









-
Macadamia, a genus of evergreen trees from the Proteaceae family, is highly valued for its unique flavor, texture, and nutritional properties. It is native to Australia but has now been introduced and widely cultivated in different parts of the world including Hawaii, South Africa, Vietnam, China, and Central and South America. Macadamia is a genus of four species M. integrifolia (Maiden & Betche), M. tetraphylla (L. A. S. Johnson), M. ternifolia (F. Muell), and M. jansenii (C.L. Gross) of which only M. integrifolia, M. tetraphylla, and their hybrids are used for commercial production of edible kernel. The other two species are non-commercial due to the high content of cyanogenic glycosides in the mature kernels[1]. Due to the absence of high-quality genomic data on Macadamia, crop improvement breeding programs have relied heavily on phenotypic characteristics, primarily from the two commercial species. This reliance poses a risk of diminishing genetic diversity[2,3]. Therefore, to enhance breeding accuracy, there is a critical need for high-quality genomic data that can provide comprehensive insights into the genetic makeup and variability within Macadamia species. Among the four species, the M. integrifolia (HAES 741) genome was the first to be sequenced using Illumina short reads[4]. This 518 Mb assembled genome was highly fragmented (N50: 4,745 bp) and incomplete having 77.4% BUSCO genes and covering only 79% of the genome[5]. HAES 741 was again reassembled using combined Pacific Biosciences (PacBio) long-read data along with the Illumina short read sequences[4]. This chromosome level assembly was more contiguous than the previous one with a size of 745 Mb, N50 of 413 kb, and 90.2% of BUSCO genes. M. jansenii was the second macadamia to be sequenced, contig level de-novo assemblies were generated using three different types of long-read sequencing methods[6]. Among the three assemblies, PacBio continuous long reads (CLR) contig assembly outperformed others in terms of contiguity (N50: 1.55 Mb). This PacBio CLR M. jansenii contig level assembly was scaffolded to chromosome level using chromosome confirmation capture (Hi-C), where 762 contigs were reduced to 219 scaffolds where 14 scaffolds were of chromosome length, the genome contiguity was improved more than 50 times (N50: 52.1 Mb) with 97% BUSCO[7].
For the first time, all four Macadamia species were sequenced and assembled using the advanced phase assembly (IPA) assembler with PacBio HiFi reads for each of the four species. This study reported that PacBio HiFi contig level assembly outperformed the earlier CLR contig and scaffold assembly[8]. A further update on the M. jansenii contig level assembly reported the possibility of achieving de novo assembly of near chromosome level from sequenced data alone[9]. Recently, a more contiguous and complete assembly of the M. integrifolia Chinese cultivar -GUIRE 1 (GR1)[10] , a Hawaiian cultivar 'kau'[11] and the M. tetraphylla genome were also reported[12]. The M. integrifolia (GR1) chromosome level genome was assembled using Nanopore sequencing, producing a genome of 807 Mb, with a scaffold N50 of 54.7 Mb and 95.7% BUSCO. The M. integrifolia (Kau) was assembled using PacBio RSII with Hi-C and generated a genome of 794 Mb, with 92% complete BUSCO. The M. tetraphylla genome was assembled with Hi-C to give a 750 Mb genome, N50 51 Mb, and BUSCO of 90%.
The available genome assemblies of macadamia, present a challenge in integrating diverse genomic data due to variability in sequencing technologies and assembly pipelines, hindering a comprehensive understanding for accurate breeding. To address this limitation, this study aimed to assemble all four genomes of Macadamia species using the same sequencing platform and assembly pipeline. This approach enables more reliable and accurate comparative genome analysis. The genomic data generated from this study will help in identifying species-specific genes and the variations among the four species. Genes for desirable characteristics present in the non-commercial species may be identified for incorporation into domesticated cultivars, to widen the gene pool of domesticated macadamia.
-
Long-read PacBio HiFi sequencing was performed on all the four species of Macadamia. The sequencing depths for each species are as follows: M. jansenii (28 X), M. integrifolia (27 X), M. tetraphylla (42 X), and M. ternifolia (37 X)[7]. The collapsed HiFiasm assemblies of the four Macadamia species resulted in highly contiguous assemblies with N50 more than 45 Mb whereas the haploid assemblies were less contiguous and slightly smaller in size as compared to the collapsed assemblies. The M. integrifolia contig assembly had the largest number of contigs, 1049 whereas M. tetraphylla had the least. The haploid 1 assembly of all the species was comparatively more contiguous and longer than the haploid 2 assembly (Supplementary Table S1). The BUCSO analysis revealed a high percentage of genome completeness, with more than 97% coverage. Among the identified BUSCO genes, the majority were found as single-copy genes, with percentages ranging from 83.3% to 84.1%. A small proportion of the BUSCOs were detected as duplicated genes (double BUSCOs), with percentages ranging from 13.4% to 14.2%. Additionally, minor percentages of fragmented BUSCOs in the assemblies, ranging from 0.6% to 0.9% was also reported. The percentage of missing BUSCOs, representing genes absent from the assemblies, were found to be low, varying from 1.4% to 2.6% (Supplementary Table S1).
Chromosome level assembly
-
The Ragtag scaffold assembly length indicated the total size of the genome assemblies for each species, which ranged from 735 to 795 Mb. The collapsed assembly was slightly larger than individual haploid assemblies and the Hap2 assembly had the smallest size, ranging from 735 to 776 Mb for each species. Among the species, M. tetraphylla had the longest collapsed assembly, while M. integrifolia had the shortest. The length of the collapsed assembly for each species reflects the total size of their merged haplotypes, providing a more complete view of their respective genomes. M. tetraphylla had the longest haploid assembly, while M. jansenii had the shortest. Among the chromosomes in the collapsed genome assemblies of the four species, chr 9 (70 to 75 Mb) and chr 10 (68 to 72 Mb) consistently exhibit the greatest lengths. On the other hand, the smallest chromosome in all collapsed assemblies was chr 7 (Supplementary Table S2). The overall BUSCO completeness scores ranged from 95.0% to 98.9%, indicating that a significant proportion of the BUSCOs were present in the assemblies. The majority of BUSCOs were found as single-copy genes, with percentages ranging from 81.6% to 84.2%, confirming the accurate representation of essential genes in the collapsed assemblies. Only a small percentage of BUSCOs appeared as fragmented or missing BUSCO genes, suggesting robust and reliable genome assembly results (Table 1, Supplementary Table S2). The N50 values for the collapsed assemblies ranged from 51.7 to 56 Mb. M. tetraphylla exhibited the highest N50 values, while M. ternifolia had the lowest. These N50 values indicate that the collapsed assemblies have relatively contiguous contigs. The N50 values for the haploid assemblies were generally smaller than those of the collapsed assemblies. The N50 values for the haploid assemblies ranged from 51.4 to 54.8 Mb. The k-mer analysis showed that M. jansenii had the smallest genome and low heterozygosity, whereas M. integrifolia and M. tetraphylla possessed larger genomes and higher heterozygosity. A substantial portion (approximately 63%−69%) of their genetic sequences was found to be unique (Supplementary Table S3 & Supplementary Fig. S1a−d). The genome size estimation by flow cytometry results showed M. tetraphylla had the largest genome size followed by M. ternfolia, which aligns with the assembled scaffolded assembly results (Supplementary Table S3).
Table 1. Chromosome level assemblies of four species of Macadamia representing assembly length, BUSCO and N50 values.
