









-
Climate change and soil erosion have provided unprecedented challenges to meet the rising demand for higher-quality food for the world's growing population. Over the past decades, maize productivity has experienced continuous improvement owing to technological innovations. The application of inorganic fertilizers has contributed to the intensity of agricultural production such as maize productivity[1]. The overuse of inorganic fertilizers, such as chemical nitrogen (N) fertilizer, can have adverse effects on both soil and groundwater[1]. Rather than relying on inorganic fertilizers derived from fossil fuels, using organic matter presents a promising alternative to remineralize depleted soil, enhance soil fertility, and ultimately increase crop productivity without adding to the environmental burden.
Humic acid (HA) is an organic matter produced and accumulated by plants and animals. It is a residual substance through the decomposition of microorganisms accompanied by geochemistry processes[2]. A previous study suggested HA fertilizer could expand the effective absorption area of roots and enhance root growth[3]. Therefore, HA fertilizer promotes the recruitment of nutritional resources such as carbon (C) and N from both the fertilizer and bulk soil. Under water stress, studies showed that HA fertilization stimulates root development, i.e., increased root length and root dry weight, thereby delaying the drought stress responses[4]. However, the connections between HA and drought resistance, especially at the rhizosphere microbiome level, remain unclear.
Recent evidence suggests that certain root-associated microbes likely mitigate the negative responses of drought stress, such as Actinobacteria and Firmicutes[5,6]. To test the hypothesis that HA delays negative drought responses in root development through enriching or depleting certain root-associated microbes, in this study, experiments were conducted to explore the effects of root-associated bacterial communities on several phenotypes during maize development and to determine how HA and drought influences the composition of root-associated microbial communities. Phenotypic traits for maize hybrids were collected during the flowering period under three different treatments (HA, drought, and control). The bacterial community was characterized from rhizospheres and bulk soils for the V4 region of the 16S rRNA gene using Illumina Hiseq2500 sequencer. A total of 18 samples (six individual plants per treatment) were collected for rhizosphere microbiome sampling, and six samples from the control were used for bulk soil microbiome sampling, resulting in ~763 million reads. Across all the samples, 40 bacterial phyla were detected in these four sample types (rhizosphere microbiomes from the control, HA, and drought group, as well as bacterial microbiomes of bulk soil from the control group).
-
The experiment was conducted on the Shandong Agricultural University research farm (Shandong, China). The soil was collected manually with sterile shovels from the farm and was then allocated equally into pots. Before planting, the soil was treated with commercial compound fertilizer (normal fertilization, N15-P15-K15) and commercial organic HA fertilizer (N15-P15-K15 + HA fertilization). Then, Xianyu335, a commercial maize hybrid, was planted with three treatments: (1) control group with normal fertilizer and conventional growth conditions; (2) HA group with HA fertilizer and conventional growth conditions; and (3) drought group with the normal fertilizer as that in the control group and moderate drought growth conditions (45% soil water content, SWC). The experiment was conducted following a completely randomized design with six replications per treatment.
Phenotypic data evaluation
-
The number of leaves (#Leaf) were counted on days 43, 49, 56, and 71 after sowing. Plant height (PH) was measured from the ground to the top of each plant, as described previously[7]. Days to silking (DTS) and days to pollen shed (DTP) were recorded for each plant.
Sample collection and DNA extraction
-
Root samples were collected manually using a sterile shovel to a depth of approximately 20−30 cm, following a previously described method[8]. Sample collection was conducted during the flowering stage. Root samples were vortexed in sterile phosphate-buffered saline (PBS) buffer (Catalog No. E607008; Sangon Biotech, Shanghai, China) for 10 min at 14,000× g and centrifuged to obtain rhizosphere soil pellet after removing the root tissue. DNA extraction was performed using the FastDNA™ SPIN Kit for Soil (Catalog No. 116560200; FastDNA™, Solon, OH, USA) following the manufacturer's protocol. In brief, up to 500 mg of soil sample were added to a Lysing Matrix E tube with Sodium Phosphate Buffer, then were homogenized and centrifuged at 14,000× g for 10 min. The supernatant was transferred to a new microcentrifuge tube with a protein precipitation solution. After mixing, the tube was centrifuged at 14,000× g for 5 min. The supernatant was transferred and resuspended with binding matrix suspension to allow the binding of DNA. Finally, after quality checking the DNA by using a NanoDrop-2000 (Therml Fisher Scientific), the purified DNA was stored at −80 °C for further analysis.
Library construction and sequencing
-
After the quality checking of the DNA sample, all the qualified DNA was used to construct the 16S library. Briefly, the qualified DNA samples were amplified using a dual-indexed primer specific to the V4 region (515F (5'-GTGCCAGCMGCCGCGGTAA-3') and 806R (5'-GGACTACHVGGGTWTCTAAT-3')), and the PCR product was converted into blunt ends with T4 DNA polymerase, Klenow fragment, and T4 polynucleotide kinase. Then, after A tailing of the 3' end of each fragment, adapters were added. Then, AMPureXP beads (Beckman-Coulter, West Sacramento, CA, USA) was used to remove fragments that were too short. Finally, libraries quality checked by a Qubit 3.0 fluorometer (Life Technologies) were sequenced with paired-end 250-bp reads using Illumina Hiseq2500.
