









-
Genetic diversity due to genetic variation is the cornerstone for ecosystem diversity and species diversity. Genetic variation is a change in the genetic material of an organism that can be passed on to future generations, including chromosomal variation, genetic recombination, genetic mutations, and the emergence of orphan genes (OGs)[1]. OGs are a group of genes in a taxonomic group for which no obvious homology can be found at the genomic level, also known as 'lineage-specific genes'[2,3]. The first report of OGs in Saccharomyces cerevisiae was released in 1996[4]. However, in the early stage, researchers knew little about OGs due to the limitation of sequencing ability to obtain the complete genome of many lineages[5]. Over recent decades, following the speedy advancement of sequencing technologies, complete genomic and transcriptomic sequences of a large number of species have been obtained quickly, accurately, and inexpensively[6]. Each freshly sequenced genome consists of a portion of OGs, and therefore, OGs in organisms such as the silkworm[7], sweet orange[8], and tea plant[9] have begun to be studied extensively. As research has continued, based on previous work on complete genomes in such species as Human, Drosophila, and Arabidopsis thaliana, researchers have proposed several mechanisms for the origin of OGs related to gene duplication and subsequent sequence divergence, transposable elements, lateral gene transfer, and de novo origination[10−14].
However, the functions of most OGs are not annotated due to the lack of homologous genes and functional domain information[15]. Although it is challenging to analyze the biological functions using comparative genomics, some preliminary explorations can be made based on their sequence structural features. OGs have a shorter generation time in comparison to non-orphan genes (NOGs), and therefore, there are some identifiable differences in gene and protein length, exon number, GC content, transcript support, and position preference on chromosomes. In sweet orange, the OGs had a lower average number of exons per gene, both shorter genes and protein length, higher GC content, as well as priority distribution on certain chromosomes, in comparison to NOGs[8]. Similar results were found in A. thaliana[15], Drosophila[16], Poaceae[17], zebrafish[18], and tea plant[9].
A considerable amount of research has now proven that OGs have a crucial impact on growth and development[19,20]. RNA interference of 200 OGs in Drosophila melanogaster induced down-regulation of their expression and was found to result in lethality mainly concentrated in the metaphase[21]. In plants, transgenic transfer of the OG (QQS), which is responsible for influencing the process of carbon and nitrogen segregation of proteins and carbohydrates in A. thaliana, into soybean was found to affect the protein content of soybean seeds similarly[22]. It is noteworthy that tissue-specific expression of OGs exists, with a preference in animals for expression in male reproductive tissues such as the testis[13], exemplified by the species-specific chimeric gene Sdic1 encoding the sperm-specific kinesin intermediate chain in D. melanogaster[23]. A similar phenomenon is present in plants, where tissue-specific expression of OGs tends to be reflected in the male reproductive system, for instance, in pollen. By way of exemplification, the orphan protein encoded by the Ms2 gene caused male sterility in wheat, barley, and phragmites[24]. Besides, OGs have been shown to play an essential part in environmental adaptation. Drought-induced over-expression of the OG (UP12_8740) in cowpea increased its tolerance to osmotic stress and soil drought[25]. Similarly, preferential expression of OGs under abiotic stress was reported in water flea[26], A. thaliana[27] and rice[28].
The Chinese rose (Rosa chinensis) is an important horticultural crop and one of the most commonly cultivated ornamentals in the world, with considerable economic value in potted and cut flowers. The bright colors of roses are visually attractive, and therefore, R. chinensis is often used as a suitable demonstration plant for the production of new flowering varieties[29]. R. chinensis is also the dominant species used for hybridization and was introduced to Europe in the 18th century as a parent of modern roses[30]. In the present study, the published R. chinensis genome[31]was used to identify OGs and a comprehensive analysis was performed, including sequence structural features, subcellular localization, gene duplication, and chromosome distribution. In addition, a weighted gene co-expression network analysis (WGCNA) was built to project the functions of these identified OGs. The differences in the expression of OGs at different times under salt and drought stress were also analyzed to explore the adaptation of OGs to the environment. In conclusion, the present results provide valuable clues to reveal the evolution, characterization, and environmental adaptation of OGs in R. chinensis.
-
Genomes and annotation information for R. chinensis were obtained from the Rosa database (
https://lipm-browsers.toulouse.inra.fr/pub/RchiOBHm-V2/ ). Predicted proteins of Rosa rugosa were downloaded from the eplant (http://eplant.njau.edu.cn ). All other Rosaceae predicted proteins were downloaded from the NCBI Datasets (www.ncbi.nlm.nih.gov/datasets ). Unique transcripts (PUT) assembled from plant mRNA sequences were downloaded from PlantGDB (https://goblinp.luddy.indiana.edu/prj/ESTCluster ), and 122 plant genomes predicted proteins were extracted from Phytozome (https://phytozome-next.jgi.doe.gov ), and 77 from eplant (http://eplant.njau.edu.cn ) were downloaded. UniProtKB was obtained from Uniprot (www.uniprot.org/uniprotkb ) and NR database were downloaded from NCBI (ftp://ftp.ncbi.nlm.nih.gov ), individually.For the prediction of the potential function of OGs, we collected RNA-seq data from publicly available materials to acquire the gene expression levels. Such data consisted of different tissues or varying stress of R. chinensis. The transcriptome data was downloaded from the National Center for Biotechnology Information database (NCBI;
www.ncbi.nlm.nih.gov ) with BioProject accession number PRJNA546486 (leaf, stem, root, pistil ovary, prickle, and stamen), PRJNA722055 (leaf imposed to drought stress: 0, 30, 60, and 90 d) and PRJNA587482 (root imposed to salt stress: 0, 2, 24, and 48 h).Identification of OGs
-
The advancement of comparative genomics has led to a much more improved investigation of the origin and evolution of OGs. A homolog-based search was performed in a pipeline to identify R. chinensis within OGs (Fig. 1). Initially, R. chinensis protein sequences were scoured against the R. rugosa proteome with the BLASTP. Any R. chinensis protein sequence with BLASTP hit with an E-value cutoff of 1e-5 was discarded once available. Homology searches were then conducted with genomes of other Rosaceae plants, Phytozome, eplant, Plant-PUTs database, Uniprot-KB database, and NR database sequentially with an E-value cutoff of 1e-5. Finally, the genes without homologs in any databases were the OGs[15,32], whereas all the alternatives with homologous were non-orphan genes (NOGs).
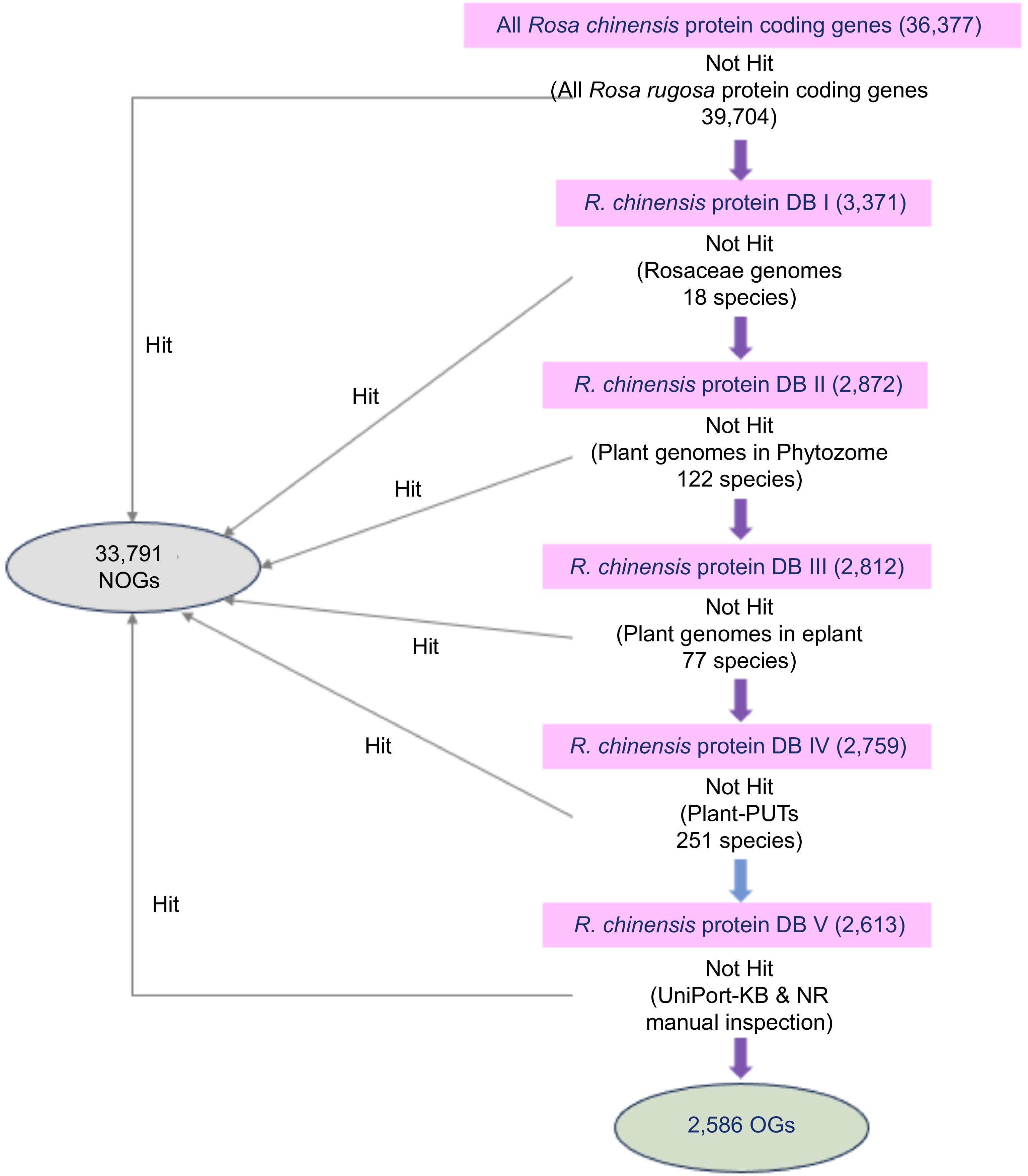
Figure 1.