M. jansenii M. ternifolia M. integrifolia M. tetraphylla Hap1 Hap2 Collapsed Hap1 Hap2 Collapsed Hap1 Hap2 Collapsed Hap1 Hap2 Collapsed Assembly length (Mb) 761 735 773 766b 748 780 748 751 775 776 775 795 Complete BUSCO 98.9% 95.0% 97.7% 97.1% 96.5% 97.7% 95.1% 94.3% 97.6% 97.4% 97.3% 97.8% Single 83.3% 82.1% 84.2% 83.8% 83.4% 84.1% 82.4% 81.6% 84.1% 83.5% 83.8% 83.7% Double 13.6% 12.9% 13.5% 13.3% 13.1% 13.6% 12.7% 12.7% 13.5% 13.9% 13.5% 14.1% Fragmented 0.6% 0.6% 0.7% 0.8% 0.8% 0.8% 0.9% 0.6% 0.6% 0.8% 0.7% 0.7% Missing 2.5% 4.4% 1.6% 2.1% 2.7% 1.5% 4.0% 5.1% 1.8% 1.8% 2.0% 1.5% N50 (Mb) 54.2 51.7 54.7 53.8 51.8 53.8 52.8 53 53.7 54 56 56 *The chromosomes were numbered according to the M. integrifolia genome which used the seven genetic linkage maps[4]. Genome structure comparison
-
The genomic structure comparison of the four Macadamia species using SyRI revealed syntenic regions, inversions, translocations, and duplications. Chromosomes 9 and 10 showed several structural rearrangements, with chr 9 exhibiting changes in the first half and chr 10 in the second half. Chr 4 also displayed genomic rearrangements at one end, while chr 12 in all four species showed several duplications in the middle (Fig. 1). Dotplots of the reference genome (M. jansenii Hi-C) against the four Macadamia species (assembled by ragtag) showed varying structural rearrangements, with M. integrifolia and M. tetraphylla having more structural differences compared to M. jansenii (Supplementary Fig. S2 & S3). Among all chromosomes, chr 9 and 10 had the majority of rearrangements. Similarly, dotplot comparison between the haploid assemblies showed M. integrifolia haploids were the most diverse, while M. jansenii haploids were the least diverse (Supplementary Fig. S3). The study showed that the genomes of different Macadamia species have different structures and arrangements, showing their unique genetic characteristics.
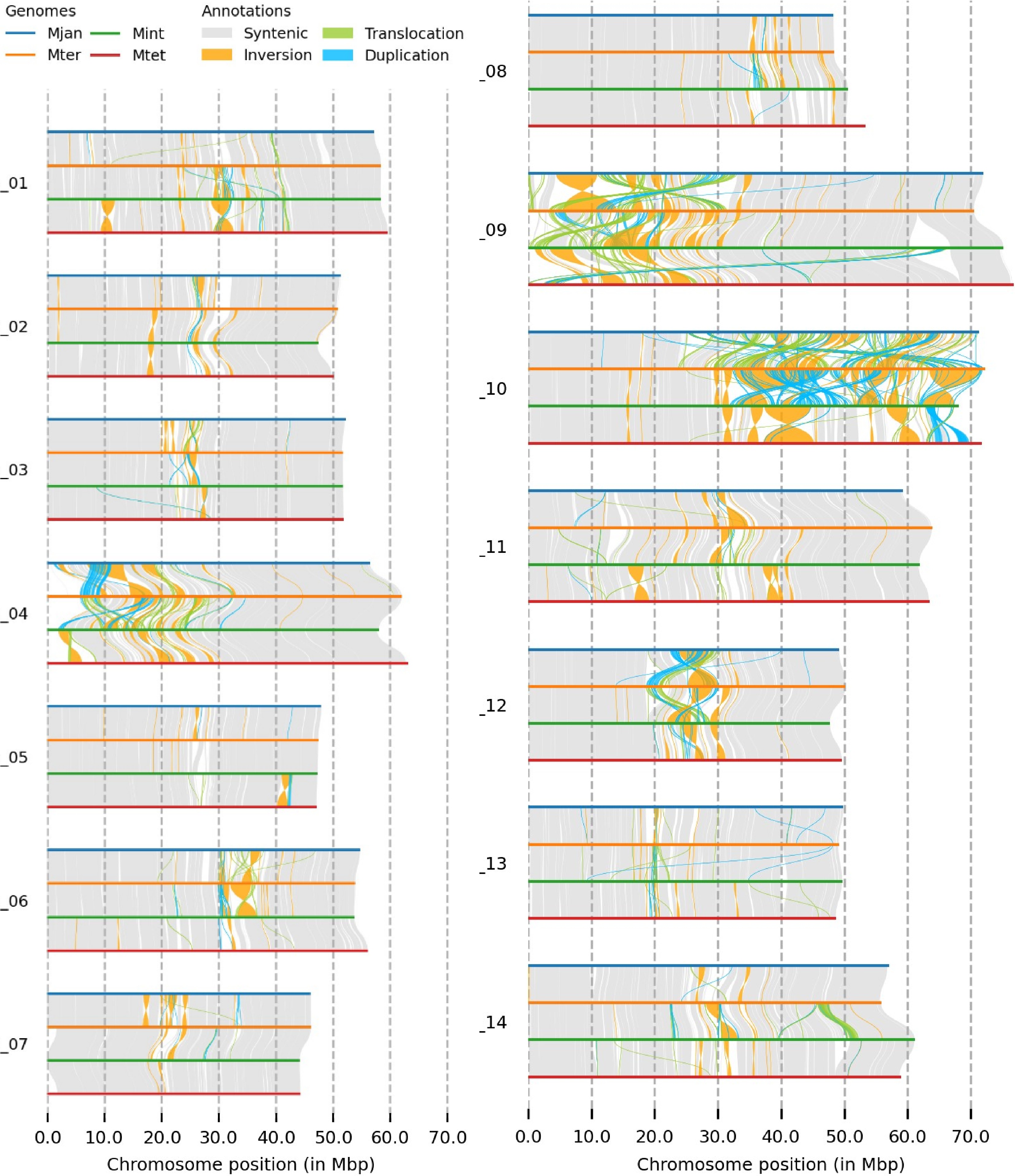
Figure 1.
The genome structure comparison of four Macadamia species, with different colours denoting each species and structural rearrangements (synteny, inversion, translocation, and duplication) as indicated on the top of the image.
