponbare all the chromosome sizes are larger and most include telomeres as compared to IRGSP.1.0 Nipponbare. (c) Whole genome dot plot of UQ Nipponbare genome vs IRGSP.1.0 Nipponbare.
Table 1. Statistics for the UQ Nipponbare haplotype resolved genome assembly.
UQ_Nip-collapsed UQ_Nip-Hap1 UQ_Nip-Hap2 Total assembly size 381,317,026 379,234,557 348,265,595 Complete BUSCOs (%) 99.30% 98.90% 94.90% Total scaffold number 12 12 12 Scaffold N50 30,712,252 30,691,512 29,307,860 Scaffold L50 6 6 6 Largest scaffold 43,960,277 43,881,444 37,312,016 GC content (%) 44 44 43 Table 2. UQ Nipponbare haplotype resolved genome assembly chromosomes sizes and telomere numbers.
UQ_Nip-Collapsed-Assembly UQ_Nip-Hap1 UQ_Nip-Hap2 Chr Size Telomere Chr Size Telomeres Chr Size Telomeres OSUQ01 43,960,277 2 OSUQ01-hap1-01 43,881,444 2 OSUQ01-hap2-01 37,312,016 2 OSUQ02 36,506,049 2 OSUQ02-hap1-02 36,408,562 2 OSUQ02-hap2-02 33,128,268 2 OSUQ03 37,404,130 2 OSUQ03-hap1-03 37,357,616 2 OSUQ03-hap2-03 35,934,962 2 OSUQ04 36,083,220 2 OSUQ04-hap1-04 35,866,358 2 OSUQ04-hap2-04 33,298,206 2 OSUQ05 30,417,698 2 OSUQ05-hap1-05 30,178,946 2 OSUQ05-hap2-05 24,819,415 2 OSUQ06 32,132,640 2 OSUQ06-hap1-06 32,049,832 2 OSUQ06-hap2-06 32,067,105 2 OSUQ07 29,813,588 2 OSUQ07-hap1-07 29,708,413 2 OSUQ07-hap2-07 28,849,953 2 OSUQ08 28,607,546 2 OSUQ08-hap1-08 28,566,876 2 OSUQ08-hap2-08 26,254,248 1 OSUQ09 23,461,744 1 OSUQ09-hap1-09 23,221,905 OSUQ09-hap2-09 21,159,188 1 OSUQ10 23,948,751 2 OSUQ10-hap1-10 23,192,984 2 OSUQ10-hap2-10 21,409,976 2 OSUQ11 30,712,252 2 OSUQ11-hap1-11 30,691,512 2 OSUQ11-hap2-11 29,307,860 1 OSUQ12 28,269,131 2 OSUQ12-hap1-12 28,110,109 2 OSUQ12-hap2-12 24,724,398 2 Comparative analysis of annotations of the new genome (UQ Nipponbare) and the IRGSP-1.0 reference revealed the presence of 3,050 additional genes, for which more than 95% had supporting transcript evidence (Supplemental Fig. S1).
These findings underscore the potential of new sequencing technologies to significantly augment reference genomes, potentially leading to more comprehensive genetic information. These results also suggest that applying advanced sequencing technologies to other established genomes may yield similar benefits, potentially enhancing our knowledge of these species. This study highlights the continuous evolution of genomics and underscore the importance of staying at the forefront of sequencing technologies for the accurate representation of complex genomes.
PacBio HiFi reads and Hi-C reads were used to generate a contig assembly with Hifiasm[14] producing a haplotype phased assembly. The contig level assembly produced single contigs for nine chromosomes, while the remaining three chromosomes were each covered by two contigs each. Hi-C data were employed to hierarchically cluster the assembled contigs into 12 pseudo-chromosomes, by using the YaHS scaffolding tool[15]. The T2T assembly had a single scaffold for each of the 12 pseudo-chromosomes and was larger in size than the corresponding IRGSP.10 genome (Fig. 1). The results of BUSCO analysis showed that the collapsed assembly covered 99.3% of the universal single copy genes with an N50 of 30.7 Mb (Supplemental Tables S1−S3). The UQ Nipponbare collapsed genome assembly is larger in size compared to the IRGSP.1.0 Nipponbare reference genome assembly. In Fig. 2, additional non-aligning regions of each chromosome in the UQ Nipponbare collapsed assembly are highlighted, along with the structural variants in the comparison of the previously published IRGSP1.0 Nipponbare reference genome assembly.
