









-
Accurate and suitable flowering time is the basis for plants to survive, and procreate as well as adapt to the local environment. Flowering in plants, which indicates the transition from the vegetative stage to the reproductive stage, is co-regulated by various external and endogenous factors, such as photoperiod, temperature, and hormones[1]. Specifically, the morphogenesis and flowering time of soybean are precisely regulated by the photoperiod, which determines yield potential and restricts soybean cultivars to a narrow latitude range. Consequently, the molecular basis for photoperiod recognition has sparked broad interest among experts from various fields. Nowadays, many photoreceptors and circadian clock homologs, such as phytochrome A (phyA), FLAVIN-BINDING, KELCH REPEAT, F-BOX 1 (FKF1), GIGANTA (GI), Pseudo Response Regulator (PRRs) and components of Evening Complex (EC: composed of ELF3, ELF4 and LUX) are identified and characterized as being involved in the photoperiod sensitivity in angiosperms, including rice, wheat, soybean, corn, and Arabidopsis, while their regulatory mechanisms dramatically vary among the different crops[1,2,3]. A recent study by Zhao et al. revealed a dynamic transcriptional and translational suppression feedback loop between FKF1/GI-EC to determine photoperiod sensitivity in soybean[4], which provides new insights for understanding latitude adaptability and night-break response in soybean.
HTML
-
In the concept of Taiji, Yin and Yang represent two antagonistic yet interdependent relationships that forms a dynamic balanced cycle. Zhao et al. described how the photoperiodic Taiji loop, E2, the homologous protein of Arabidopsis GI, interacts with a E3 ligase FKF1 in vitro and in vivo confirmed by pull-down, yeast two-hybrid, and co-immunoprecipitation, then targets J/ELF3 protein for degradation, symbolizing Yang as it occurs during the daytime. When night arrives, the expression of E2 not FKF1 increased in j and lux1 lux2 mutants compared with Williams 82. Furthermore, J protein forms EC complex with GmLUX, reciprocally repressing the transcription of E2 through binding to its genomic region, which was confirmed by ChIP-qPCR assay, symbolizing Yin as it occurs at night. Simultaneously, both E2 and EC functions genetically rely on each other. The balance between Yin and Yang is crucial for soybean photoperiod sensitivity[4]. When the function of components is lost from either part of Yin or Yang, the balance will be disturbed, leading to a reduction in sensitivity to changes in photoperiod. For example, fkf1a fkf1b, and e2 e2la e2lb mutants stand to outcompete Yin, causing plants to be insensitive to photoperiod and exhibit early flowering time phenotypes. Notably, E3/GmphyA3 and E4/GmphyA2, which encode phytochrome A homologs are previously shown to promote the degradation both of J and LUX protein[5,6], thus the early flowering phenotype of e3 e4 mutants show outcompeting of Yin is inconsistent with the Taiji loop[4]. Accordingly, the loss of function of GmEID1, the positive regulator of J protein will lead to late flowering[5]. Therefore, it is exciting to investigate whether the phenotype of other factors involved in regulating the activity of FKF1, E2, and EC are in conformity with the photoperiodic Taiji loop. If it matches, the photoperiodic Taiji loop will provide an easy way to speculate the approximate flowering time of soybean.
-
To survive, plants have to evolve complex mechanisms to rapidly respond to their daily and seasonal changing environments. In Arabidopsis, the circadian component GI interacts with ZTL, FKF1, and COP1 in a time-dependent manner to control the stability of CO protein from different layers, to finely regulate the transcriptional level of FT[2]. Importantly, the coincidence of GI with FKF1 is regulated by photoperiod, leading to differential degradation of CDF1 (CYCLING DOF FACTOR 1) and differential transcriptional levels of CO between LD and SD conditions, which is a way for Arabidopsis to recognize photoperiod[7]. Interestingly, there is an OsGI/OsFKF1-OsCDF1 molecular module in rice, however, this module appears to be opposite to that of Arabidopsis. Firstly, the downstream target genes of the module in rice are specific, namely Ehd2, Ghd7, and Ehd1[8]. Secondly, OsCDF1 may help the OsFKF1/OsGI complex promote flowering instead of antagonizing its function[9]. Excitingly, Zhao et al.[4] revealed that the homologs of GI, namely E2, E2La, and E2Lb, collectively determine photoperiod sensitivity in soybean by repressing the expression of the gene E1. Mechanically, E2 can recruit GmFKF1a and GmFKF1b, and target EC complex component J/ELF3 protein for degradation under non-inductive LD photoperiod. Genetically, the flowering phenotype of fkf1a fkf1b e2 triple mutant is similar to the e2 single mutant, suggesting FKF1s act upstream of E2. Therefore, J/ELF3 is identified as a specific target of the GmFKF1s/E2 complex in soybean, as GI didn't participate in regulating ELF3 stability in Arabidopsis. Interestingly, Zhao et al. analyzed the daily expression of J protein in E2 and e2 background as well as in FKF1a FKF1b, and fkf1a fkf1b backgrounds through immunoblot assay, and found that J protein accumulates only in LD conditions in e2 mutants but accumulates under both LD and SD conditions in fkf1a fkf1b background, indicating E2 functions as a scaffold linking GmFKF1 with J and promotes the degradation of the latter only under LD conditions. Nevertheless, their study did not give a clear explanation of E2's effect on J protein under SD conditions[4], which awaits being fully explored in the future.