Genome annotation
-
The repeat content analysis of the four species identified a total of 61% to 62% across both haploid and collapsed assemblies. This indicates that a major portion of the genomes is composed of repetitive elements. Among the different repeat types, Long Terminal Repeat (LTR) elements were the most prevalent, comprising around 22.1% to 23.8% of the genomes, followed by Long interspersed nuclear elements (LINE) elements. Other repeat types, such as DNA elements, unclassified elements, small RNA elements, satellites, and simple repeats, contributed to a smaller fraction of the total repeat content, ranging from 4.13% to 6.51% (Supplementary Table S4). The consistency of the total repeat content between haploid and collapsed assemblies suggests that the repetitive landscape is preserved even after haplotype merging. Comparing the collapsed assemblies with their respective haplotypes, for the number of predicted genes, it can be seen that the gene content remained relatively stable. Among the collapsed assemblies, M. integrifolia exhibits the highest number of genes, 40,534 while M. jansenii exhibits the lowest number of genes, 37,198. In the haploid assemblies, the number of genes ranges from 36,465 to 47,388. The number of genes distribution across the chromosomes, showed chr 9 and 10 have more genes than the other chromosomes (Table 2). The higher number of CDS and protein sequences identified by Braker3 compared to the gene count is because some genes produce multiple transcripts through alternative splicing. The telomere analysis revealed that the collapsed assemblies generally exhibited 'telomere to telomere' arrangements for most chromosomes. However, a few exceptions were observed, where telomere was present only at one of the ends, suggesting missing or ambiguous telomeric sequences on some chromosome ends (Supplementary Table S5). The functional annotation of the CDS sequences showed a majority of the similarity hits with Telopea, the only other member of the Proteaceae with a high-quality genome sequence. All the species showed similarity with Telopea followed by Nelumbo nucifera and Tetracentron sinense (Supplementary Fig. S4). The pathway analysis of the annotated CDS sequences, identified a consistent number of pathways among the four species, M. jansenii and M. tetraphylla each identified 580 pathways, 578 pathways in M. ternifolia, and M. integrifolia exhibited 581 pathways. The top five pathways, namely purine and thiamine metabolism, response to drought, biosynthesis of cofactors, and starch and sucrose metabolism, were found in all four species. This suggests that these pathways play crucial roles in the biological processes and responses shared by all four species.
Table 2. Distribution of genes across the 14 chromosomes of Macadamia species.
M. jansenii M. ternifolia M. integrifolia M. tetraphylla Hap 1 Hap2 Collapsed Hap 1 Hap2 Collapsed Hap 1 Hap2 Collapsed Hap 1 Hap2 Collapsed Chr_01 2483 2543 2474 2455 2484 2612 2483 2389 2665 2643 2521 2631 Chr_02 2666 2514 2608 2774 2666 2739 2453 2613 2699 2664 2735 2786 Chr_03 2802 2868 2844 3007 2943 3053 2837 2771 2974 2949 2917 3017 Chr_04 2780 2670 2718 2832 2706 2931 2833 2746 3078 3142 2782 2813 Chr_05 2800 2783 2798 2798 2636 2911 2755 2569 2814 2746 2780 2866 Chr_06 2607 2579 2568 2623 2465 2683 2585 2616 2667 2702 2731 2709 Chr_07 2790 2702 2696 2764 2699 2836 2810 2587 2623 2711 2578 2712 Chr_08 2768 2768 2677 2742 2671 2770 2509 2802 2878 3062 2869 2837 Chr_09 2870 2897 2878 2915 2874 3053 3373 2816 3842 3626 2978 3137 Chr_10 2402 2359 2428 2301 2209 2463 2699 2367 3103 3710 2295 2392 Chr_11 2820 2896 2812 2910 2845 3001 2917 2879 3087 2888 3024 2935 Chr_12 2590 2567 2517 2642 2408 2721 2576 2092 2538 2617 2430 2566 Chr_13 2766 2627 2732 2641 2716 2790 2684 2723 2875 2694 2663 2724 Chr_14 2560 2409 2448 2598 2474 2626 2446 2495 2691 2634 2534 2608 Total no. of genes 37704 37182 37198 38002 36796 39189 37960 36465 40534 40788 37837 38733 Number of mRNA 43510 43098 43092 44506 43016 45694 44527 43010 47301 47184 44490 45519 Number of CDS 43510 43098 43092 44506 43016 45694 44527 43010 47301 47184 44490 45519 Gene family analysis
-
Anti-microbial gene analysis: The homologs of an anti-microbial gene was identified in all four species of Macadamia by using a BLAST search. Only one gene was identified in all four species on chr 9. The sequence alignment of the reference gene MiAMP-2 with copies in all four species revealed a high degree of homology (Supplementary Fig. S5). This protein sequence alignment clearly shows four repeated segments with a four cysteine motif C-X-X-X-C-(10 ± 12)-X-C-X-X-X-C.
Fatty acid pathways
-
The number of FatA and FatB genes, essential for fatty acid production varied between species. M. integrifolia had the highest number of both genes, 10 and 11, respectively, suggesting the potential of this species for robust fatty acid synthesis. SAD (Stearoyl-ACP Desaturase) genes, which are mainly responsible for converting stearic acid (C18:0, SA) to oleic acid (C18:1, OA)[13], were present in high numbers across the four species, indicating their active involvement in the desaturation processes. This supports the observations of Hu et al.[14]. The conversion of C16:0 to C18:0 through elongation is a more efficient process compared to the conversion of C16:0 to C16:1 and the desaturation of C18:0 to C18:1 appears to be more effective than the desaturation of C16:0 to C16:1[14]. KAS (Ketoacyl-ACP Synthase) genes, crucial for fatty acid chain elongation, are notably absent in M. integrifolia, potentially indicating a unique fatty acid metabolism pathway in this species. In contrast, the other three species possess KAS genes, particularly M. jansenii and M. ternifolia (10 each), highlighting their capacity for elongating fatty acid chains (Supplementary Table S6a).
Cyanogenic glycoside pathway
-
CYP 79, which catalyses the first step in the biosynthesis of cyanogenic glycosides by acting on amino acids and converting them into aldoximes[15] was found to be present in M. integrifolia and M. tetraphylla and absent in M. jansenii and M. ternifolia, indicating a potential deviation from the typical cyanogenic glycoside biosynthesis pathway in these species. In contrast, CYP71, responsible for further converting aldoximes into cyanohydrin[16], was uniformly present among all the species. The number of BGLU and UGT genes, which are responsible for the detoxification and the glycoside modification was found to vary across the four species, reflecting differences in detoxification capabilities in the cyanogenic pathway. M. tetraphylla lacks UGT genes entirely, potentially indicating unique detoxification mechanisms (Supplementary Table S6b).
WRKY genes
-
The WRKY gene family, known for its key role in plant development and stress responses[17], revealed varying protein counts ranging from 58 to 61 among the four Macadamia species (Supplementary Table S6c). These findings align with the prior discovery of 55 WRKY proteins within the M. tetraphylla genome as reported by Niu et al. in 2022[12].
Orthologous and phylogenetic analysis
-
Orthologous clusters were generated across the four Macadamia species using Telopea as the outgroup, to identify genes that have been conserved across different species and may have similar functions. The clustering patterns of gene families across five plant species: T. speciosissima and the four Macadamia species revealed a total of 195,004 proteins grouped into 34,696 gene clusters. Among all the clusters only 31 clusters showed overlaps among two or more of the plant species and 8,217 single-copy clusters indicated conserved genes among the five species (Supplementary Table S7). A total of 30,111 (15.4%) singleton or species-specific gene were found in 2,090 unique gene clusters, where Telopea contains the maximum number of unique gene clusters (902). Among the Macadamia species, M. integrifolia had the maximum (403) whereas M. jansenii the lowest number of singleton gene clusters (201) (Fig. 2 & Supplementary Fig. S3). The Gene Ontology (GO) enrichment analysis of these unique gene clusters holds great promise in providing valuable insights into the distinct biological functions and potential adaptations of each species.