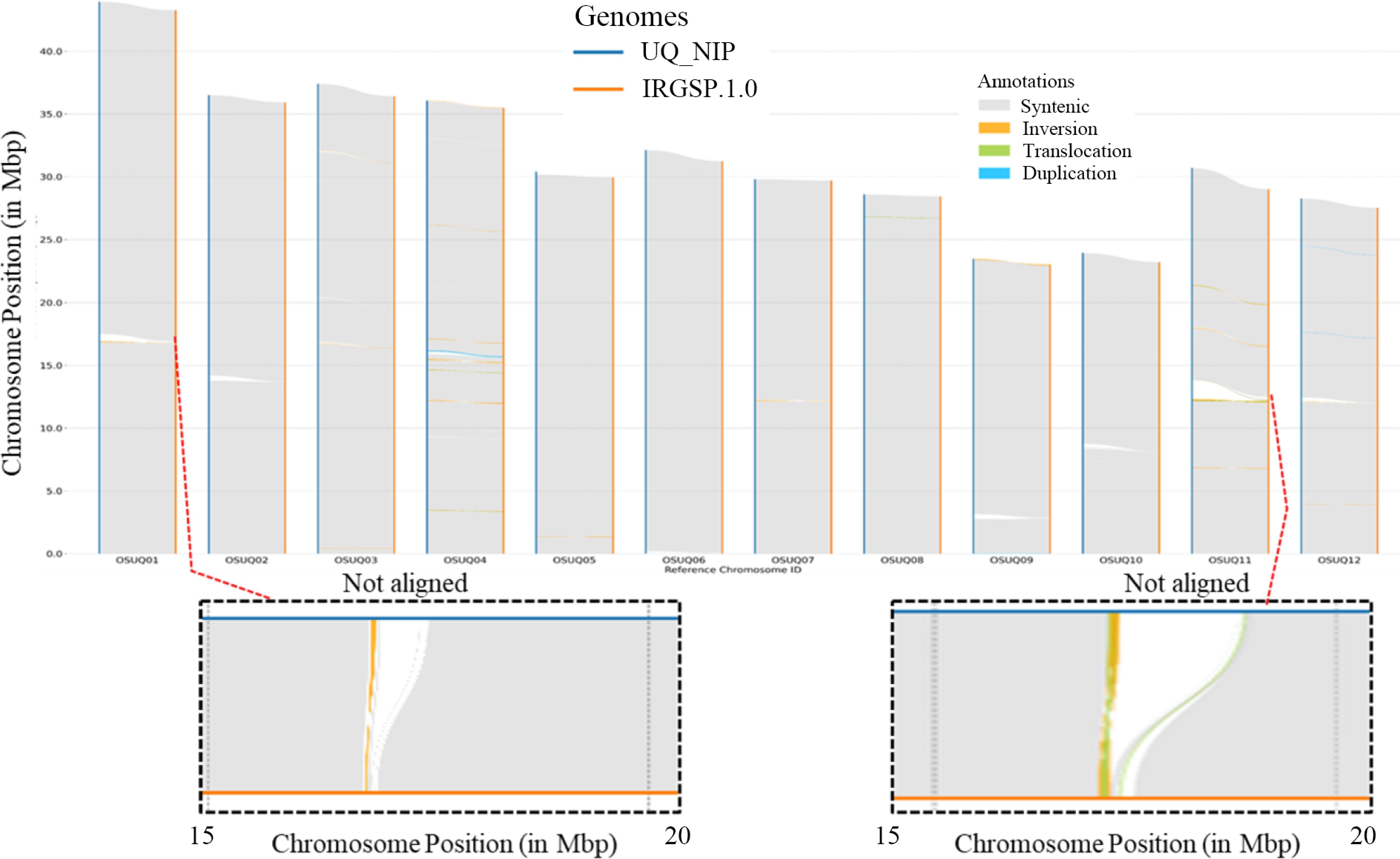
Figure 2.