Data processing
-
After sequencing, the raw data were cleaned by removing low-quality reads and reads contaminated by adapters, with a maximum of three bases mismatch allowed. The paired-end reads with overlap were merged into tags using FastLength Adjustment of Short reads (FLASH, v1.2.11)[9] with a minimal overlap length of 15 bp and the mismatching ratio of an overlapped region less than or equal to 0.1. Tags were then clustered into the operational taxonomic unit (OTU) using USEARCH (v7.0.1.1090)[10]. Principal component analysis (PCA) was then used to summarize factors mainly responsible for the differences of OTU composition in different samples, the similarity is high if two samples are closely located. Based on the OTU abundance information, the relative abundance of each OTU in each sample was be calculated, and the PCA of OTU was carried out with the relative abundance value. The Bray-Curtis distance matrix was used to assess the dissimilarity between samples. The software used in this step was package 'ade4' of the software[11] in R. To assess the statistical significance of the clustering observed in PCA, a PERMANOVA (Permutational Multivariate Analysis of Variance) was performed using the 'vegan' package[12] in R.
Statistical analyses
-
Student's t-test for phenotypic traits was used to determine the difference between different groups. The abundance differences of microbial communities between groups were detected by Wilcoxon signed-rank test and the p values were adjusted using the FDR approach[13]. Meanwhile, Linear Discriminant Analysis Effect Size (LEfSe) was incorporated to identify key taxa contributing to differences between conditions.
-