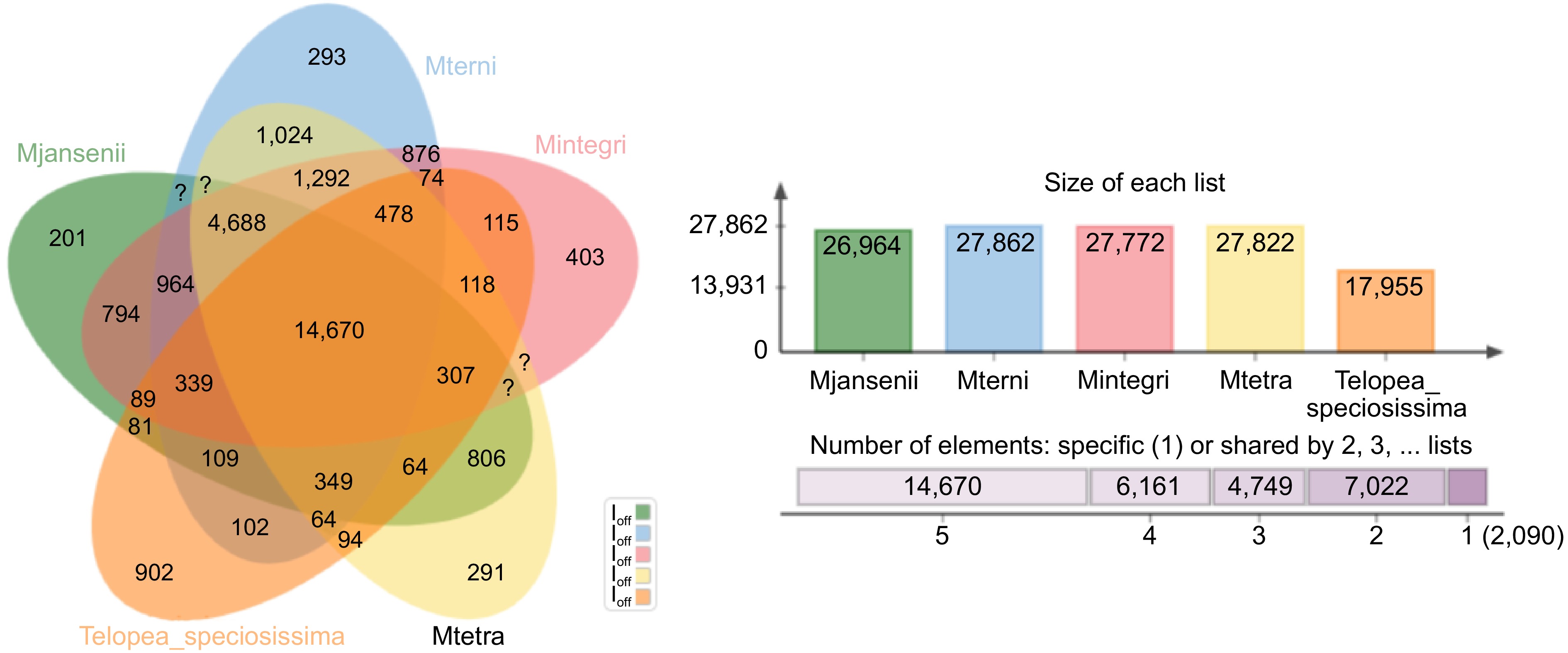
Figure 2.
A Venn-diagram showing clusters of orthologous groups of genes (OGs) for the four Macadamia species and T. speciosissima. Number of orthologous groups (OGs) belonging to core genome (OGs common among all five species- union of all circles), number of singletons (unique genes—outer area of each circle), and the common ones of remaining different combination of all five species (in between the core and the periphery of the diagram) are described.
A phylogenetic tree was constructed to investigate the genetic divergence and evolutionary distances among the Macadamia species, with Telopea as the outgroup. The tree has two main branches. One branch includes M. integrifolia and M. tetraphylla, indicating a shared genetic lineage. The other branch comprises M. jansenii and M. ternifolia, highlighting their distinct genetic lineage. (Supplementary Fig. S6 & S7).
WGD and synteny
-
The analysis of ks values in all four species of Macadamia genomes revealed a distinctive peak at ks ≈ 0.32 (Fig. 3). The Telopea genome exhibited a peak at ks ≈ 0.28. This comparison of the peaks in Macadamia and Telopea suggests a more recent whole-genome duplication (WGD) event in Telopea compared to Macadamia. In some WGD studies, WGD and divergence time estimation have been based solely on ks values. However, in recent years, there has been growing research cautioning against exclusively relying on ks plot analysis for these estimations. Instead, additional sources of evidence are recommended to ensure more robust WGD assessments[18,19].
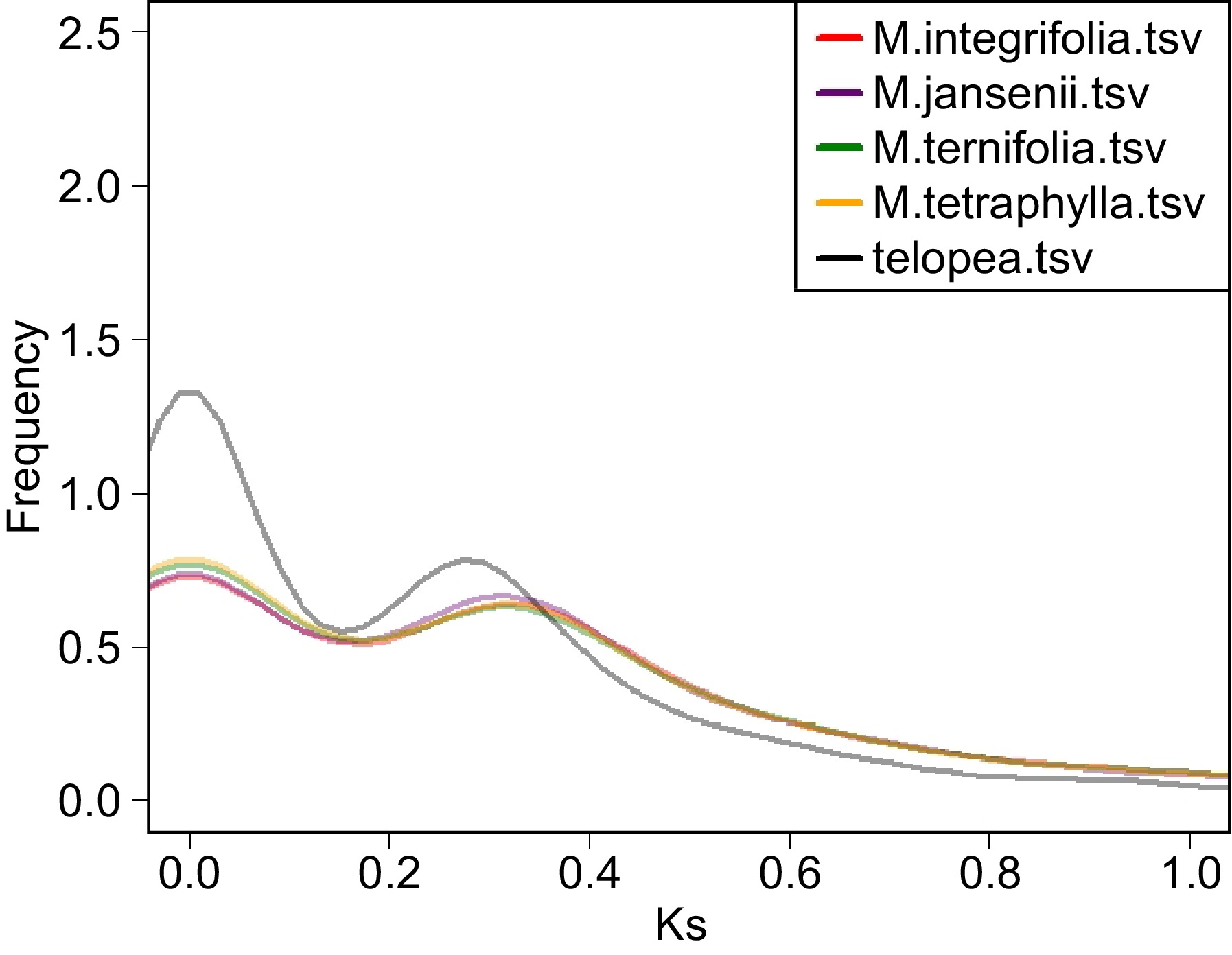
Figure 3.