Procedure for identifying the orphan genes in R. chinensis genome. The purple arrows represent a homolog-based search by BLASTP with an E-value cutoff of 1e-5. The blue arrow represents a homolog-based search by TBLASTN with an E-value cutoff of 1e-5. Genes without homologs in any databases were identified as OGs (2,586), while genes with homologs were classified as NOGs (33,791).
Genic features
-
To visualize the structural characteristics of the OGs, a genome-wide profile of R. chinensis was applied. DAMBE7 software was employed to evaluate the isoelectric points of OGs and NOGs[33]. Discrepancies among OGs and NOGs, including gene size, length of the protein, size of the exons and introns, number of the exons, and content of GC were computed with the use of in-house Python scripts. The Wilcox rank-sum test was then used to identify the significant difference across distinct groups for OGs and NOGs. Information on chromosome localization was retrieved from chromosome sequences and plotted using MapGene2Chrom (
http://mg2c.iask.in/mg2c_v2.0 ). A final BUSCA (Bologna Unified Subcellular Component Annotator) was applied to predict the OGs subcellular localizations[34].Gene duplication analysis
-
Based on previous studies, there are varied models interpreting the origin of OGs[3,14], of which gene duplication has been considered the dominant mechanism underlying the emergence of OGs[35]. The present work started with a BLASTP search for homologous genes with an E-value cutoff of 1e-8, followed by the identification of different types of gene duplication using MCscanX with was capable of detecting WGD, tandem duplication, proximal duplication, transposon duplication, and dispersed duplication[36].
Gene expression analysis
-
To analyze tissue expression, growth and development, and ability to adapt to the environment of R. chinensis, the transcriptome data was downloaded. The raw RNA-seq data was in turn filtered using the Trimmomatic program[37]. With the aim of identifying differentially expressed genes (DEGs) between treatments, we used the default settings of the abundance_estimates_to_matrix.pl, run_DE_analysis.pl (DESeq2) and analyze_diff_expr.pl modules of the Trinity package. Significant differences in gene expression were ascertained using the |log2FC| ≥ 1 with a false discovery rate (FDR) < 0.05 as thresholds, and RSEM implemented in the Trinity package was employed to calculate FPKM (fragments per kilobase of exon per million fragments mapped)[38]. A cluster analysis was conducted using R software targeting particular expressed genes based on RNA-seq data and following functional validation was selected. It was assumed that genes with FPKM value > 0.02 were already expressed[39]. Besides, genes that were exclusively expressed in specific tissue were determined by PaGeFinder software with specificity measure (SPM)[40], and once the SPM value was ≥ 0.9, the gene in that tissue was identified specifically.
Weighted gene co-expression network analysis (WGCNA) and function annotation
-
Following the discard of genes with FPKM < 1, WGCNA was constructed and the genes were grouped into modules aided by the WGCNA package in R software[41]. The automatic network builder function block-wise Modules were utilized to construct the network with default parameters. After that, eigengene values were computed for individual modules for every tissue and the module with the highest correlation coefficient, while fulfilling a p-value < 0.05, was picked to serve as the tissue-specific module for further analysis. The candidate with the most significant representative gene in each module was assumed to be the module eigengene. Module membership[10] and gene importance[32] were calculated for every ME in each tissue-specific module, and once MM > 0.95 and GS > 0.85, the gene was deemed to be the central gene of the module. KEGG enrichment analysis was carried out on an online platform, OmicShare (
www.omicshare.com ). -
The OGs of R. chinensis were characterized according to the methodology employed in recent studies (Fig. 1) with the recently published database resources[15,42,43]. A total of 36,377 annotated protein-coding genes within the R. chinensis genome were used for BLASTP with all R. rugosa protein-coding genes (39,704) presented in this study. During this procedure, there were a total of 33,006 genes with significant similarity (E-value < 1e-5), and 3,371 genes (DBI) were kept for the follow-up analysis. The NOGs indicating homology were eliminated, and a further scan of the remaining genes were carried out alongside the published genomes of the Rosaceae family. Altogether 2872 genes (DBII) were kept for further analysis at this step. The NOGs displaying homology were dropped and the remaining genes were matched with 122 plant genomes in Phytozome for an additional search, yielding 2,812 genes being retained for the following step (DBIII). After comparing these 2,812 genes with the 77 plant genomes in the eplant database, 2,759 genes were not found as homologs (DBIV). Of these 2,759 genes subsequently matched against 251 PlantGDB-assembled Unique Transcripts (PUTs) sequences and no homologs were found for 2,613 genes (DBV). The last step, was to completely erase the impact of false positives from the analysis, leftover genes were examined in the context of UniProt-KB and NR databases, an action that finally left 2,586 genes. These leftover 2,586 genes were labelled as OGs in the R. chinensis genome, representing 7.11% of the entire genome of R. chinensis (Supplementary Table S1), as opposed to these remaining 33,791 genes whose similarity to the databases was defined as NOGs.
Features of OGs
-
Aiming at clarifying any significant differences between OGs and NOGs, the analysis highlighted and compared the sequence structural features between 2,586 OGs and 33,791 NOGs found in the present study. The results showed a significantly smaller gene size (Wilcox rank sum test, p < 2.2e-16) and protein size (Fig. 2a, Wilcox rank sum test, p < 2.2e-16) for OGs than for NOGs (Table 1), where 1,512.34 bp was the OGs gene size and 72.5 amino acids (aa) was the OGs protein size, 2868.43 bp for NOGs gene size and 378.4 aa for NOGs protein size, which suggested that the NOGs protein was 5.22-fold lengthening than OG protein (Table 1). An in-depth analysis on the structural components of the genes revealed that the shorter protein size was predominantly attributed to the lower number of exons (Fig. 2b, Wilcox rank sum test, p < 2.2e-16). The exon size (Fig. 2c, Wilcox rank sum test, p = 7.789e-15) and intron size (Fig. 2d, Wilcox rank sum test, p < 2.2e-16) of OGs, however, were both remarkably bigger than NOGs. The GC content and the isoelectric point of OGs were further comparatively examined with those of NOGs. It was shown that the GC contents of OGs were lower than that of NOGs (Table 1, Fig. 2e, Wilcox rank sum test, p < 2.2e-16). In contrast, the isoelectric point of OGs (8.53) was higher than that of NOGs (7.42) (Table 1, Fig. 2f, Wilcox rank sum test, p < 2.2e-16). In general, these results suggested that there existed significant differences in genetic features of OGs and NOGs.