To investigate the potential effect of drought on plant growth and root-associated microbiomes, the plants were subjected to drought treatment (45% SWC). The rhizosphere and bulk soil samples were collected from maize hybrids to characterize microbial communities using next-generation sequencing (Fig. 1a). The phenotypic results revealed dramatic differences in plant height and root traits between control and drought conditions (Fig. 1b). The weights of both fresh (p < 0.01) and dry roots (p < 0.05) were significantly greater in the control group than drought group at the same stage (Fig. 1b bottom left, Fig. 1c & d). The plant height of the drought group was significantly shorter than that of the control group during the flowering stage (Fig. 1b middle left, Fig. 2a). Plant height was measured on days 43, 49, and 56 after planting. The results indicated that plant height at stages d49 and d56 was significantly decreased due to drought stress (Fig. 2a, p < 0.001). Meanwhile, the number of leaves were measured on days 43, 49, 56, and 71. The number of leaves were significantly higher in the control group at the earlier stage, however, more leaves were observed at the later stage in the drought group, and DTP were significantly delayed in the drought group (Fig. 2a, p < 0.05), which may indicate compensatory and delayed growth of plants due to drought stress.
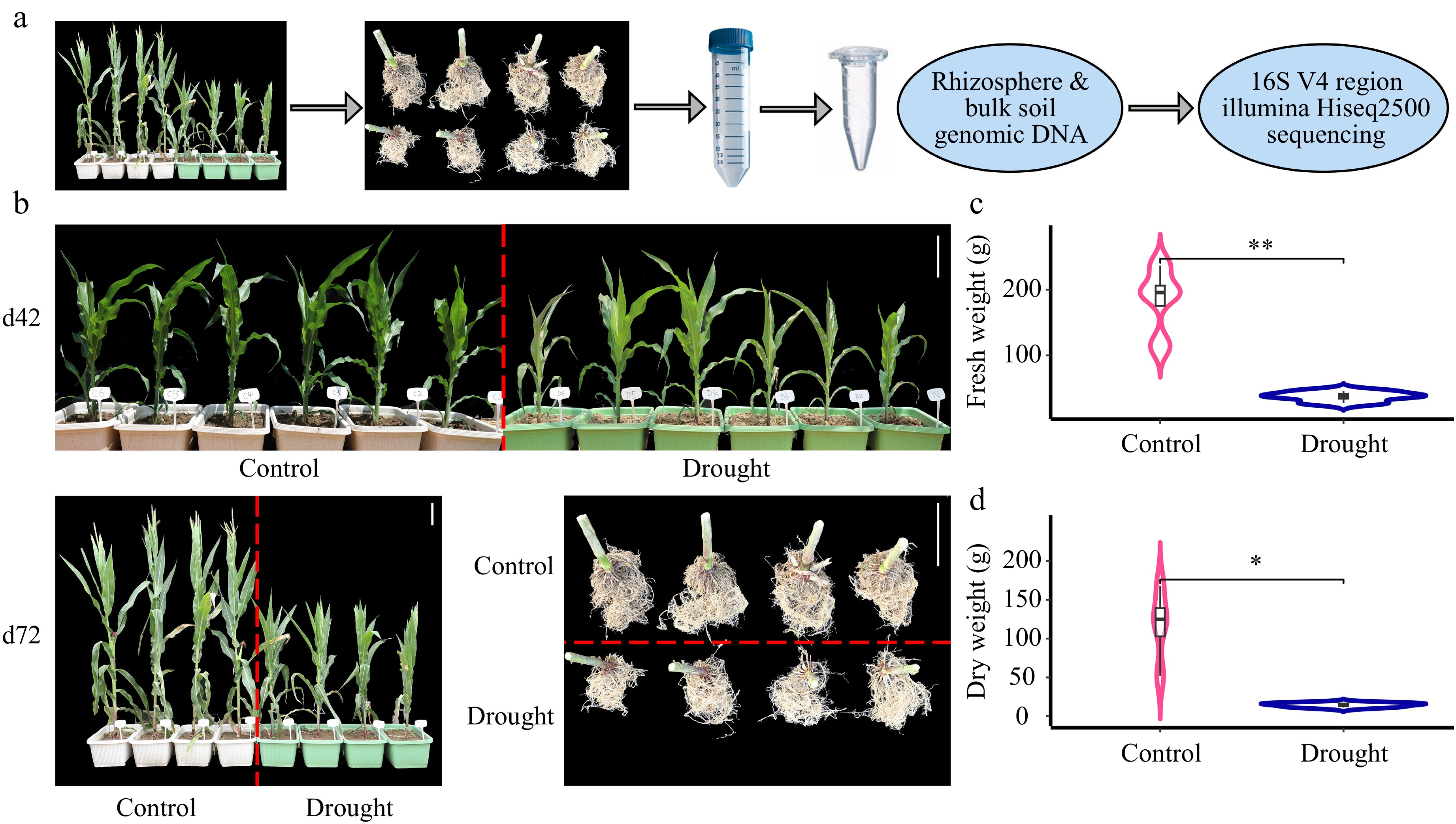
Figure 1.
Root performs significantly differently under drought stress vs normal conditions. (a) Flowchart of the study performed from the field work to the next generation sequencing. (b) The plant growth at days 42 (d42) and harvest at d72, as well as the root harvest at d72 under control and drought conditions, the scale bar is 20 cm. Statistics of the weight of (c) fresh roots, and (d) dry roots using R/Student's t test between each two of the control and drought groups. ** indicates p < 0.01, * indicates p < 0.05.
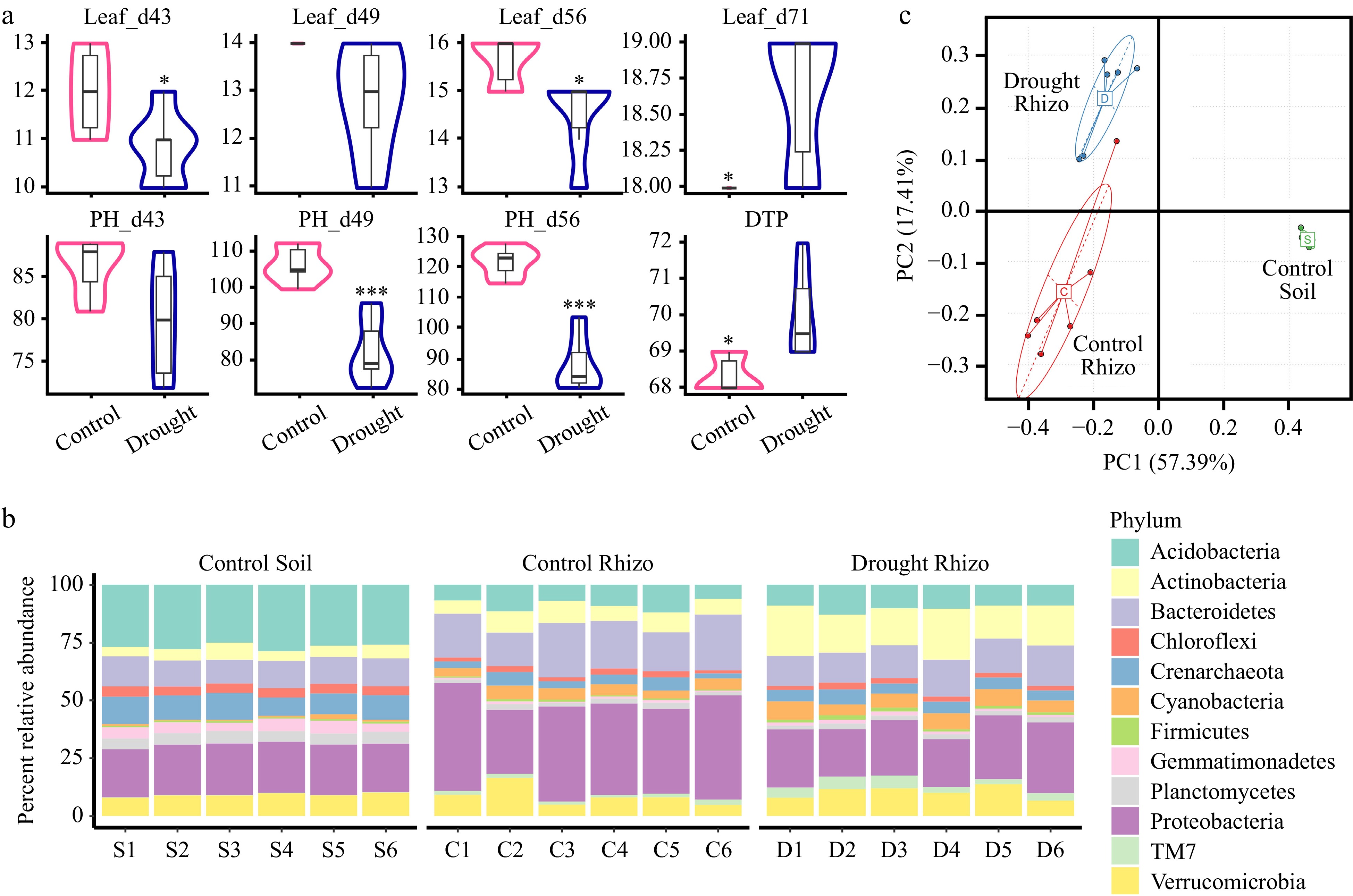
Figure 2.