Ks distribution plot of the four Macadamia species and Telopea. The colour code of each species is provided on the top left corner.
The duplication events were further verified using the synteny plots which highlighted the duplicated genetic regions and genes. Synteny analysis revealed extensive genetic similarity within the species and among the four species, particularly on chromosomes 9 and 10 (Fig. 4 & Supplementary Fig. S8)
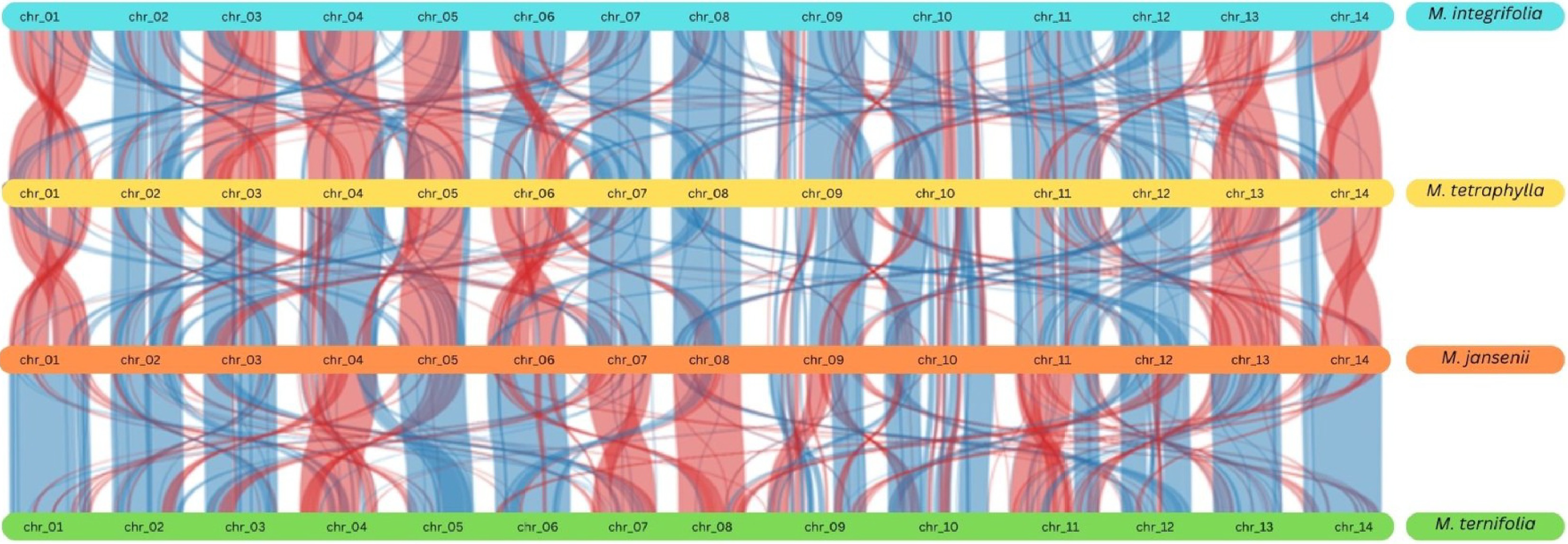
Figure 4.
Synteny plot across all the four Macadamia species. The vertical lines connect orthologous genes across the four species. The blue coloured ribbons represent the regular conserved regions while the red ribbons represent the inverted regions.
Expansion-contraction of gene families
-
The study of differences in protein families among the annotated species revealed significant differences between the groups. The protein family size varied notably between the Macadamia species and Telopea. A total of 613 different protein clusters were contracted and only 21 protein family clusters showed expansion in Macadamia as compared to Telopea. Among the two clades of Macadamia, the edible, species (M. integrifolia and M. tetraphylla) exhibited more expansion- contraction (+18/−140) than the bitter non-edible species (+0/−5) (Fig. 5). Among five contracted clusters of the bitter species, one cluster belonged to Xanthotoxin 5-hydroxylase CYP82C4, which is expressed in roots under iron-deficient conditions.
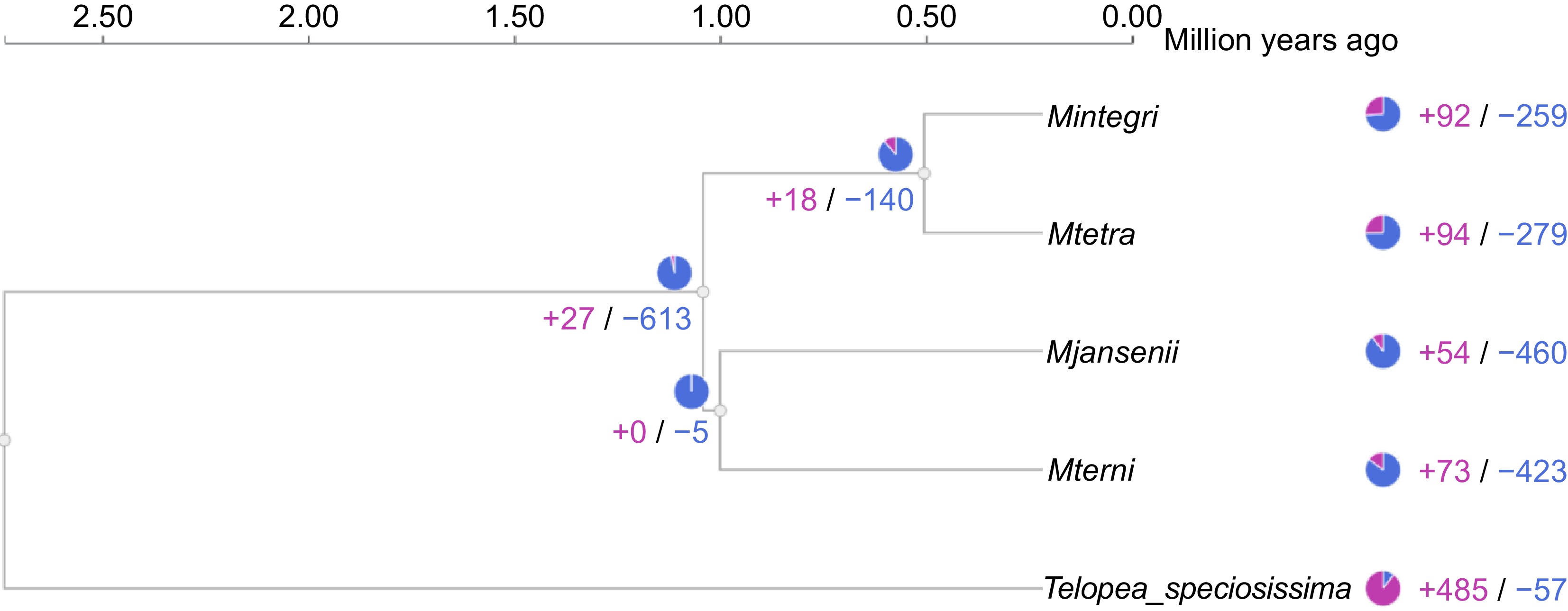
Figure 5.