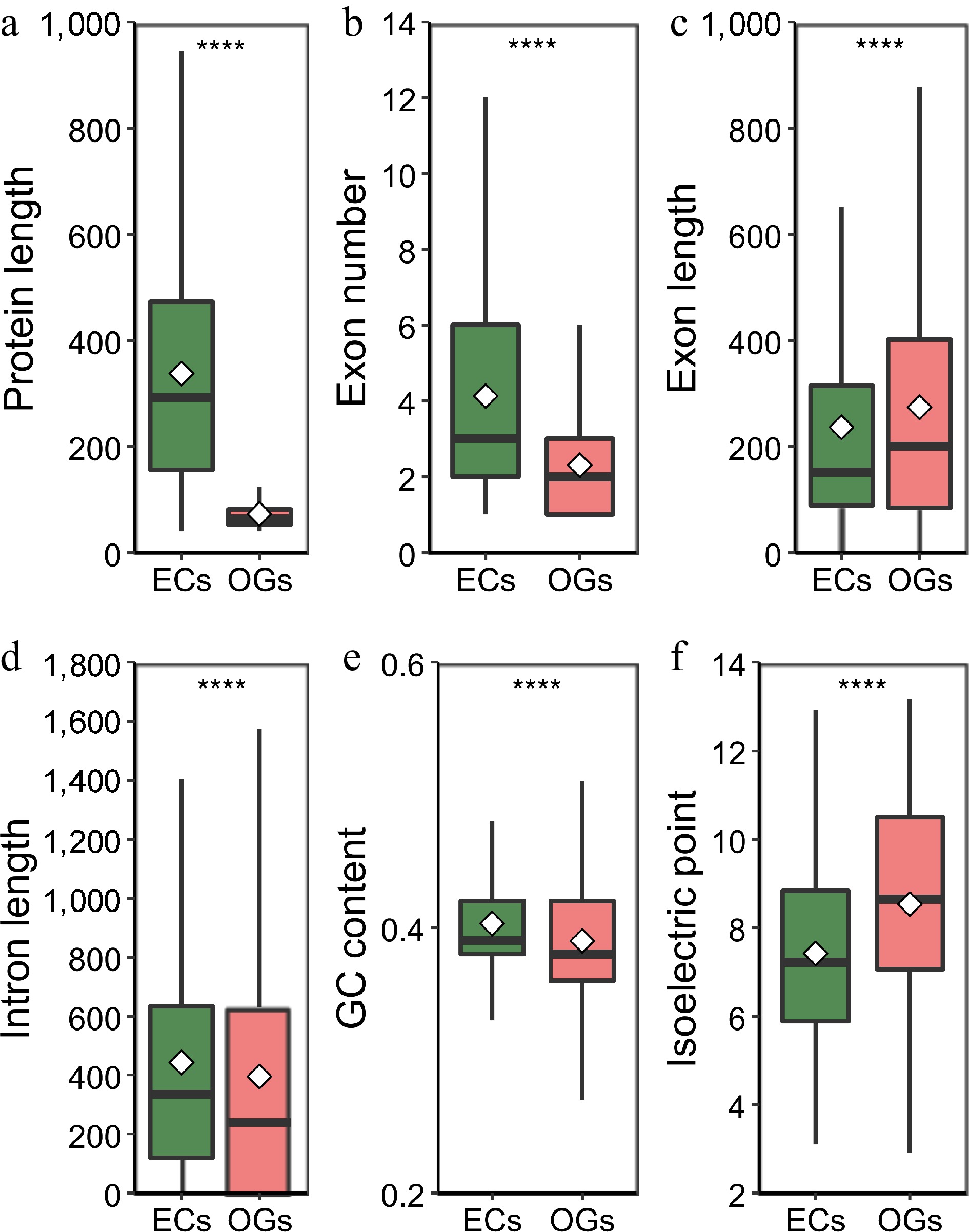
Figure 2.
Analysis and comparison of the structural characteristics of orphan genes (OGs) and non-orphan genes (NOGs). (a) Box-plot comparisons of protein length. (b) Exon number per gene. (c) Exon length. (d) Intron length. (e) GCs content. (f) Isoelectric point. White squares represent the mean value. **** indicate significance levels at p < 0.0001.
Table 1. Genic features of orphan genes (OGs) compared with non-orphan genes (NOGs).
Items OGs NOGs Wilcox rank sum test
probabilityMean (SE) Median Mean (SE) Median Gene size (bp) 1,512.34 (1,736.26) 880 2,868.43 (2,590.26) 2,239 < 2.2e-16 Protein size (aa) 72.5 (32.17) 63 378.4 (306.51) 306 < 2.2e-16 Exons per gene 2.34 (1.99) 2 4.82 (4.74) 3 < 2.2e-16 Exon size (bp) 347.84 (385.78) 225.5 322.22 (423.62) 164 7.789e-15 Intron size (bp) 571.3 (998) 270 556.31 (731.19) 356 < 2.2e-16 Gene GC content (%) 39 (4.85) 38.05 40.31 (4.06) 39.46 < 2.2e-16 CDS GC content (%) 41.9 (6.47) 41.46 45.08 (4.54) 44.35 < 2.2e-16 Isoelectric point 8.53 (2.34) 8.64 7.42 (1.94) 7.21 < 2.2e-16 For the analysis of OGs' genomic distribution, the OGs were plotted over the chromosomes of R. chinensis based on the available information from genome annotation (Supplementary Table S1). A total of 2560 OGs were spread over seven chromosomes. A maximum number of OGs per chromosome was in the order of Chr2 (465), Chr5 (423) and Chr1 (395). The percentages of OGs distributed on the seven chromosomes were 7.97%, 7.01%, 6.84%, 7.37%, 6.91%, 6.41%, and 7.29%, respectively (Fig. 3b), demonstrating that there was no chromosomal preference in the distribution of the OGs in R. chinensis. Besides, the distribution density of OGs was higher near the telomeres, and the distribution was relatively balanced on the chromosomes apart from the aggregation phenomenon in some chromosomal regions (Fig. 3c). Generally, the spread of OGs on these seven chromosomes was reasonably uniform.
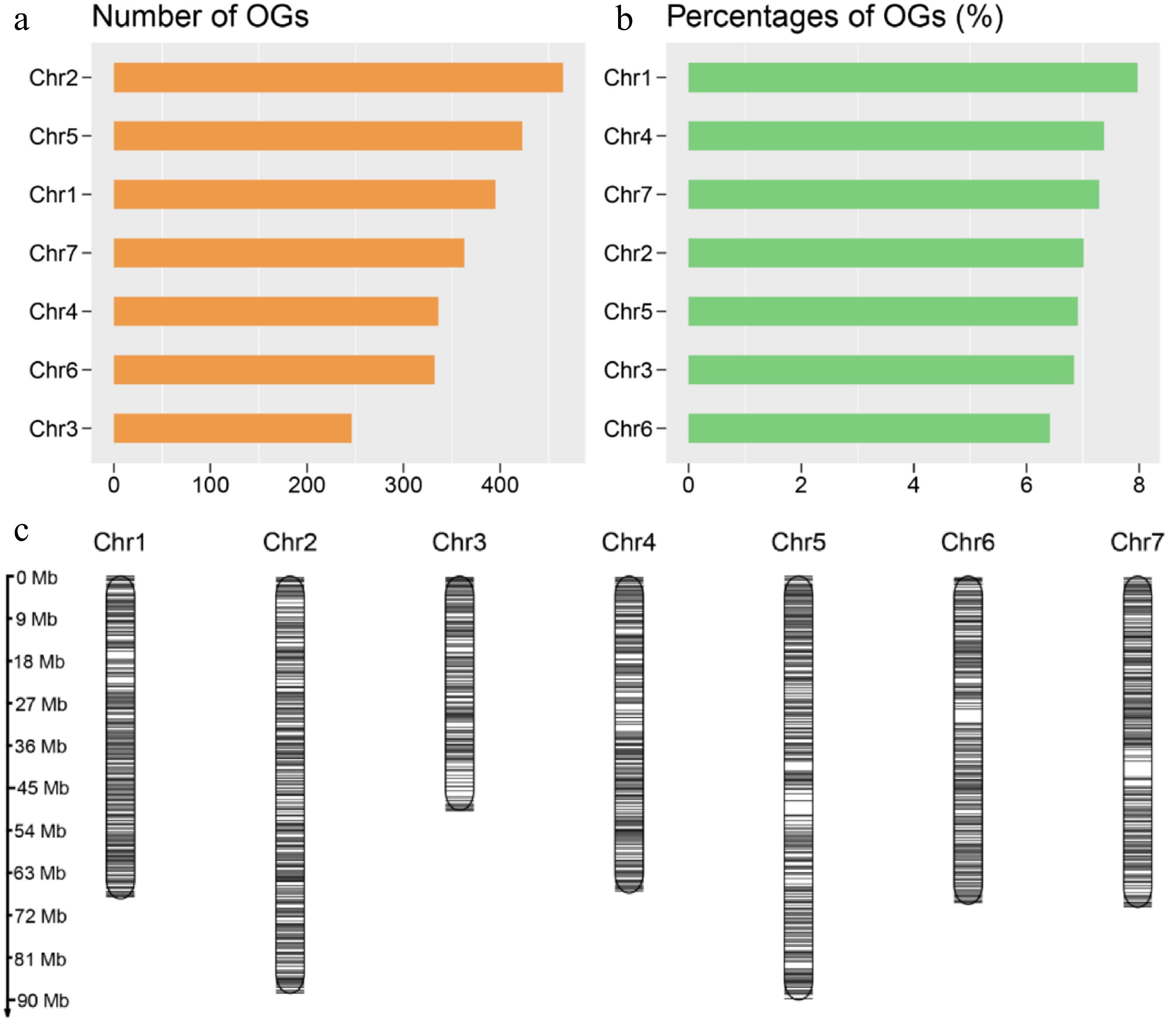
Figure 3.