Sequence collinearity and structural variants, including inversions, translocations, duplications, and non-aligning regions, were analysed between the UQ Nipponbare genome assembly and the IRGSP-1.0-Nipponbare genome assembly. The two assemblies were aligned using minimap2, and the resulting BAM file was indexed with samtools. Detection of structural variations between these two genomes was performed using SyRI[26−28]. The non-aligning regions of chromosomes 1 and 11 are highlighted in the bottom section of the figure.
For the two phased haplotypes (T2T) of UQ Nipponbare; haplotype 1 covered 98.9% of the single copy orthologs with an N50 of 30.6 Mb, whilst haplotype 2 covered 94.9% single copy orthologs with an N50 of 29.3 Mb (Supplemental Tables S4−S9 & Supplemental Figs S2 & S3). The haplotype 1 chromosomes were larger than the haplotype 2 chromosomes (Fig. 3). This first haplotype resolved Nipponbare genome incorporated 3,050 new genes compared to IRGSP-1.0, and is expected to be a valuable and significant resource for rice researchers, and these additional genes had a wide range of functions (Supplemental Table S11). Of these additional genes, 58 genes fell in new regions that were missing in the IRGSP genome, but most genes were in regions that were not new due to the improved accuracy of sequencing.
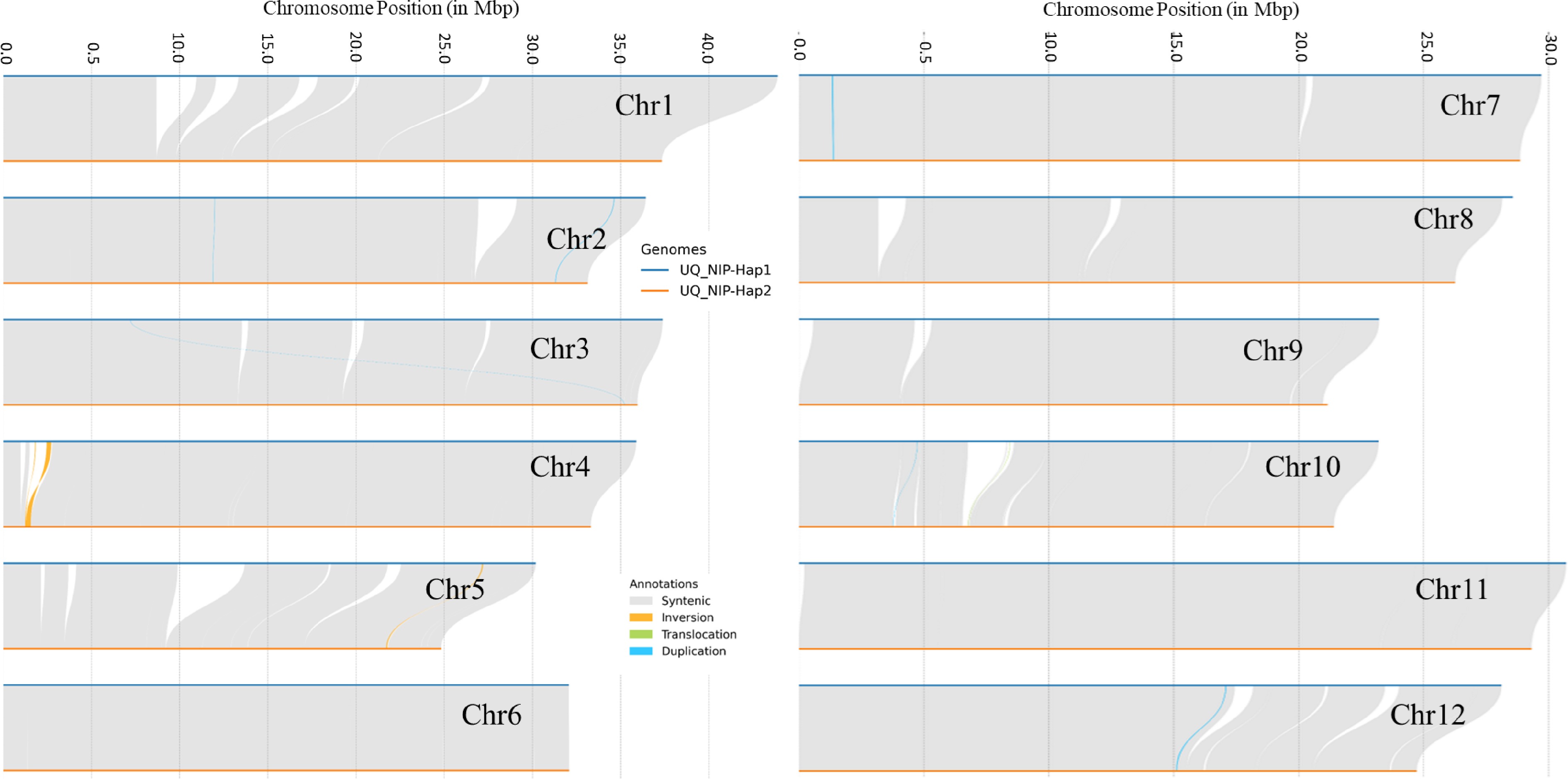
Figure 3.