Summary statistics of phenotypes and relative abundance of the top 12 of the most abundant bacterial phyla. (a) Summary statistics of number of leaves (#Leaf) on days 43, 49, 56, and 71 after sowing, plant height (PH, cm) at days 43, 49, and 56 after sowing, and days to pollen shed (DTP, days) using Student's t test between cotrol and drought groups. * indicates p < 0.05, *** indicates p < 0.001. (b) Percent relative abundance of the most abundant phyla for control, drought treatments in rhizospheres and for control in bulk soils. All individuals were arranged in order within each group along the x axis. (c) Principal component analysis across the phyla from the Control Soil, control rhizosphere (Control Rhizo), and drought rhizosphere (Drought Rhizo) microbiome data.
Microbial diversity under drought stress
-
A previous study on sorghum reported that drought delayed the development of the root microbiome and led to a higher abundance and greater activity of monoderm bacteria[13]. In this study, the effect of drought on root-associated microbial communities in maize was explored. As expected, the bulk soil microbiomes and maize rhizosphere microbiomes showed different patterns (Supplementary Fig. S1). The present results suggested that the composition of the rhizosphere at the phylum level caused by drought treatment differed significantly from that of the control (Figs 2b, 3; Supplementary Fig. S2). PCA was used to explore the differences in OTU composition across the samples. The PCA plot (Fig. 2c) showed a clear clustering of the samples based on their OTU profiles. Samples that were more similar in the drought group are located closer together, and similar patterns were observed in the control rhizosphere group, and the control soil group. The PERMANOVA results confirmed that the clustering observed in the PCA was statistically significant (p < 0.001). Consistent with the previous report on sorghum[13], the relative abundance of Actinobacterial and Firmicutes were significantly enriched and the relative abundance of the Proteobacterial and Fibrobacteres were significantly depleted in the rhizosphere microbiome of the drought group compared to the controls (Fig. 3a, p < 0.01). In addition, compared with the control group, some other microbes at the phylum level were significantly over-represented in the rhizosphere of the drought group, such as Euryarchaeota, Chlorobi, while some microbes at the phylum level were significantly depleted, such as Tenericutes (Fig. 3a, p < 0.01). LEfSe analysis was further conducted to identify key microbial taxa that were differentially abundant between the control and drought groups. These taxa, including Actinobacterial and Firmicutes, were visualized using a cladogram (Fig. 3b). The results indicated that these specific microbial groups were more prevalent under drought conditions, suggesting their potential role in shaping the microbial community structure in response to drought conditions.