Orphan genes (OGs) distribution on chromosomes. (a) The numbers of OGs on each chromosome of R. chinensis. (b) Percentage of OGs on each chromosome of R. chinensis. (c) Chromosomal distribution of the identified OGs. Black horizontal lines represent OGs.
Subcellular localization and gene duplication
-
Protein functions typically are, to a certain extent, inferred from their subcellular localization. In this study, among the 2,586 OGs that were identified, 788 were positioned in extracellular space, 652 in the chloroplast, 534 in the nucleus, 261 on the organelle membrane, 232 in the endomembrane system, 86 on the plasma membrane, 27 in the mitochondria, and all but eight in the chloroplast thylakoid lumen (Fig. 4).
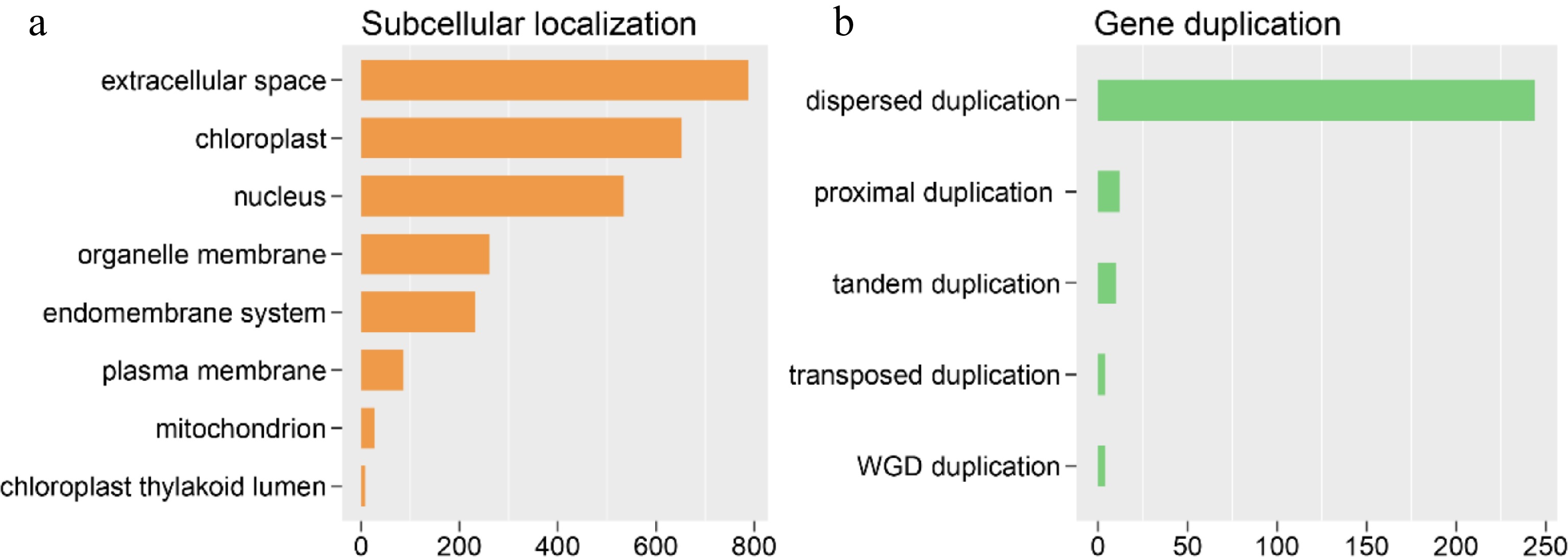
Figure 4.
Orphan gene (OGs) subcellular and gene duplication analysis. (a) OGs assigned to different subcellular locations. (b) The OGs number of different duplication types.
The origination of OGs is essential in the evolution of the genome. OGs often arise from a combinatorial mix of diverse mechanisms of origin. While gene duplication is believed to be the predominant model for the origin of OGs, the gene duplication theory consists primarily of the generation of the new gene via differentiation following duplication. 2,586 OGs derived from the genome of R. chinensis were detected in this study, including 274 OGs derived from gene duplication, accounting for approximately 10.6% of the whole OGs ((Supplementary Table S2). There were altogether four OGs originating from whole-genome duplication (WGD). Besides, the number of OGs resulting from tandem duplication, transposed duplication, proximal duplication, and dispersed duplication were 10, 4, 12, and 244 (Supplementary Table S2), respectively.
Expression profiles of OGs
-
The gene expression pattern for one gene across different tissues must enlighten the corresponding biological function. Preceding transcriptomic data on six tissues subjected to normal growth conditions were reanalyzed. It was found that the transcriptional data contained 2,088 (80.74%) OGs and 29,572 (87.51%) NOGs with FPKM > 0.02. Typically, the expressed amount of OGs is relatively lower than that of NOGs. Of these, in pistil ovary and stamen, there were the most OGs expressed (Fig. 5a). In addition, 1,139 OGs were identified to be represented across all six tissues (FPKM > 2 in a minimum of one tissue), and 346 OGs were hyper-expressed in all six tissues (FPKM > 2 in all of them). The amount of actively expressed OGs (GeneSpring normalized expression value > 0) followed a generally parallel trend throughout the six tissues and the highest expression level in stamen (Fig. 5b). It was further observed that 214 OGs were specifically expressed in six tissues, out of which 24 were specifically expressed in root, 20 in the stem, 29 in leaf, 21 in prickle, 90 in stamen, and 30 in the pistil ovary (Fig. 5c), such genes may have specific roles within the corresponding tissues. It was obvious that OGs showed more potential expression in stamen (Fig. 5d). A tissue preference for the expression of most OGs was seen according to the expression abundance in each tissue (Fig. 6).
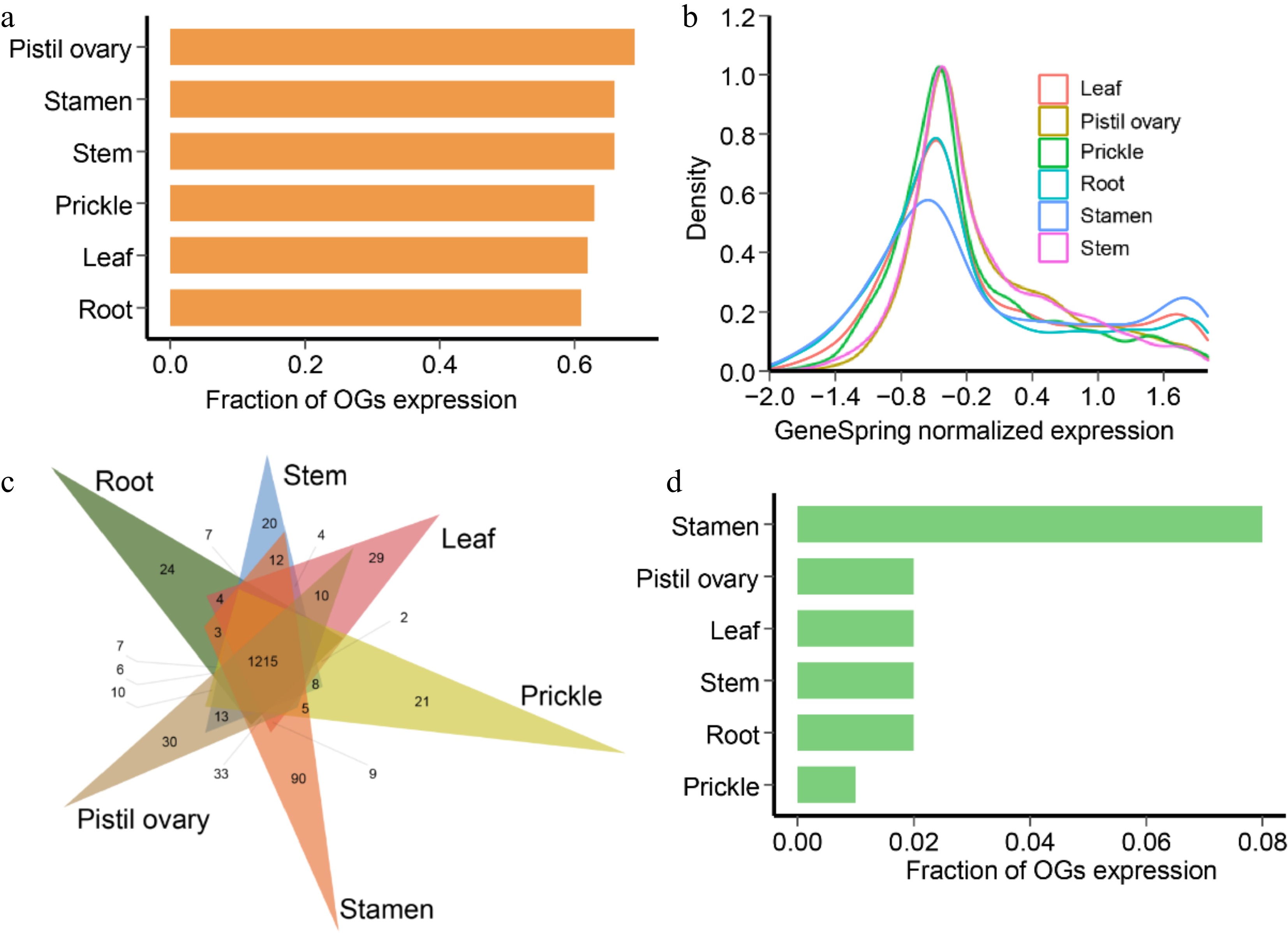
Figure 5.