Taken together, it seems that there is a conserved FKF1-GI interaction in different species. However, the function and downstream targets which determine flowering time are considerably different.
-
In summary, Zhao et al. have established a novel transcriptional-translational suppression feedback loop in soybean to determine photoperiod sensitivity, which enriches the photoperiodic flowering network (Fig. 1). Zhao et al. focused on the function of E2 and its two homologs in the photoperiod-mediated flowering pathway and raised the focus of E2 in other undefined biological processes. Moreover, they surprisingly found that E2 can physically interact with J but not with LUX, and promotes the degradation of J. This provides a new regulatory factor on EC activity. Subsequently, they detected and proved the interaction of E2 with FKF1s acting like the complex of GI-FKF1 in Arabidopsis, which are blue light-activated and photoperiod regulated[7]. Also, the expression of E2 is regulated by EC in LD. Together, the two parts of the FKF1/E2-EC photoperiod feedback loop can responsd to environmental light signals.
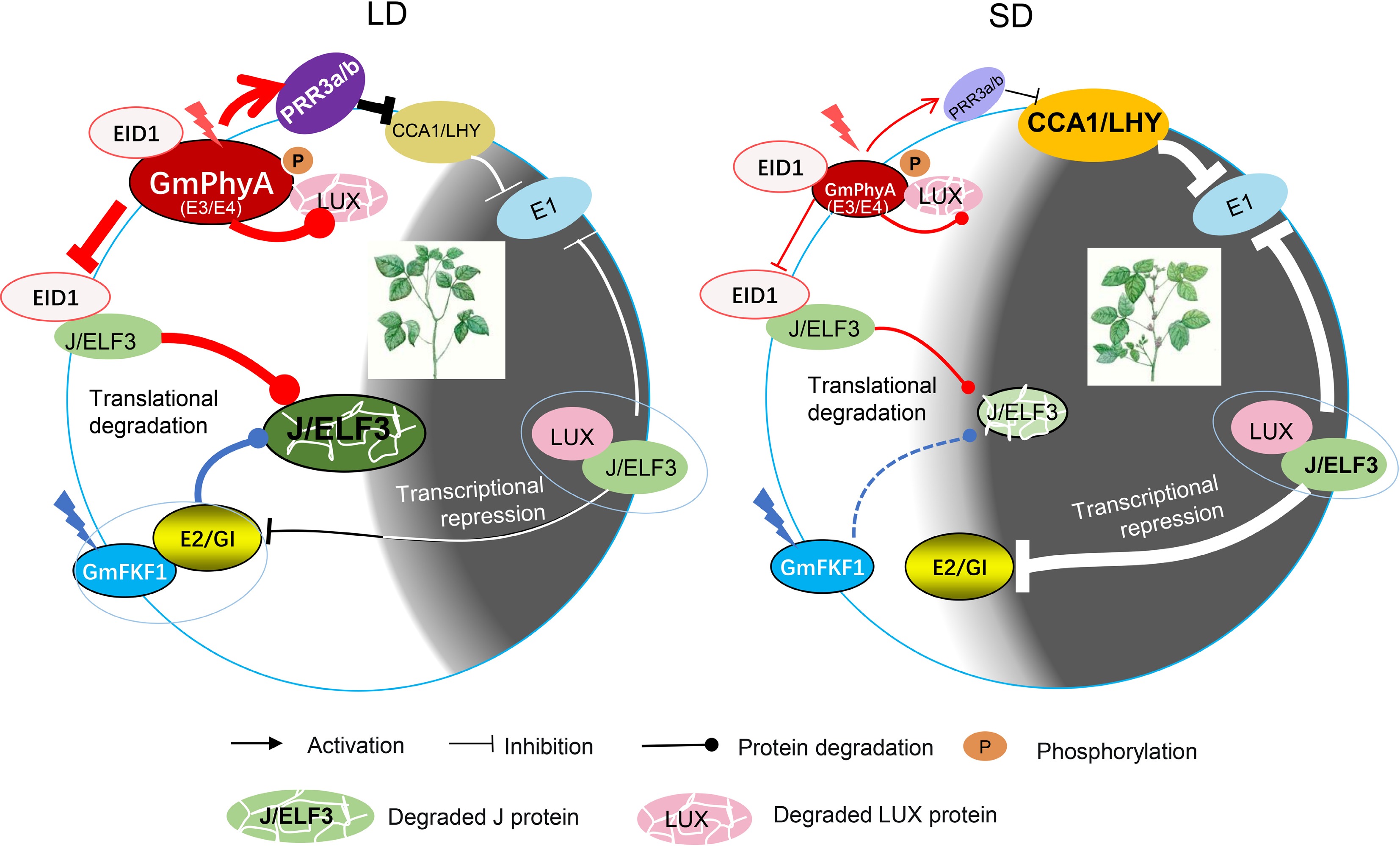
Figure 1.
Photoperiodic flowering network in soybean. Under LD conditions, photoexcited FKF1 interact with E2 and target J protein to degradation. Also, photoexcited E3/E4 interact with GmEID1, acting as a competitive interaction inhibitor of GmEID1 and J, which also leads to degradation of J. Photoexcited E3/E4 also interact and promote the degradation of LUX. E3/E4 can promote the expression of PRR3a/PRR3b to inhibit CCA1 and LHY, the repressor of E1. Together, E1 is highly expressed and delays flowering of soybean under LD conditions. Under SD conditions, the functions of E3 and E4 are compromised, which may indirectly promote the accumulation of J, LUX and CCA1/LHY, therefore effectively inhibit the expression of E1. Moreover, although FKF1 can somehow promote the degradation of J protein, the degradation of J by E2 and the transcription regulation of E2 by EC disappear. Hence, E1 is highly repressed and promotes flowering under SD condition.