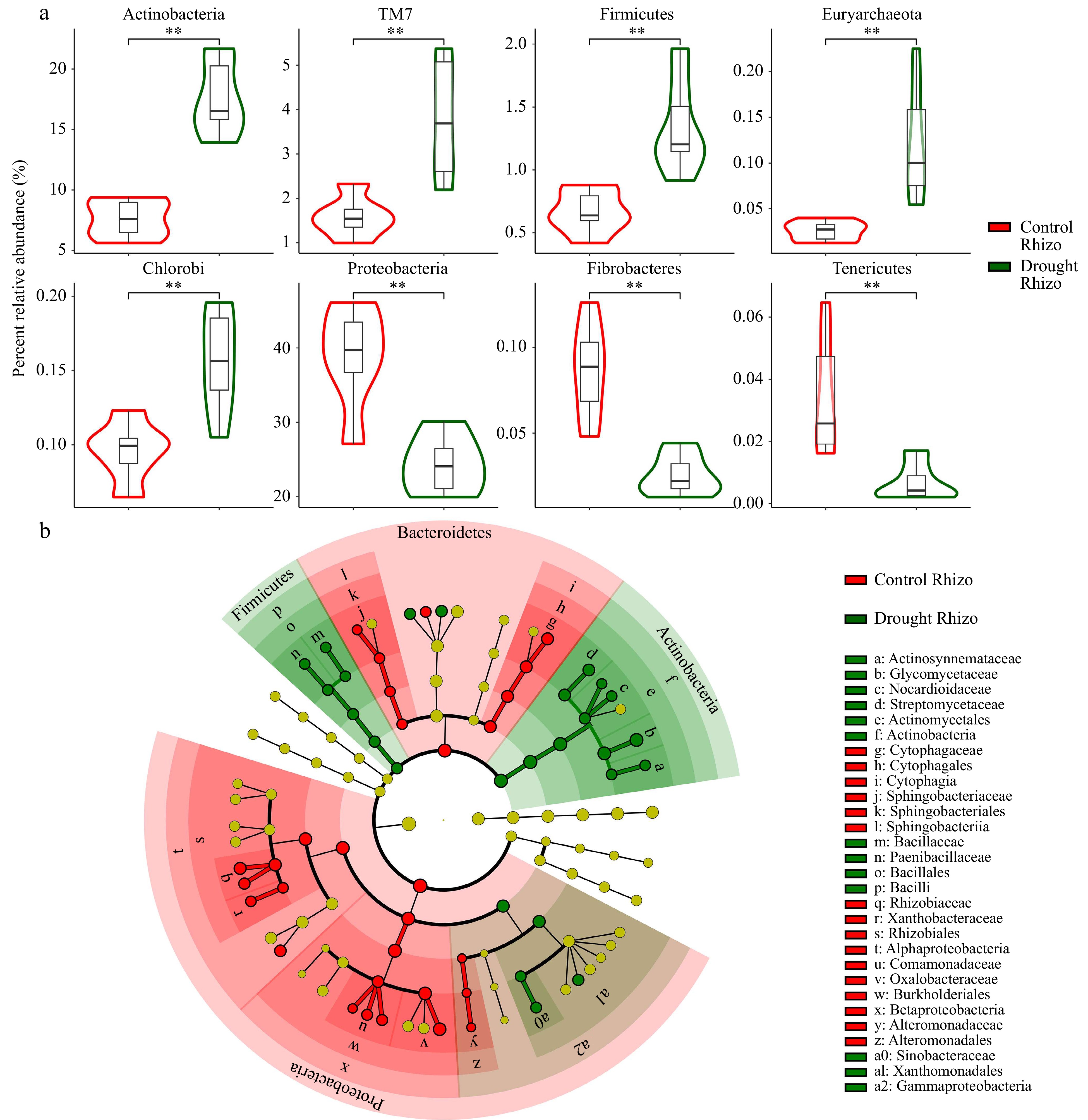
Figure 3.
(a) Wilcoxon Signed Rank Test for relative abundance of the abundant bacterial phyla, and (b) LEfSe analysis in control (red) and drought (darkgreen) groups. ** indicates p < 0.01.
Plant growth and microbial diversity response to humic acid fertilizer
-
Previous studies suggested that HA regulates plant growth by altering the root exudation profile[14] and played a role in protecting plants against water stress when co-inoculated with bacteria[15]. To further test the hypothesis that HA has a greater influence on the development of the root microbiome compared to the control during the flowering period, commercial compound fertilizers and organic HA fertilizer treatments were utilized. During the flowering stage, the HA group exhibited better-developed lateral roots compared to the control (Fig. 4a bottom left), although there were no statistically significant differences in both fresh and dry root weights between the two groups (Fig. 4b & c). Meanwhile, most of the phenotypes detected in this study showed minor differences between the control and HA group, except that the plant height at the earlier stage was significantly higher in the HA group (Supplementary Fig. S3). This result indicates that HA may play a potential role on belowground root traits but have little effect on aboveground traits.