Gene expression patterns of R. chinensis orphan genes (OGs). (a) Fraction of OGs having expression in different tissues. (b) GeneSpring normalized expression levels of OGs in different tissues. (c) Fraction of OGs having tissue-specific expression in different tissues of adult stage. (d) Venn diagram showing the number and relationships of expressed OGs in root, stem, leaf, prickle, stamen, and pistil ovary.
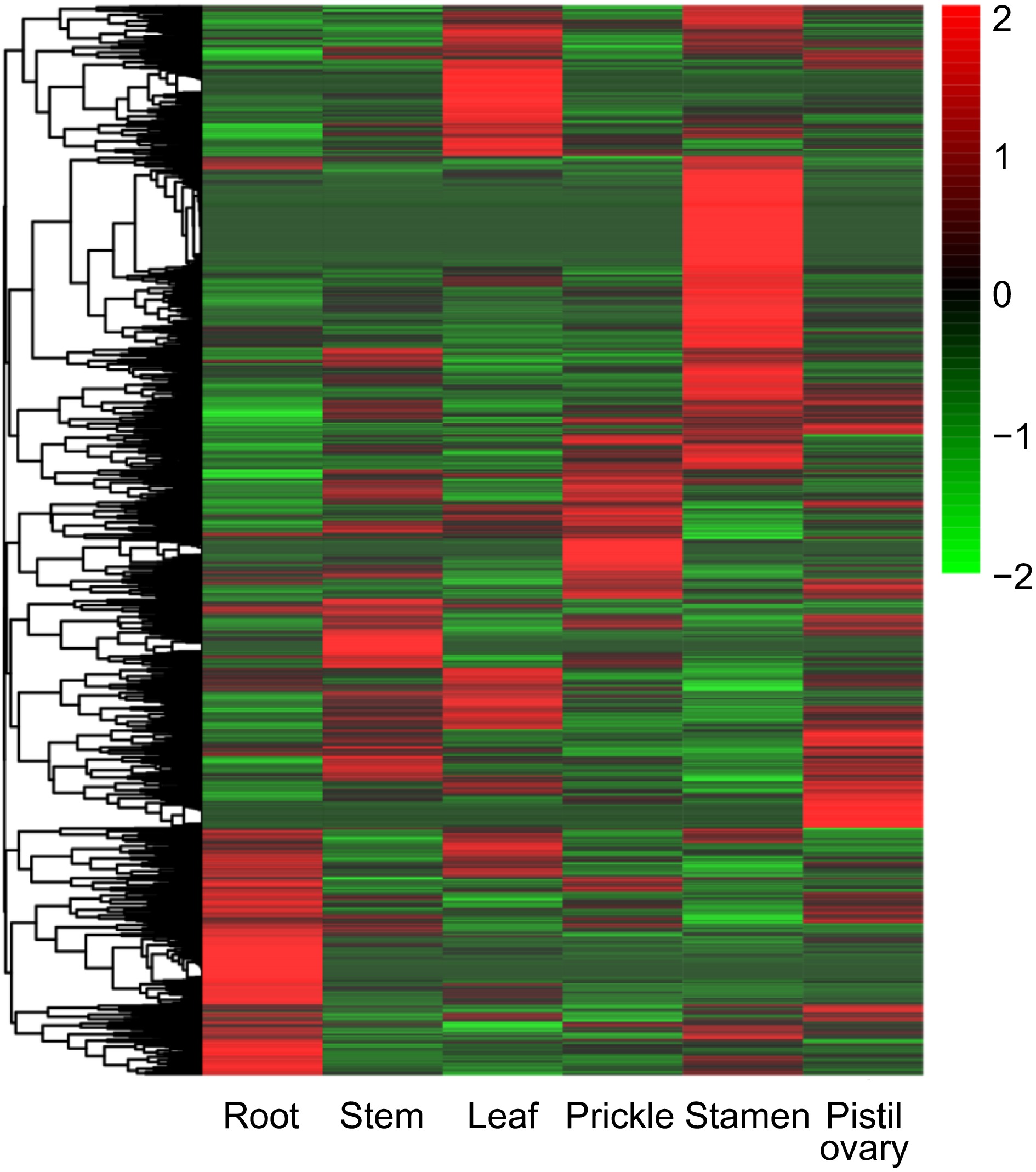
Figure 6.
Expression pattern of orphan genes in different tissues includes root, stem, leaf, prickle, stamen, and pistil ovary of R. chinensis.
Functional inference of OGs
-
As the function of OGs cannot be inferred from homologous genes, however, OGs were exclusively expressed in diverse tissues (Fig. 5). The potential functions of OGs were further profiled employing WGCNA, a tool for determining synergistic gene modules. Fifteen modules were defined. Considering different tissues as traits, the modules associated with the optimization of characteristic vector genes and phenotypes were filtered and mapped the heat map of module-trait relationships. Five modules were eventually settled on that had an extremely strong positive correlation to the trait (Fig. 7a). Subsequently, 5,218 hub genes were screened and confirmed in five modules, consisting of 217 OGs. In MEblue (leaf), there were 1,454 hub genes, containing 31 OGs. In the MEbrown model (root), 1,436 hub genes were present, covering 49 OGs. In the MEgreen model (pistil ovary), a total of 250 hub genes existed, including 11 OGs. Among the MEred model (stem), with 159 hub genes that included one OG. In the MEturquoise model (stamen), as many as 1,919 hub genes were found, comprising 125 OGs (Supplementary Table S3). These five modules were followed up with an analysis of KEGG enrichment immediately (p-value < 0.05). In MEblue (leaf), it was predominantly enriched in photosynthesis (ko00195), porphyrin, and chlorophyll metabolism (ko00860), carbon fixation in photosynthetic organisms (ko00710), carotenoid biosynthesis (ko00906), and plant hormone signal transduction (ko04075). In the MEbrown model (root), it was primarily affluent in glutathione metabolism (ko00480), phenylpropanoid biosynthesis (ko00940), MAPK signaling pathway (ko04016), and plant hormone signal transduction (ko04075). In the MEgreen model (pistil ovary), linoleic acid metabolism (ko00591), alpha-Linolenic acid metabolism (ko00592), and plant hormone signal transduction (ko04075) were enriched. In the MEred model (stem), it was mainly enriched SNARE interactions in vesicular transport (ko04130) and isoflavonoid biosynthesis (ko00943). In the MEturquoise model (stamen), pentose and glucuronate interconversions (ko00040), phosphatidylinositol signaling system (ko04070), glycerophospholipid metabolism (ko00564), glycolysis/gluconeogenesis (ko00010), galactose metabolism (ko00052) and ether lipid metabolism (ko00565) (Fig. 7b).