Gene family expansion and contraction across the Macadamia species and Telopea. The blue colour represents contraction and pink presents expansion of gene clusters.
All the four species of Macadamia individually displayed more contraction than expansion. The expansion ranges from 259 to 423 clusters of protein, where M. jansenii showed the highest number of contractions, followed by M. ternifolia, and M. tetraphylla. Whereas only 54−94 protein clusters were expanded, and M. tetraphylla displayed the highest expansion of proteins (+94), one of these expanded clusters was associated with the GO term 'rejection of self-pollen' However, for Telopea the opposite was found with more expansion than contraction (+485/−57) of protein clusters (Fig. 5). Both the edible species show similar changes and the gene enrichment analysis of both also showed a similar pattern, and the same held true for the non-edible species.
-
In this study, high-quality reference genomes and annotations were created for the four species of Macadamia. The gene model set completeness, as measured by BUSCO, suggested that the annotation pipeline used was suitable for comprehensive capture of protein-coding genes. The comparison of genome assemblies of the already available genomes of M. jansenii, M. integrifolia, and M. tetraphylla with those generated in this study revealed notable improvements in the assembly statistics. For M. jansenii, the newly assembled genome demonstrated an increase in length (from 758 to 773 Mb), improvement in N50 value from 52 to 55 Mb, and slight improvement in BUSCO as compared to the already available M. jansenii Hi-C assembly[7]. This study has greatly improved the M. integrifolia (cultivar 741) genome with a longer assembly length of 775 Mb and a significantly higher BUSCO of 97% and N50 value of 53 Mb as compared to previous assemblies by Nock et al., in 2016 (N50: 4.7 kb)[5] & 2020 (N50: 413 kb)[4]. Similarly, the M. tetraphylla genome showed great improvement in terms of N50 56 Mb and 98% BUSCO as compared to the already available M. tetraphylla genome[12]. The genome assemblies generated in this study provide enhanced continuity, higher BUSCO completeness, and increased gene identification compared to previous versions, providing a robust basis for genome comparison. Additionally, the genome assemblies attained complete chromosome coverage from telomere to telomere for most of the chromosomes, which has not been reported in the previous studies.
The comparison of collapsed assembly statistics of four Macadamia species revealed M. tetraphylla assembly stood out with the longest genome length. The M. jansenii has the shortest assembly length among the four. The gene content comparison across the four species revealed that M. integrifolia assembly exhibited the highest number of genes, followed by M. ternifolia and M. tetraphylla. These variations in gene counts may be attributed to species-specific genomic features. Haploid-resolved assemblies are essential in genomics research, as they facilitate accurate gene phasing, improved annotation, and enhanced insights into genetic diversity[20−22]. Heterozygosity between the haplotypes in diploids can complicate the genome assemblies. The low heterozygosity of M. jasnenii and high heterozygosity of M. integrifolia and M. tetraphylla[4,7,10,12] was also supported by k-mer analysis, haploid assembly statistics and dotplot comparisons. The dot plot comparison of the two M. jansenii haploid assemblies, showing minimal differences between the two. On the other hand, the highly heterozygous species, M. integrifolia and M. tetraphylla, exhibit significant differences in the dot plots, gene numbers, structural rearrangements and individual chromosome lengths. These findings highlight the genomic variations at haploid levels among the different Macadamia species, providing valuable insights into their genetic diversity.
Antimicrobial proteins (AMP) are essential components of plant innate immunity, exhibiting diverse activities such as antibacterial, antifungal, insecticidal, and antiviral effects, enabling effective defense against pathogens and pests[23,24]. Comparative analysis of the gene encoding a well-known AMP protein across the four macadamia species, showed that the gene location remained conserved on chr 9 across all the species and the sequence alignment revealed a highly conserved motif of cysteines, however, the amino acid sequence was variable. These results aligned with earlier reports of these novel proteins[23−25]. The present study reveals the sequences of the genes and confirms a high level of conservation across the Macadamia species.
The variable distribution of CYP79, across the four species, may indicate potential deviations from the conventional cyanogenic glycoside biosynthesis pathway in the two bitter species, M. jansenii and M. ternifolia. In contrast, CYP71's uniform distribution across all species, indicating its essential role. The differential counts of detoxifying enzymes, BGLU and UGT, underscores species-specific strategies, with lack of UGT genes in M. tetraphylla suggesting a different detoxification mechanism. These insights will be important in breeding new varieties making use of genotypes from all four species. Breeding new varieties with smaller trees for intensified production will require introgression of smaller plant stature from the two bitter species (M. ternifolia and M. jansenii). This will require avoidance of the transfer of the cyanogenic glycoside genes associated with bitterness.
A unique feature of Macadamia is the novel fatty acid composition. The analysis of fatty acid pathway genes showed M. integrifolia stands out with the highest numbers of both FatA and FatB genes, signifying a strong capability for fatty acid production and may explain the domestication of Macadamia being based mainly on this species. Additionally, the higher abundance of SAD genes across the four species suggests their active role in desaturation, as confirmed by Hu et al.[14], highlighting the efficiency of C18:0 to C18:1 conversion. The absence of KAS genes in M. integrifolia suggests a potential uniqueness in its fatty acid metabolism pathway, distinct from the other three species, which possess KAS genes (especially M. jansenii and M. ternifolia with 10 each), highlighting their capacity for extending fatty acid chains. Variations in WRKY protein counts (ranging from 58 to 61) across Macadamia species supporting their roles in development and stress responses. This analysis has greatly expanded knowledge of fatty acid biosynthesis in Macadamia and identified significant species differences. This suggests the need for further studies of the differences in lipid composition of Macadamia.
Utilizing long-read assemblies in this study of Macadamia gene families significantly increased the accuracy of results for expansion and contraction events. This accuracy is crucial for identifying essential genes and gene families involved in important biological processes and hence the accurate interpretation of expansion-contraction (CAFE) analysis. Remarkably, the edible macadamia species demonstrated a higher incidence of expansion-contraction, while the bitter species exhibited fewer changes. This observation implies potential differences in the distribution of gene families between the two groups, suggesting distinct evolutionary trajectories. Understanding the factors behind the expansion of particular gene families in edible Macadamia species could provide valuable clues about the evolution of Macadamia and be harnessed for the development of improved cultivars with desirable traits. Moreover, the presence of common ks peak events in the four Macadamia species suggest significant evolutionary events that have shaped their genomes. Comparison of the ks plot between the Macadamia and the Telopea genomes, suggests that Telopea has also undergone a duplication event. This may be a separate event but the possibilities of different rates of sequence evolution make conclusions difficult. Synteny analysis further highlights the conservation of genetic regions and genes within each species and reveals intriguing similarities among the different species, particularly on chromosomes 9 and 10. These findings emphasize the importance of whole genome duplication in shaping the genetic landscape of macadamia and provide valuable insights into the evolutionary dynamics of this economically important crop. The analysis of orthologous clusters and gene families among the four Macadamia species and Telopea provided valuable insights into the conservation and divergence of genes in these plants. Among the 195,004 proteins grouped into 34,696 gene clusters, only 31 clusters showed overlaps among two or more species, while 8,217 clusters contained conserved single-copy genes across the five species. These unique gene clusters hold great promise for uncovering distinct biological functions and potential adaptations of each species. The phylogenetic tree, with Telopea as the outgroup, demonstrates two main branches: one containing M. integrifolia and M. tetraphylla and the other comprising M. jansenii and M. ternifolia, illustrating the genetic relationships among the Macadamia species. The core orthologous genes, as expected included gene families related to categories like cell growth, DNA replication and repair, metabolism, and cell cycle regulation.