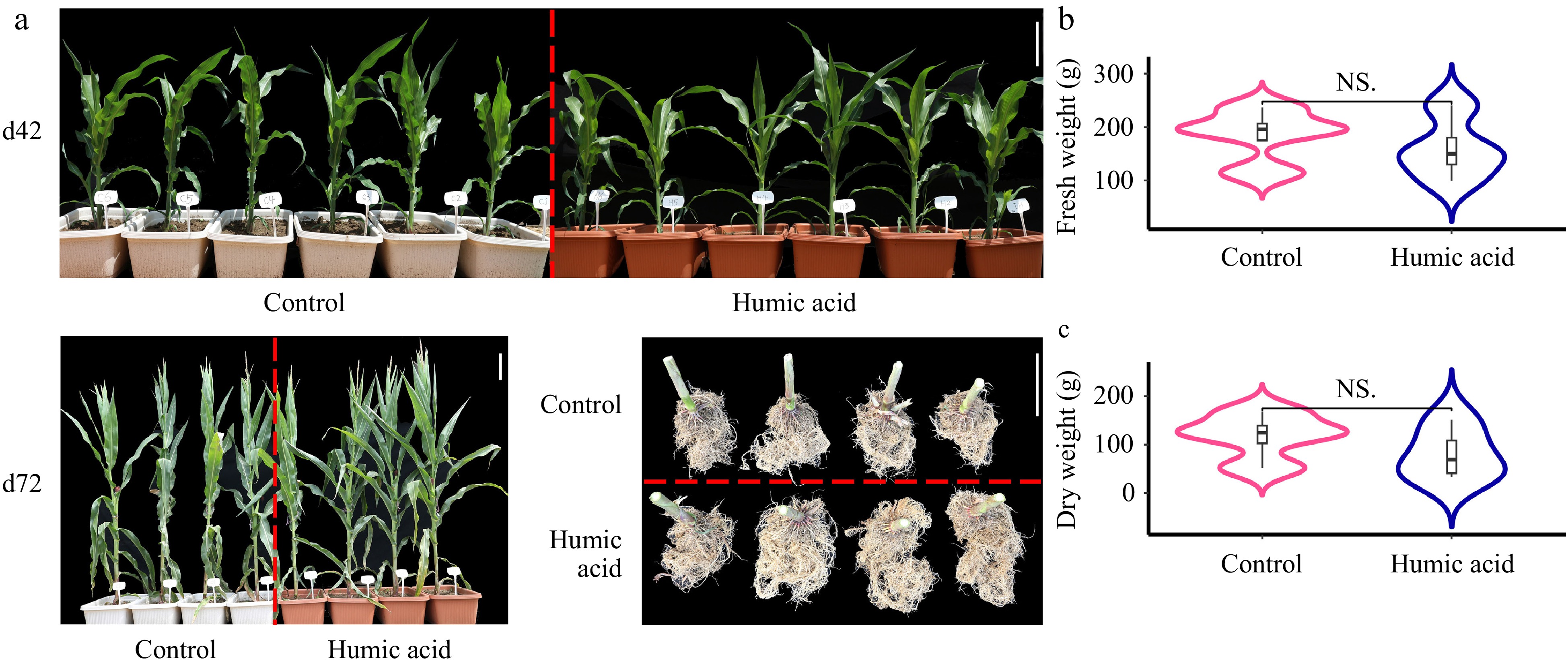
Figure 4.
Humic acid potentially benefits root growth vs normal conditions. (a) The plants grown on days 42 (d42) and harvested at d72, as well as the roots harvested on d72 under control and humic acid conditions, the scale bar = 20 cm. Statistics of the weight of (b) fresh roots, and (c) dry roots using R/Student's t test between the control and humic acid conditions. 'NS' indicates not significant.
To better understand the potential role of HA fertilization on the root microbiome, the root microbiome under HA fertilization was determined. Phylum-level relative abundance of Acidobacteria, Acitinobacteria, Bacteroidetes, Crenarchaeota, Cyanobacteria, and Proterobacteria revealed that the rhizosphere microbiomes exhibited a different composition compared to the control (Fig. 5a, 'Control Rhizo' and 'Humic Acid Rhizo'; results of statistical analyses were shown in Supplementary Fig. S4). While the above results suggest a different pattern for an abundance of these rhizosphere microbiomes on the phylum level during the development of the root microbiome under HA-treated environments. Evidence of the root rhizosphere microbiome is observable in these microbial lineages when a finer taxonomic resolution was used, as illustrated by family-level changes (Fig. 5b; results of statistical analyses are shown in Supplementary Fig. S5).