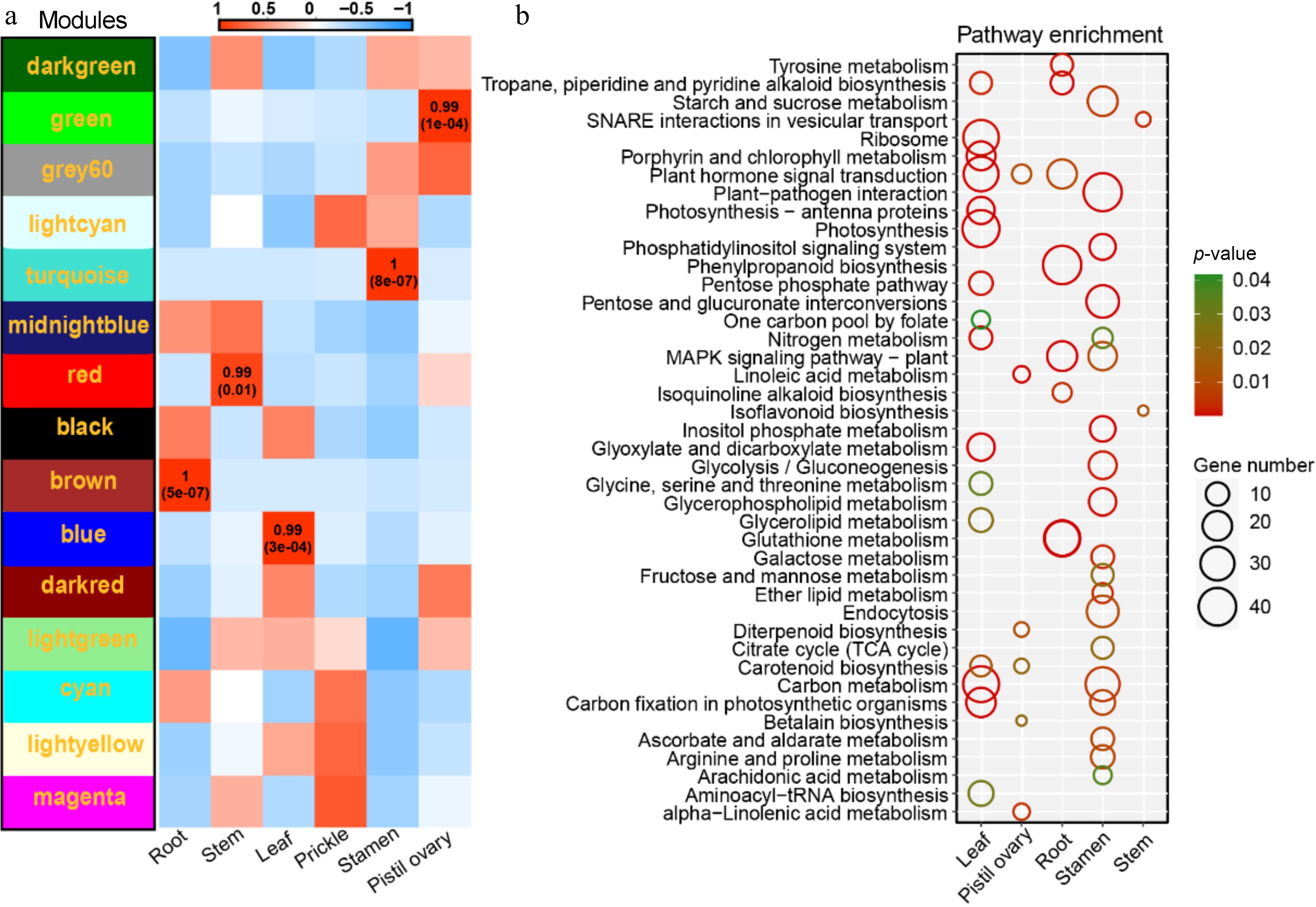
Figure 7.
Co-expression network analyses. (a) Heat map of module-tissue relationship. (b) KEGG enrichment analysis of five tissue-specific modules, include KEGG enrichment analysis result of MEblue module genes (leaf). KEGG enrichment analysis result of MEgreen module genes (pistil ovary). KEGG enrichment analysis result of MEbrown module genes (root). KEGG enrichment analysis result of MEturquoise module genes (stamen). KEGG enrichment analysis result of MEred module genes (stem).
In addition, with the aim of probing the potential link between OGs and environmental adaptation, the expression of OGs were reanalyzed in roots under salt stress (0, 2, 24, and 48 h under salt stress) and leaves under drought stress (0, 30, 60, and 90 d under drought stress) using published RNA-seq data. At salt treatment, compared with the control group (0 h under salt stress), 21 (up-regulated: 7, down-regulated: 14), 116 (up-regulated: 56, down-regulated: 60) and 56 (up-regulated: 91, down-regulated: 39) identified OGs in 2 h vs 0 h, 24 h vs 0 h, and 48 h vs 0 h were differentially expressed, correspondingly (Supplementary Table S4). A total of 201 OGs overlapped (201/2586, 7.78%) that were salt responsive in roots, of which 112 OGs (112/201, 55.72%) were up-regulated and 89 OGs (89/201, 44.28%) were down-regulated (Fig. 8a). Fuzzy c-means clustering analysis of all salt-associated DEGs (including OGs and NOGs) was further divided into six Clusters of gene co-expression patterns (Fig. 8b). Two Clusters were focused on with increasing (Cluster 2), and decreasing trends (Cluster 4) of gene expression levels with increasing salt treatment time. Genes with Memberships > 0.7 in Cluster 2 and Cluster 4 were screened for subsequent functional enrichment analysis. There was a total of 440 DEGs in Cluster 2, including 15 OGs, and 336 DEGs in Cluster 4, including 7 OGs (Supplementary Table S5). Nineteen OGs were engaged and enriched in GO terms, consisting 'cellular hyperosmotic salinity response', 'cellular response to hydrogen peroxide', and 'response to salt stress' (p-value < 0.01) (Supplementary Table S6). KEGG enrichment results showed that ABC transporters and brassinosteroid biosynthesis pathway were remarkably enriched (p-value < 0.05) (Table 2).
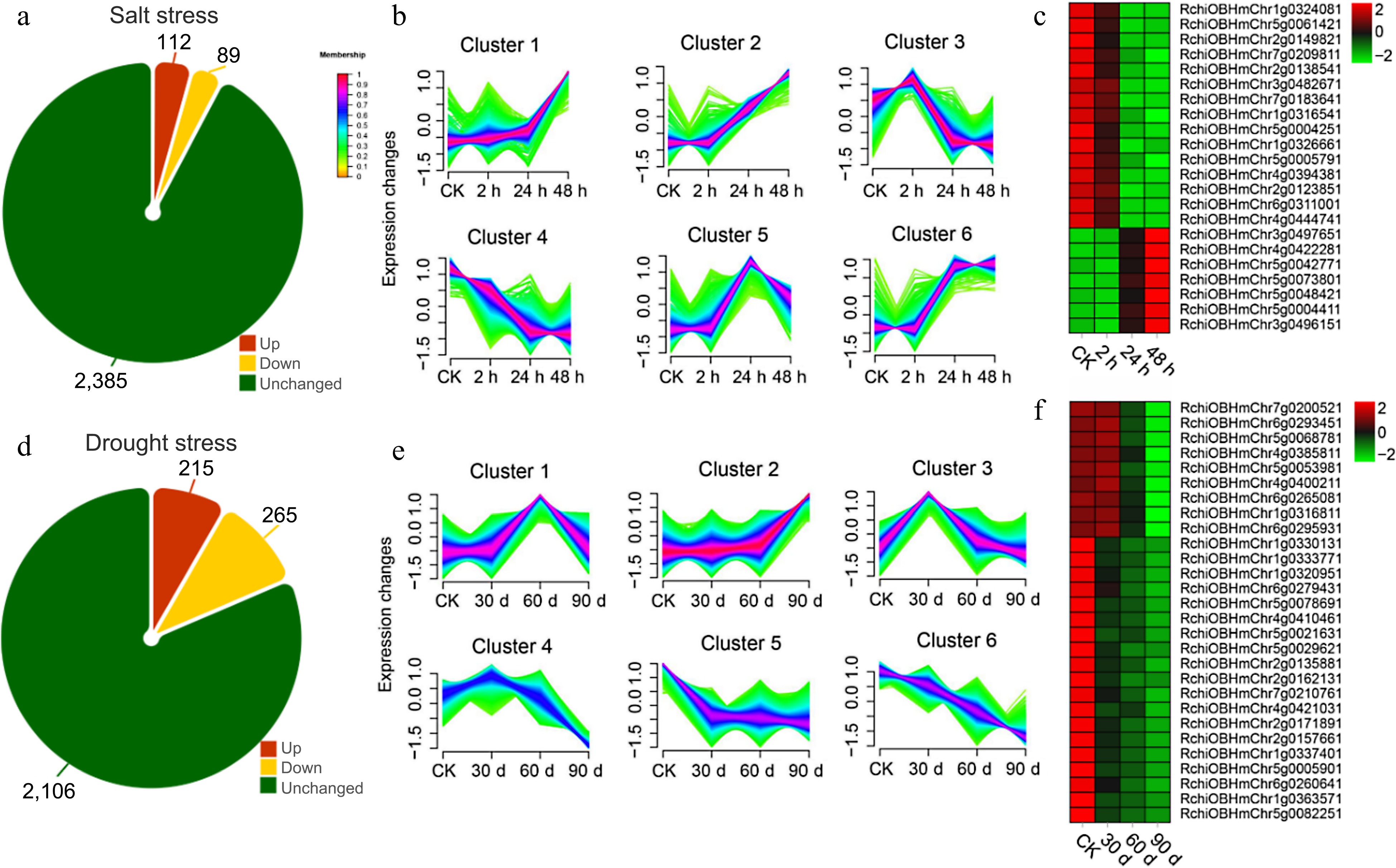
Figure 8.