Sequence collinearity and structural variants, including inversions, translocations, duplications, and non-aligning regions, were examined between the UQ Nipponbare haplotype 1 genome assembly and the UQ Nipponbare haplotype 2 genome assembly. The same method used in Fig. 2 was used for this analysis.
-
The CTAB method[19] was used to extract DNA from young leaves of a rice (Oryza sativa cv Nipponbare) plant grown in a glasshouse at the University of Queensland (Australia). The high quality DNA extracted was sequenced using a PacBio (Pacific Biosciences) Sequel II platform to produce HiFi sequences.
Genome assembly
-
Approximately 54.9 Gb of HiFi reads were obtained. HiC reads (59.6 Gb) were downloaded from the NCBI Sequence Read Archive database (SRR6470741). De novo haplotype-resolved assembly of these reads was performed using hifiasm with parameters '--write-ec --write-paf -l0'[14]. The contig assemblies were scaffolded using the YaHS tool[15]. QUAST and BUSCO were used to evaluate the quality and completeness of a genome assembly[16,17]. A telomere identification toolkit (tidk) was used to search for tandem repeats of the telomeric sequence 'TTAGGG' and 'TAAACCC' and the exact location in the Nipponbare collapse, haplotype-1 and haplotype-2 assembly (
https://github.com/tolkit/telomeric-identifier ).Genome annotation
-
Repetitive DNA sequences were obtained from a Oryza repeat database[18] and used to mask the genome with the Repeatmasker soft masking option[19]. Protein sequences of Viridiplantae from OrthoDB v.11[20] and RNA-sequencing (RNA-seq) reads from the NCBI Sequence Read Archive database (SRR23560402, SRR23560417, SRR23560416, SRR23560419, SRR23560418, SRR23560409, SRR23107175, SRR23107177, SRR23107178, SRR8051554, SRR7974062, SRR8051550) were obtained. Quality and adapter trimmed RNA-seq reads were aligned to the masked genomes using HISAT2[21]. Annotation of protein-coding genes in Nipponbare was conducted using a combination of homology-based prediction, de novo prediction, and transcriptome-based prediction methods using Braker[22]. BUSCO was used to assess the genome annotation completeness. The Large Gap Mapping tool (length fraction; 0.9 similarity fraction; 0.9) of CLC was used to identify the new genes with the comparison of IRGSP-1.0 Nipponbare genes (CLC Genomics Workbench 23.0.05, QIAGEN, USA,
www.clcbio.com ) and further transcript evidence for these new genes was estimated (Supplemental Table S12). The functional annotation of the identified additional genes was performed using OmicsBox 2.2.4[23]. CDS sequences were subjected to a BLASTX analysis with a specific e-value of 1.0E-10 against the non-redundant protein sequences database, utilizing Viridiplantae taxonomy. Subsequently, the CDS sequences were processed through InterProScan, and GO terms were extracted for all matches acquired via the BLAST search, employing Gene Ontology mapping with the Blast2GO annotation tool. The annotations generated from InterProScan and Blast2GO were then harmonized by merging the respective GO terms (Supplemental Table S11). -
The authors confirm contribution to the paper as follows: experiment interpretation: Abdullah M; project supervision: Furtado A, Masouleh AK, Henry RJ; data analysis: Abdullah M, Furtado A, Masouleh AK; draft manuscript preparation and data interpretation: Okemo P; study conception: Henry RJ. All authors approved the final version of the manuscript.