The comparative genomics and experimental study, presented here, allows for the first time a genus-wide view of the biological diversity of the Macadamia, which provides a strong foundation for genome-wide analysis.
-
Leaf samples of all four species were freshly collected from trees in ex-situ collections, with specimens gathered from both Nambour and Tiaro locations, operated by the Department of Agriculture and Fisheries. Accessions: M. jansenii (ANAM82-5-11), M. integrifolia (741), M. tetraphylla (GTIARO1-17-7) and M. ternifolia (GTIARO1-2-14). Leaf tissue was ground under cryogenic conditions, utilizing a mortar, pestle, and Tissue Lyser. DNA isolation was conducted on all accessions, following a modified extraction method[26] excluding phenol. The HiFi sequencing data of the four Macadamia species[8] was used for this study. RNA sequence data for M. jansenii was used from Sharma et al.[8]. Total RNA for M. ternifolia and M. tetraphylla were extracted from fresh leaf tissues using the RNA isolation method of Rubio-Piña & Zapata-Pére[27] along with the Qiagen kit method and sent for short read sequencing at Macrogen Oceania, NSW, Australia. RNA Seq data for young leaves of M. integrifolia (HAES 741) was downloaded from NCBI SRA data SRR10897159.
Genome assembly
-
The HiFi reads of four species were assembled using HiFiasm to generate both the collapsed and the haploid assemblies[8,22]. The contig assembly generated from HiFiasm was then scaffolded using a reference-guided approach with the RagTag tool[28] using M. jansenii Hi-C as the reference[7]. The chromosomes were numbered according to the M. integrifolia genome[5]. The contigs of more than 1 Mb in size were used as input for the reference-guided approach. To assess the completeness of the assembles, the Benchmarking Universal Single-Copy Orthologs (BUSCO) (version 5.4.6)[29] was used with the eudicots_odb10 dataset. The genome completeness was evaluated using the quality assessment tool QUAST[30].
Genome estimation (flowcytometry and k-mer) and dot plots
-
For flow cytometry methods nuclei were extracted from leaf tissue by mechanical dissociation as described by Galbraith et al.[31] with modifications for woody plant species. Briefly, 40 mg of young macadamia leaf was co-chopped with 15 mg of the internal standard Oryza sativa ssp. Japonica cv. Nipponbare, in 0.4 mL of ice-cold nuclear isolation buffer in a 5 cm polystyrene Petri dish. For M. tetraphylla and M. integrifolia, Arumuganathan & Earle[32] nuclear isolation buffer was used; while MB01[33] nuclear isolation buffer was used for M. ternifolia and M. jansenii. Samples were chopped for approximately 10−12 min, first into fine longitudinal strips with new parts of a sharp razor blade and then into perpendicular slices. The resulting homogenates were gently filtered through a pre-soaked 40-μm nylon mesh into a 5 mL round bottom polystyrene tube. Homogenates were then stained with 50 μg/mL of propidium iodide (PI) (Sigma, P4864-10ML) and 50 μg/ml of RNase A (Qiagen, 19101) for 10 min on ice. The BD Biosciences LSR II Flow Cytometer and FlowJo software package was used to analyze the homogenates. Briefly, fluorescence was collected using a 488 nm excitation laser tuned to 514.4 nm and a 610/20 nm bandpass filter. Instrument settings were kept constant across and throughout experiments: forward scatter voltage at 199, side scatter voltage at 300, fluorescence intensity voltage at 500, with a slow flow rate (20−50 events/s). Three biological replicates were performed on three different days. For each biological replicate, a minimum of 1,500 PI-stained events were collected per PI-stained peak. Nuclear DNA content was calculated as previously described[34] using 388.8 Mb at 1C for the assumed size of O. sativa[35].
Genome estimation using K-mer analysis was performed by Jellyfish's Version 2.3.0[36] count and histo commands. The histo file was visualized in genomescope[37]. Dot plots for the assembly comparisons were plotted using the Chromeister[38] tool available at Galaxy Australia (
https://usegalaxy.org.au ).Genome annotation
-
The identification and classification of the de novo repeat elements in all the collapsed assemblies of all four species was performed using the RepeatModeler (version 2.0.2a) (
www.repeatmasker.org/RepeatModeler ). The repeats identified were then masked by repeatmasker (version 4.0.9) (www.repeatmasker.org ). The gene models in the masked assemblies were identified using an ab initio method along with RNA-seq evidence Braker3 version 3.0.3[39]. To prepare the input files for the Braker3 run, the masked assemblies were first aligned with RNA-seq using HISAT2 version 2.1[40], then the output aligned .sam file was converted to a .bam file using samtools[41]. The soft masked genome assembly file along with the sorted bam file was used as input files for the Braker3 pipeline. The protein and coding sequence (CDS) fasta files generated from Braker3 contain multiple transcripts therefore a Python script was used to keep only one transcript per gene. The filtered protein and CDS fasta was then used for the downstream analysis. Tidk version 0.2.31 (Telomere identification toolkit) tool (https://github.com/tolkit/telomeric-identifier ) was used to identify the telomere region in the genome assemblies using 'search' and 'plot' commands.Functional annotation of the gene set identified for each of the four genomes was performed through Omicx box (version 3.0.27) (OmicsBox, 2019). This pipeline consists of BLAST2GO[42] and Interproscan[43]. For BLAST2GO, the 'blastx-fast' feature was used with NCBI non-redundant protein sequences (nr v5) database and the e-value was set at 1e-10 with 10 blast hits. The taxonomy filter was set at 33090 Viridiplantae. For Interproscan all the available databases such as families, structural domains, sites, and repeats databases were selected. For the pathway analysis: Plant reactome (Gramene)[44] and KEGG pathway[45] were performed using Omics box.
Gene family analysis: Anti-microbial genes were identified across the four species by conducting a BLAST homology search, looking for transcripts resembling M. integrifolia's antimicrobial cDNA (MiAMP2). Sequence alignment using Clone Manager ver 9.0 was performed with an alignment parameter scoring matrix of Mismatch (2), Open Gap (4), and Extension-Gap (1). To identify genes involved in cyanogenic glycoside, fatty acid metabolism, and WRKY gene across the four genomes, BLAST was performed and the top hits based on sequence similarity were selected.
Orthologous and phylogenetic analysis
-
Orthologous and phylogenetic analysis was performed using Orthofinder (V2.5.5)[46] using the protein sequences of all the four Macadamia species along with data for Telopea. The common and unique set of orthologous protein sequences among the five species were plotted using the UpSet plot and the venn diagram of the Orthovenn3[47]. The core or single copy orthologs obtained from Orthofinder were used to construct the phylogenetic tree using Orthovenn3.
Whole genome duplication
-
Whole genome duplication (WGD) analysis was performed to compute the whole set of paralogous genes in the genome using WGD tool version 1.1.2[19,48]. Ancient WGDs were calculated by examining the distribution of synonymous substitution per site (Ks) within a genome or Ks distribution. WGD analysis of all four species of Macadamia was performed to estimate the origin and diversification. Wgd 'dmd' and 'ksd' commands were used to generate the Ks distribution plot.