Transcriptome analysis of orphan genes (OGs) under salt and drought stress. (a) Number of differentially expressed OGs under salt stress in leaves of R. chinensis. (b) Trends in the expression of differentially expressed genes at different time points under salt stress. (c) Heat map of the expression of OGs under the trend of pattern Cluster 2 and Cluster 4. (d) Number of differentially expressed OGs under drought stress in roots of R. chinensis. (e) Trends in the expression of differentially expressed genes at different time points under drought stress. (f) Heat map of the expression of OGs under the trend of pattern Cluster 1 and Cluster 3.
Table 2. Enriched KEGG pathway for R. chinensis orphan genes.
Type Cluster KEGG pathway p-value Salt stress Cluster 2 Alanine, aspartate and glutamate metabolism 0.011861 Base excision repair 0.013442 Pentose and glucuronate interconversions 0.016805 Ubiquinone and other terpenoid-quinone biosynthesis 0.032602 Cluster 4 Glutathione metabolism 0.006557 ABC transporters 0.007274 Brassinosteroid biosynthesis 0.014615 MAPK signaling pathway 0.016903 Terpenoid backbone biosynthesis 0.023847 Glycine, serine and threonine metabolism 0.035604 Drought stress Cluster 1 Carbon metabolism 4.02E-06 Ubiquinone and other terpenoid-quinone biosynthesis 5.04E-05 Carbon fixation in photosynthetic organisms 5.22E-05 Riboflavin metabolism 0.000115 Pentose phosphate pathway 0.00284 Glyoxylate and dicarboxylate metabolism 0.008393 Steroid biosynthesis 0.03522 DNA replication 0.036059 Thiamine metabolism 0.043981 Cluster 3 Pyrimidine metabolism 0.002402 Aminoacyl-tRNA biosynthesis 0.019032 Purine metabolism 0.037451 Upon drought treatment, in contrast to the control group (0 day under drought stress), 101 (up-regulated: 46, down-regulated: 55), 204 (up-regulated: 76, down-regulated: 128) and 352 (up-regulated: 152, down-regulated: 200) OGs in 30 d vs 0 d, 60 d vs 0 d, and 90 d vs 0 d were differentially expressed, respectively (Supplementary Table S7). Altogether, 480 OGs overlapped (480/2586, 18.56%) in response to drought in roots, with 215 OGs (215/480, 44.79%) being up-regulated and 265 OGs (265/480, 55.21%) being down-regulated (Fig. 8d). An analysis of fuzzy c-means clustering centered on two Clusters (Cluster 1: increasing and Cluster 3: decreasing) of gene expression levels as the duration of drought treatment increased. Individual genes with Memberships > 0.7 in Cluster 1 and Cluster 3 were filtered out for follow-up functional enrichment analysis. 344 DEGs, including nine OGs, were enriched in Cluster 1, and 348 DEGs in Cluster 3 including 19 OGs (Supplementary Table S8). A total of 28 OGs were engaged and enriched in GO terms, namely 'photosynthesis', 'reductive pentose-phosphate cycle' and 'regulation of defense response' (p-value < 0.01) (Supplementary Table S9). The results of KEGG enrichment revealed that carbon metabolism and pentose phosphate pathway were markedly enriched (p -value < 0.05) (Table 2). Strikingly, for 107 OGs a response to both categories of stresses was also discovered (Supplementary Table S10), indicating that these genes likely function significantly in stress tolerance.
Petal expansion is the principle procedure by which roses open. Interestingly, it was found that 158 OGs were differentially expressed during petal expansion from the results of Han et al. (Supplementary Table S11)[71]. In the results of Han's study, they used WGCNA to identify two modules MEyellow and MEgreenyellow that may be involved in adaxial–abaxial regulation in rose petals. In the MEyellow model, with 383 hub genes, comprising nine OGs. Within the MEgreenyellow model, a total of 52 hub genes were identified, covering two OGs (Supplementary Table S12). Functional analysis of the hub genes of both modules showed that they mainly include transcription factors such as MYBs and WUSCHEL. They also encode various enzymes such as laccase, cellulose synthase, and trehalose-6-phosphate synthase. All in all, 11 OGs were identified that may participate in adaxial–abaxial regulation and have a significant effect on the flowering open procedure.
-
The burgeoning field of comparative genomics has accelerated the exploration of the emerging field of OGs, with OGs potentially important for the development, function, and evolution of living organisms being identified in successive species[7,44−46]. The accurate identification of OGs is an important prerequisite for their functional prediction and analysis. In the present study, 2,586 OGs from the genome of the rose species R. chinensis were identified, representing approximately 7.11% of the genome, a proportion consistent with the typical percentage of OGs in organisms[47]. Similar to the present results, Lin et al.[15] identified 1,324 OGs in the A. thaliana genome covering approximately 4.9% of the entire genome, Zhao and Ma[9] characterized a total of 1,789 OGs in tea plant accounting for about 3.37% of the genome, and Guo et al.[28] identified 1,926 OGs in the rice genome representing approximately 4.9% of the whole genome. At present, the authentication of OGs relies primarily on comparing the target genome with published genomes of its homologous species. The more reference genomes are available, the richer the annotation information and the smaller the genome gaps of the target genome. Consequently, the number of reference genomes will affect the number and accuracy of OGs identified, where more reference genomes will likely result in fewer OGs and higher accuracy. However, the limitations of currently available identification tools lead to the possibility that the present study may not reflect the fully authentic OGs in the R. chinensis genome. Future improvements in the exclusion of pseudogenes from identification tools and evolutionary analysis of non-conserved genes may further improve the accuracy of identification.
Distinctive gene structure of OGs
-
The shorter origin of OGs relative to NOGs has led to differences in sequence structural features, including gene size, protein size, number and size of exons and introns, GC content, and isoelectric point. The sequence structures between OGs and NOGs were analyzed and compared to reveal whether these general differences exist in the R. chinensis genome. Typically, NOGs have a larger gene size compared with OGs[17,18,42], and the results were consistent with this (Table 1). The short protein length and few exons of OGs (Fig. 2) were similar to the general characteristics of plant families Poaceae[17], Brassicaceae[27], Rutaceae[8], and Camellia[9]. The decrease in the number of exons in OGs may be an important factor contributing to the reduction in their average size (Fig. 2), as the average length of exons is somewhat constant[47]. Consequently, even if the length of exons and introns of OGs was significantly higher than that of NOGs, it had little effect on average size. De novo origination might be another reason accounting for this shorter gene size of OGs, due to their short evolutionary time[48]. In addition, it was speculated that another possible contributor to the difference was the higher proportion of intron-less OGs. Kaessmann suggested that recurrent line-specific expansion may lead to a dramatic enrichment of intron-less genes and the creation of new genes by retrotransposition, a phenomenon that has been demonstrated in zebrafish[13]. On the other hand, in the R. chinensis genome, the GC content of OGs was noticeably less than that of NOGs, in line with Aegiceras corniculatum[49], and the wheat genome[39]. Notably, the GC content of OGs and NOGs tended to be highly variable across species. In contrast to the present results, the GC content of OGs was markedly higher than that of NOGs in sweet orange[8], Bombyx mori[7], zebrafish[18], and tea[9]. The selection and adaptation of organisms to the external environment and genetic recombination are important drivers of changes in GC content[50,51]. The isoelectric point is intimately associated with protein function, and its alteration is thought to be a modifying effect on protein function, important for solubility, subcellular localization, and protein interactions[52]. It was revealed that the isoelectric point of OGs was distinctly above that of NOGs, which may also be driven by selection[53]. For example, in prokaryotes, adaptation to the environment has led to changes in protein isoelectric points[54].
Expression characteristics of OGs
-
During evolution, new genetic elements acquired by the genome, such as OGs, are one of the important sources of functional and phenotypic diversity[55]. The expression patterns of OGs on different tissues for the prediction and understanding of their biological functions are accessible using RNA-seq[56]. OGs are inconsistently expressed on different tissues, and in general, OGs are highly expressed in the reproductive system of plants and animals[8,18,57,58]. In the present study, 214 OGs were expressed tissue-specific, of which 90 and 30 OGs were expressed only in reproductive organs, stamen, and pistil ovaries, respectively; in addition, 24, 20, 29, and 21 OGs were expressed in nutritional organs, including roots, stems, leaves, and prickle (Fig. 5). The specific expression of more than 50% of R. chinensis OGs on reproductive organs implied their important role in reproductive development, which was largely in line with the expression profile of other plants' OGs[9,28,49], and more detectable expression was found in stamen (42.06%). Many studies have shown that OGs, or young genes as some researchers call them, were more inclined to be expressed in the male organs. In 2015, Cui refined this doctrine to 'new genes out of the male' and hypothesized that new genes have an important role in reproductive isolation and species differentiation[1]. The present results also support this hypothesis. In a word, the specific expression of OGs in R. chinensis provides important data on their resistance mechanisms, which is resistant to herbivores, pathogens, or mechanical damage, and also prevents water loss, which suggests an important role in the evolution of habitat adaptation in R. chinensis.