Except for photoperiod sensitivity, as discussed by Zhao et al., the photoperiodic Taiji loop also contributes to understanding of latitude adaptability. In SD conditions, J represses the expression of E1, releasing the repressive activity of E1 on FT, thereby promoting flowering in soybean. Consequently, the natural mutation of J will result in a relatively long vegetable phase and enhance yield, which is helpful for soybean cultivars possessing mutated J, adapted to low latitude regions. Soybean cultivars possessing e2 are usually adapted to high latitudes[3,10]. Tof11 and Tof12, two homologs of the PRR3 protein, have been shown to be beneficial in promoting the expansion of soybean to middle and high latitudes mainly by repressing the expression of LHY homologs, a repressor of E1[11]. It is an open question whether Tof11 and Tof12 participate in regulating both E2 and EC as PRRs family proteins do in Arabidopsis.
Meanwhile, the model explains the night break as light intervals at night will stimulate the formation of the E2-FKF1 complex and activate phyA, thereby degrading J and LUX proteins, releasing E1 and delaying flowering[5,6]. Although the delayed flowering phenotype makes a steady fit with the explanation, it is controversial whether light intervals can make enough E2-FKF1 complex to degrade J protein, since it is also unclear the effect of light intervals on the expression pattern of E2 and FKF1.
-
The authors confirm contribution to the paper as follows: draft manuscript preparation: Wang X, He Y, Wang L; diagram drawing: Wang X, Xu H; manuscript revise: Wang L. All authors approved the final version of the manuscript.
-
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
This work was supported by the National Key Research and Development Program of China (2023YFF100300) and Science & Technology Specific Projects in Agricultural High-tech Industrial Demonstration Area of the Yellow River Delta (2022SZX13) to Wang L.
-
The authors declare that they have no conflict of interest.
- Copyright: © 2024 by the author(s). Published by Maximum Academic Press on behalf of Hainan Yazhou Bay Seed Laboratory. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
Wang X, He Y, Xu H, Wang L. 2024. Taiji loop composed of circadian component mediates photoperiodism in soybean. Seed Biology 3:e014 doi: 10.48130/seedbio-0024-0013


|