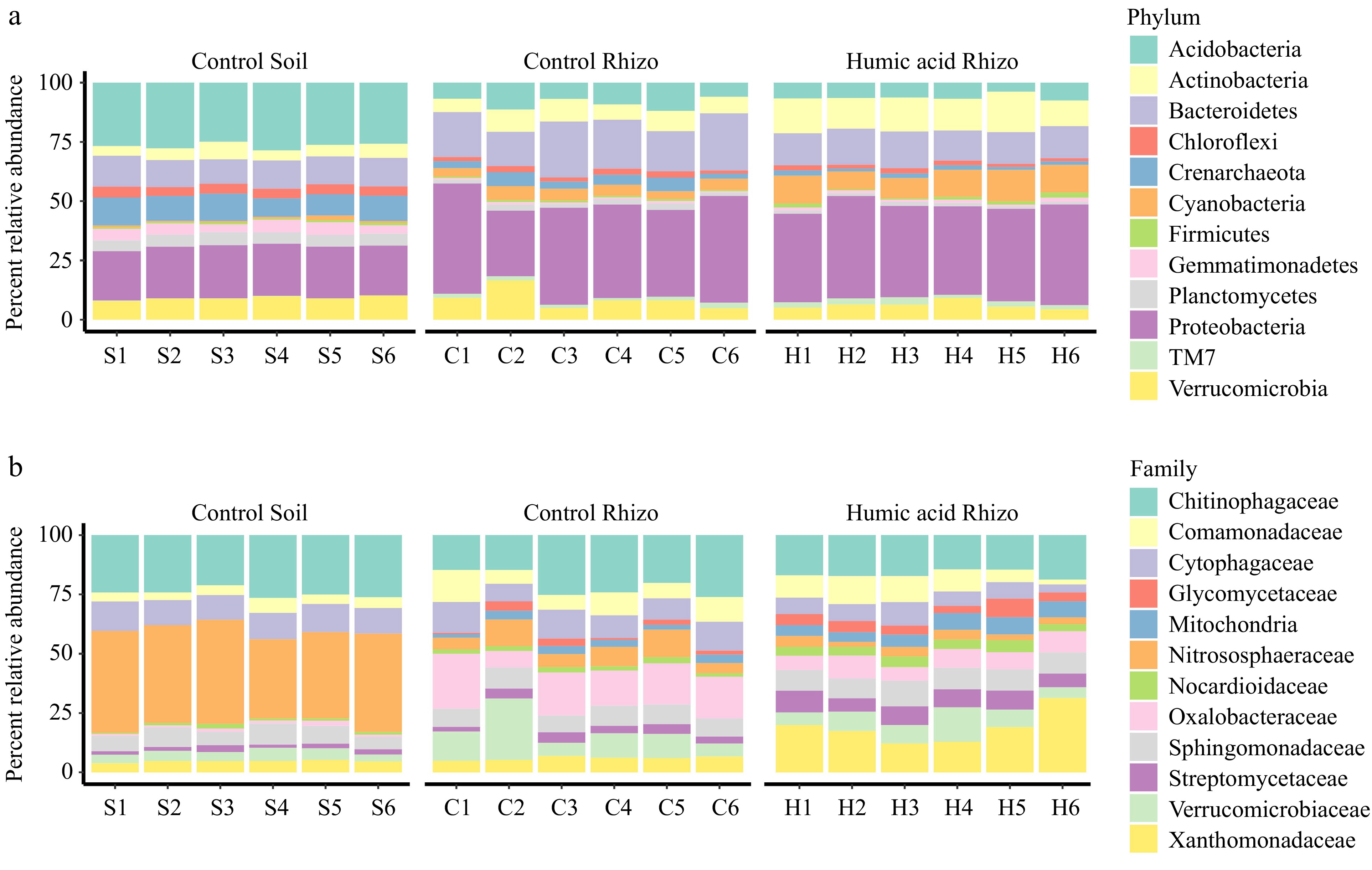
Figure 5.
Relative abundance of the top 12 of the most abundant bacterial phyla and families. Percent relative abundance of the (a) most abundant phyla, and (b) family for (c) control, humic acid (H) treatments in rhizospheres and for control in bulk soils (S). All individuals were arranged in order within each group along the x axis.
-
This study identified 40 bacteria phyla across soil, control rhizosphere, drought-induced, and HA-induced rhizosphere bacteria. The diversity of 12 out of 40 bacterial communities at the phyla level significantly varied in their resistance to drought stress (Supplementary Fig. S2). Among these, Actinobacteria and Bacteroidetes were notably enriched and depleted, respectively, in HA fertilizer-treated samples (Supplementary Fig. S4), which was consistent with a previous study[16]. In this study, the increased abundance of Actinobacteria might be caused by the HA increase in the N content in the soil, further enhancing the rate of nitrogen cycling[16]. Actiomyces are known for their ability to produce enzymes that degrade complex organic matter, which could enhance the availability of nutrient uptake. This nutrient enrichment might synergize with HA, thus improving plant growth and resistance under stress conditions. While the present study primarily focused on the most abundant bacterial phyla and families, it is crucial to consider the role of low-abundant yet core microbial taxa. These taxa, although present at lower relative abundances, often contribute significantly to ecosystem stability and plant health. Low-abundance microbial communities can serve as keystone species, providing essential functions such as nutrient cycling, production of growth-promoting hormones, and suppression of pathogenic organisms. Targeted approaches in further studies, such as metagenomics or metatranscriptomics, could help elucidate the specific roles of these low-abundant microbial taxa in shaping plant phenotypes and contributing to overall plant fitness under varying environmental conditions.