Functional inference of OGs
-
It is not feasible to use homology comparisons to infer the possible expression characteristics and functions of OGs, as they are unique in each species. Under this circumstance, co-expressed gene modules rich in biological information become a dependable vehicle for inferring the biological processes that maybe involved in OGs, as co-expressed genes usually exhibit significant functional similarities[59]. In the present study, WGCNA was used to identify 217 OGs distributed in five modules (Supplementary Table S3). KEGG analysis revealed that these co-expressed gene modules were engaged in a variety of biologically important processes. These mainly included linoleic acid metabolism, pentose and glucuronate interconversions, photosynthesis, and plant hormone signal transduction. In all, the involvement of OGs in important physiological processes such as growth and development, signal transduction, and metabolism in roses demonstrates that OGs can be functional and are likely to be essential.
OGs involved in pentose and glucuronate interconversions may contribute to male sterility, given the crucial role of anthers in male gametogenesis, while male sterility mutants usually exhibit genetic disruption linked to anther and pollen development[60,61], which can result from abnormal carbohydrates metabolism[62,63]. Carbohydrates, such as pentose and glucuronate, play multiple roles in pollen development, serving as a major source of energy for plant metabolism and as important signaling molecules for regulating growth and development[64,65]. β-glucuronidase, a glycosyl hydrolase, is mainly responsible for the lysosomal degradation of mucopolysaccharides, dermatopoietin, and keratin sulfate[66]. In addition, β-glucuronidase is involved in the metabolism of various endogenous substances, including pentose, glucuronides, porphyrins, starch, and sucrose[67]. The pentose and glucuronide interconversion pathway, enriched with OGs, is highly correlated with male sterility in studies of cabbage (Brassica oleracea L. var. capitata)[68] and cotton (Gossypium hirsutum L.)[69,70] transcriptome analysis, suggesting the involvement of key genes in this pathway.
Rose flower opening is dependent on petal expansion. The adaxial-abaxial regulation of petals led to a heterogeneous distribution of auxin, and the transport and signaling of the phytohormone auxin were involved in the entire development of flowers[71], and the OGs might be active in determining and maintaining the adaxial-abaxial polarity of petals. On the other hand, floral fragrance is an important feature of ornamental roses, providing sensory pleasure to humans, and monoterpenes are one of the main classes constituting the fragrance of roses[72]. Usually, monoterpenes are mainly produced in plastids and their substrates are derived from the methylerythritol 4-phosphate pathway[73]. However, the lack of photosynthesis in rose petals may lead to a reduced flux of substrates produced through the methylerythritol 4-phosphate pathway. Therefore, photosynthesis not only provides roses with energy for growth and development, but also plays a role in the production and dispersal of floral fragrances. Fatty acid derivatives are prominent compounds in the leaves and sepals of roses[74]. An important class of enzymes involved in the formation of fatty acid-derived volatiles is lipoxygenase (LOX), the enzymatic oxidation of linoleic acid by LOX to produce hexanal. In addition, cleavage of linoleic acid at the 12−13 double bond yields the C12 precursor of jasmonic acid[75]. It has been shown that increased levels of jasmonic acid methylation led to a delayed degradation of carotenoids and affected the carotenoid content of the yellow rose cultivar R. hybrida 'Frisca'[76].
Rose plants usually grow in subtropical climates where environmental stresses such as drought and salinity are important factors limiting their growth and productivity. It has been shown that OGs were preferentially expressed under abiotic stress, as in yeast[77] and rice[26,28]. Using available RNA-seq data, the expression patterns of OGs were screened under salinity and drought stresses, respectively, and observed 201 and 480 OGs stimulated individually, suggesting the possible important role of these stress-responsive OGs in adaptation to extreme environmental stresses (Fig 8). The increased stimulation of OGs under drought is probable to be highly correlated with their native environment, exemplified by the Middle East Taif region, where Damask rose (Rosa damascena Mill.) originated[78]. The roots serve as the primary site of plant perception of soil water deficit, and subsequently, drought response signals from the roots are transmitted to the leaves[79]. The enrichment of OGs in phytohormone signaling pathways suggested that they may have an essential position in drought sensing and signaling. Surprisingly, a total of 107 OGs responding to both types of stress were found (Supplementary Table S10). RhEXPA4 is a rose expansion protein gene that regulates leaf growth and bestows drought and salt tolerance in Arabidopsis[80] This suggests that these 107 OGs may be critical candidates for further studies of environmental adaptations in roses.
In conclusion, even without direct functional evidence, the analysis of co-expressed gene modules implies that OG genes maybe involved in important biological processes such as developmental regulation, signal transduction, metabolism, and stress adaptation in roses.
-
The authors confirm contribution to the paper as follows: study conception and design: Mao J; data analysis, draft manuscript preparation: Ma D, Ding Q; critical manuscript revision: Mao J, Ma D, Zhao Z, Han X. All authors read and approved the final manuscript.
-
The data that support the findings of this study are available in the Rosa database repository:
https://lipm-browsers.toulouse.inra.fr/pub/RchiOBHm-V2/ . We appreciate Laboratoire Reproduction et Developpement des Plantes for providing their valuable databases to the public. This work was financially supported by Fundamental Research Funds for the Central Universities (JUSRP124005).
-
The authors declare that they have no conflict of interest.
-
accompanies this paper at (https://www.maxapress.com/article/doi/10.48130/tp-0024-0036)
-
Received 4 July 2024; Accepted 26 August 2024; Published online 23 October 2024
-
# Authors contributed equally: Dongna Ma, Qiansu Ding
- Supplementary Table S1 List of OGs.
- Supplementary Table S2 The list of OGs originate from gene duplication.
- Supplementary Table S3 List of genes in the six tissue-specific modules in OGs.
- Supplementary Table S4 List of OGs differentially expressed under salt treatment.
- Supplementary Table S5 Trend analysis of differentially expressed OGs under salt treatment.
- Supplementary Table S6 Significant enrichment of GO of Cluster 2 and Cluster 4 co-expressed genes under salt stress.
- Supplementary Table S7 List of OGs differentially expressed under drought treatment.
- Supplementary Table S8 Trend analysis of differentially expressed OGs under drought treatment.
- Supplementary Table S9 Significant enrichment of GO of Cluster 1 and Cluster 3 co-expressed genes under drough stress.
- Supplementary Table S10 OGs that were differentially expressed under both drought and salt treatments.
- Supplementary Table S11 Differential expression information of 158 OGs across six samples.
- Supplementary Table S12 List of genes of OGs of the 11 candidate adaxial–abaxial patterning gene modules.
- Copyright: © 2024 by the author(s). Published by Maximum Academic Press on behalf of Hainan University. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Ma D, Ding Q, Zhao Z, Han X, Mao J. 2024. Orphan genes are involved in environmental adaptations and flowering process in the rose. Tropical Plants 3: e036 doi: 10.48130/tp-0024-0036 

Orphan genes are involved in environmental adaptations and flowering process in the rose
- Received: 04 July 2024
- Revised: 11 August 2024
- Accepted: 26 August 2024
- Published online: 23 October 2024
Abstract: Orphan genes (OGs) are genes with no obvious homology compared to other species and play a key part in new function generation, phenotypic changes, and adaptive evolution. Rosa chinensis is an important horticultural variety and the most popular cut flower worldwide, with high economic and ornamental benefits. Herein, 2,586 OGs were identified in the R. chinensis genome, accounting for approximately 7.11% of all protein-coding genes. Genetic structure analysis indicated that the OGs had a shorter protein size, fewer exons, lower GC content, and a higher isoelectric point compared to non-orphan genes (NOGs). Transcriptomic analyses revealed that OGs had a stronger tissue-specific expression, with more than 50% specifically expressed in reproductive organs. Weighted gene co-expression network analysis (WGCNA) resulted in 215 OGs distributed in five modules. The co-expression genes of these OGs were engaged in a variety of important biological processes, including photosynthesis, pentose and glucuronate interconversions, linoleic acid metabolism, and phytohormone signaling. It was found that 107 OGs were significantly up- or down-regulated across responses to both abiotic stresses (salt and drought). Fuzzy c-means clustering identified 50 OGs (salt: 22 and drought: 28) had increasing and decreasing expression patterns, suggesting their potential function in interactions with the environmental adaptation. In addition, there were 11 OGs involved in the flowering process of roses. The present study provides the first systematic identification of OGs in R. chinensis as well as a comprehensive analysis of the characteristics and potential functions, resulting in valuable clues and new insights into the importance of these new genes.
-
Key words:
- Rosa chinensis /
- Orphan genes /
- Transcriptome /
- Gene expression /
- Environment adaptation