-
The whole genome sequence data reported in this paper have been deposited in the Genome Warehouse in National Genomics Data Center[24,25], Beijing Institute of Genomics, Chinese Academy of Sciences / China National Center for Bioinformation, under accession number GWHEQBP00000000 that is publicly accessible at
https://ngdc.cncb.ac.cn/gwh . This research was supported by the ARC Centre of Excellence for Plant Success in Nature and Agriculture (Grant No. CE200100015).
-
The authors declare that they have no conflict of interest. Robert J. Henry is the Editorial Board member of Tropical Plants who was blinded from reviewing or making decisions on the manuscript. The article was subject to the journal's standard procedures, with peer-review handled independently of this Editorial Board member and the research groups.
-
accompanies this paper at (https://www.maxapress.com/article/doi/10.48130/tp-0024-0007)
-
Received 7 December 2023; Accepted 5 February 2024; Published online 3 April 2024
- Supplemental Table S1 BUSCO statistics of the UQ-NIP-collapsed assembly.
- Supplemental Table S2 QUAST statistics of UQ-NIP-collapsed assembly.
- Supplemental Table S3 UQ-NIP-Collapsed assembly chromosome sizes and number of telomeres.
- Supplemental Table S4 BUSCO statistics of UQ-NIP-Hap1-assembly.
- Supplemental Table S5 QUAST statistics of the UQ-NIP-haplotype1 assembly.
- Supplemental Table S6 BUSCO statistics of UQ-NIP-haplotype 2 assembly.
- Supplemental Table S7 QUAST statistics of the UQ-NIP-haplotype 1 assembly.
- Supplemental Table S8 UQ-NIP-Haplotype 1 chromosome sizes and number of telomeres.
- Supplemental Table S9 UQ-NIP-haplotype1 chromosome sizes and number of telomeres.
- Supplemental Table S10 Summary of sequence data used for genome assembly.
- Supplemental Table S11 Function of the newly identified genes 3050 genes (6423 CDS).
- Supplemental Table S12 RNA-seq support evidence for new genes.
- Supplemental Fig. S1 The Large Gap Mapping tool of CLC was used to identify the new genes by comparison of IRGSP-1.0 Nipponbare with UQ-Nipponbare. The genes from the UQ-Nipponbare were initially mapped to the IRGSP-1.0 Nipponbare genome sequence. After this initial mapping, both the mapped and unmapped genes were then subjected to another round of mapping, this time against the IRGSP-1.0 Nipponbare gene annotation.
- Supplemental Fig. S2 Dotplot of the chromosomes of the UQ_Nipponbare collapsed assembly against those of the UQ_Nipponbare haplotype 1 assembly.
- Supplemental Fig. S3 Dot plot of UQ_Nipponbare collapsed assembly chromosomes against UQ_Nipponbare haplotype 2 assembly.
- Copyright: © 2024 by the author(s). Published by Maximum Academic Press on behalf of Hainan University. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Abdullah M, Furtado A, Masouleh AK, Okemo P, Henry RJ. 2024. An improved haplotype resolved genome reveals more rice genes. Tropical Plants 3: e009 doi: 10.48130/tp-0024-0007 

An improved haplotype resolved genome reveals more rice genes
- Received: 07 December 2023
- Revised: 20 January 2024
- Accepted: 05 February 2024
- Published online: 03 April 2024
Abstract: The rice reference genome (Oryza sativa ssp. japonica cv. Nipponbare) has been an important resource in plant science. We now report an improved and haplotype resolved genome sequence based upon more accurate sequencing technology. This improved assembly includes regions missing in earlier genome sequences and the annotation of more than 3,000 new genes due to greater sequence accuracy. This phased genome will be a useful resource for rice research.
-
Key words:
- Haplotypes /
- Genomes /
- Rice /
- Sequencing