To improve maize drought resistance and harness the benefits of HA for agricultural productivity, manipulating the root microbiome provides a promising opportunity. However, it is essential to understand the causes and consequences of shifts in the soil and root microbiome induced by drought and HA. In the current study, drought delayed plant development, measured by the number of leaves and plant height. However, as plants under normal conditions began reproductive growth and experience senescence, the drought-treated plants showed a significantly higher leaf count, consistent with previous observations[17]. Drought conditions were found to alter the microbial diversity, leading to an increase in drought-resistant phyla such as Actinobacteria and Firmicutes and a decrease in drought-sensitive phyla such as Bacteroidetes and Proteobacteria (Supplementary Fig. S2). These shifts could significantly impact crop performance by influencing nutrient availability and stress resistance. Understanding these microbial shifts could provide valuable insights into how the microbiome could contribute to adaptation to environmental changes. A previous study on physiological and molecular mechanisms of HA influencing drought resistance in maize, demonstrated that the application of HA under drought conditions improved the proportion of soil macro-aggregates (e.g., P, K, Fe, and Mg), increased ATP synthase activity, enhanced the content of IAA, and the concentration of osmotically active solutes[18]. This previous study also showed that maize yields were improved with HA treatments under drought stress. However, how HA fertilization impacts belowground root traits and microbiome diversity remains largely unknown. In this study, HA demonstrated a potential positive effect on root architecture, leading to more extensive root development. However, it is important to understand why these changes in root structure did not translate to statistically significant differences in root biomass. A possible reason may be due to the physiological mechanisms influenced by HA. Previous studies[4,18] suggest that HA can enhance root elongation and branching without necessarily increasing total root weight. The improvements in root architecture may be related to changes in root surface area, which facilitates nutrient and water uptake, rather than an increase in root mass. This improvement may not significantly affect overall root weight within the short-term duration of the present study.
The current study did not perform the HA treatment under drought stress. Whether these potentially beneficial microbiomes will be recruited with HA treatments under drought conditions needs to be tested with additional experiments. Nevertheless, the results indicate that HA affects the abundance and composition of rhizosphere microbiome and underscore the importance of root-associated microbiomes in responding to drought stress (Supplementary Fig. S6).
We would like to thank Dr. Fupeng Song for assistance in soil water content determination. We appreciate the constructive suggestions from anonymous reviewers. This study was funded by the National Key Research and Development Program of China to H. Liu (2022YFD1201700), the National Natural Science Foundation of China to X. Yang (32101705), and the Youth Innovation team project to X. Yang.
-
The authors confirm contribution to the paper as follows: study conception and design: Yang X; experiment conduction: Sun M, Zhang L, Kang C, Li X, Sun Y, Zeng X, Dong L; data analysis: Yang X, Zhu H, Liu H; draft manuscript preparation: Yang X, Liu H. All authors critically revised and provided final approval of this manuscript.
-
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
-
The authors declare that they have no conflict of interest.
-
# Authors contributed equally: Mingfei Sun, Lin Zhang
- Supplementary Fig. S1 Heatmap of the bacteria phylum (A) as well as Shannon’s diversity (B&C) represented in the control soil (S), control (C), drought (D) and HA (H) rhizosphere microbiome.
- Supplementary Fig. S2 Wilcoxon Signed Rank Test for percent relative abundance of the abundant bacterial phylum in Control Soil, Control rhizosphere (Control Rhizo) and drought rhizosphere (Drought Rhizo) groups.
- Supplementary Fig. S3 Summary statistics of phenotypes.
- Supplementary Fig. S4 Wilcoxon Signed Rank Test for relative abundance of the abundant bacterial phyla in control soil, control rhizosphere and Humic Acid rhizosphere groups.
- Supplementary Fig. S5 Summary statistics of relative abundance of the top 12 of the most abundant baterial families.
- Supplementary Fig. S6 Summary analyses of LEfSe among control soil (S), control (C), drought (D) and HA (H) rhizosphere conditions.
- Copyright: © 2024 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Sun M, Zhang L, Zhu H, Kang C, Li X, et al. 2024. Responses of microbial communities to drought stress and humic acid fertilizer in maize. Genomics Communications 1: e006 doi: 10.48130/gcomm-0024-0006 

Responses of microbial communities to drought stress and humic acid fertilizer in maize
- Received: 19 September 2024
- Revised: 26 November 2024
- Accepted: 28 November 2024
- Published online: 23 December 2024
Abstract: Humic acid (HA) fertilizer was previously considered to be able to enlarge the effective absorption area of roots and promote plant root growth. Recent evidence suggests that certain root-associated microbes might be able to mitigate the negative responses of drought stress. In this study, the abundance and diversity of root-associated bacterial communities under HA fertilizer and drought stress treatments were explored. Rhizosphere microbiomes of three groups (HA, drought, and control) and microbiomes from bulk soil were collected during the flowering period in maize. Using Illumina Hiseq2500, the bacterial data for the V4 region of the 16S rRNA gene was obtained. Analysis of the sequencing data from 40 bacteria phyla revealed the abundance levels of the 12 bacterial communities were significantly different in the rhizosphere of the drought-treated samples compared to the controls. Two of these phyla, Actinobacteria and Bacteroidetes, were also significantly enriched or depleted in HA fertilizer-treated samples. The results underscore the dynamics of the rhizosphere microbiomes under drought stress and indicate the association between HA and rhizosphere microbiomes. However, more studies are warranted to understand the effect of HA fertilizer and beneficial microbiomes in responding to drought.
-
Key words:
- Humic acid (HA) /
- Drought /
- Rhizosphere /
- Microbiome /
- Maize