Conservation of gene order and genomic regions
-
A pairwise whole-genome comparison was performed using SyRI[49] to find the structural and sequence differences between the two genomes. The genomes were first aligned using the minimap2[50] and samtool[41] was used to index the alignment BAM file. The BAM file was then used to run the SyRI tool, the same output file was then passed through the visualisation tool plotSR[51] using default parameters to visualize the synteny and the structural rearrangements between the Macadamia species.
Collinearity and expansion-contraction of gene families
-
The degree of collinearity within and between the genomes of the four Macadamia species were calculated by using MCScanX[52]. The protein fasta file of all four species were combined and used as input for the all-vs-all homology search with the Blastp algorithm with e-value set at 1e-10, max target sequences at 5, and output format 6. The resulting tabular blastp file along with the combined gff file was then fed into MCScanX using default parameters. For self synteny MCScanX was run with default settings with the blastp output and the gff file of individual species. The web-based tool - SynVisio[53] was used to visualize collinearity. The CAFE5 tool of Orthovern3 was used to perform the expansion and contraction of the gene families. All default parameters were used.
-
The authors confirm contribution to the paper as follows: project design and supervision: Henry RJ, Masouleh AK, Furtado A, Topp B ; genome assembly, annotation and downstream analysis: Sharma P, Masouleh AK; flow-cytometry analysis: Constantin L; draft manuscript preparation: Sharma P, Constantin L; data deposition: Sharma P. All authors reviewed the results and approved the final version of the manuscript.
-
The genome sequencing data from PacBio has been submitted under NCBI bioproject PRJNA694456. The genome assemblies and annotation of four Macadamia species have been deposited in the Genome warehouse under the bioproject: PRJCA020274. NCBI genome submission ids for M. integrifolia: SUB14785838, M. tetraphylla: SUB14787551, M. jansenii: SUB14787648 & M. ternifolia: SUB14786002.
This project was funded by the Hort Frontiers Advanced Production Systems Fund as part of the Hort Frontiers strategic partnership initiative developed by Hort Innovation, with co-investment from The University of Queensland, and contributions from the Australian Government. RH was funded by the Australian Research Council (CE200100015). We thank the Research Computing Centre (RCC), University of Queensland for support and providing high performance computing resources. We are also thankful to Virginia Nink and the Queensland Brain Institute Flow Cytometry Facility for technical assistance with flow cytometry.
-
The authors declare no conflict of interest. Robert J. Henry is the Editorial Board member of Tropical Plants who was blinded from reviewing or making decisions on the manuscript. The article was subject to the journal's standard procedures, with peer-review handled independently of this Editorial Board member and the research groups.
-
accompanies this paper at (https://www.maxapress.com/article/doi/10.48130/tp-0024-0029)
-
Received 11 February 2024; Accepted 25 March 2024; Published online 21 October 2024
- Supplementary Table S1 HiFiasm contig assembly statistics and Benchmarking Universal Single Copy Gene (BUSCO) completeness in four Macadamia species.
- Supplementary Table S2 Chromosome-level assemblies for four Macadamia species, indicating the length of each chromosome and the overall assembly length.
- Supplementary Table S3 Genome estimation statistics of four Macadamia species through K-mer analysis (using Jellyfish tool) and flow cytometry.
- Supplementary Table S4 Repeat element distribution across Macadamia species.
- Supplementary Table S5 Telomere distribution across all the four macadamia assemblies.
- Supplementary Table S6 Distribution of gene families (Fatty acid, cyanogenic and WRKY) across the four species of Macadamia.
- Supplementary Table S7 Distribution table of Orthologous gene clusters across the four Macadamia species and Telopea.
- Supplementary Fig. S1 (a): K-mer profile (k = 21) spectrum analysis to estimate genome size of M. jansenii generated from short read sequence data using Jellyfish and GenomeScope. (b): K-mer profile (k = 21) spectrum analysis to estimate genome size of M. ternifolia generated from short read sequence data using Jellyfish and GenomeScope. (c): K-mer profile (k = 21) spectrum analysis to estimate genome size of M. integrifolia generated from short read sequence data using Jellyfish and GenomeScope. (d): K-mer profile (k = 21) spectrum analysis to estimate genome size of M. tetraphylla generated from short read sequence data using Jellyfish and GenomeScope.
- Supplementary Fig. S2 Dotplots illustrating the genomic comparison of M. jansneii Hi-C assembly (used as reference) against all the four assembled Macadamia genomes.
- Supplementary Fig. S3 Dotplots illustrating the genomic comparisons between the haploid assemblies of each Macadamia species.
- Supplementary Fig. S4 Species distribution graph of coding sequences of M. jansenii.
- Supplementary Table S6a
- Supplementary Fig. S5 Multiple sequence aligmnet of Antimicrobial protein across the four Macadamia species. 01, 02, 03, 04: represents AMP protein sequence from M. jamsenii, M. ternifolia, M. integrifolia and M. tetraphylla, respectively.
- Supplementary Fig. S6 Distribution of unique and common orthologous gene clusters across the Macadamia species and Telopea.
- Supplementary Fig. S7 Phylogenetic tree of Macadamia species with Telopea, with number of orthogroups corresponding to each species in purple.
- Supplementary Fig. S8 Self synteny of four Macadamia species, showing the collinearity of genes across the genome assemblies.
- Copyright: © 2024 by the author(s). Published by Maximum Academic Press on behalf of Hainan University. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Sharma P, Masouleh AK, Constantin L, Topp B, Furtado A, et al. 2024. Genome sequences to support conservation and breeding of Macadamia. Tropical Plants 3: e035 doi: 10.48130/tp-0024-0029 

Genome sequences to support conservation and breeding of Macadamia
- Received: 11 February 2024
- Revised: 15 March 2024
- Accepted: 25 March 2024
- Published online: 21 October 2024
Abstract: Macadamia, a genus native to Eastern Australia, comprises four species, Macadamia integrifolia, M. tetraphylla, M. ternifolia, and M. jansenii. Macadamia was recently domesticated largely from a limited gene pool of Hawaiian germplasm and has become a commercially significant nut crop. Disease susceptibility and climate adaptability challenges highlight the need for a wider range of genetic resources for macadamia production. High-quality haploid resolved genome assemblies were generated using HiFiasm to allow comparison of the genomes of the four species. Assembly sizes ranged from 735 to 795 Mb and N50 from 53.7 to 56 Mb, indicating high assembly continuity with most of the chromosomes covered from telomere to telomere. Repeat analysis revealed that approximately 61% of the genomes were repetitive sequences. The BUSCO completeness scores ranged from 95.0% to 98.9%, confirming good coverage of the genomes. Gene prediction identified 37,198 to 40,534 genes. The species shared a common whole genome duplication event. Synteny analysis revealed a high conservation and similarity of the genome structure in all four species. Differences in the content of genes of fatty acid and cyanogenic glycoside biosynthesis were found between the species. An antimicrobial gene with a conserved cysteine motif was found in all four species. The four genomes provide reference genomes for exploring genetic variation across the genus in wild and domesticated germplasm, targeting conservation of genetic resources and supporting plant breeding.
-
Key words:
- Genome assembly /
- Genome annotation /
- Comparative genomics /
- Wild species