









-
Benefiting from the development of next-generation sequencing (NGS) technologies, metagenomic sequencing has become a powerful tool in the last two decades to reveal the taxonomic and functional inventories of various environmental microbiomes[1,2]. The NGS platform, however, typically generates a large number of short reads by sequencing artificially produced fragmented DNA, which is referred to as the shotgun sequencing approach[3]. According to a standard operating procedure for metagenomic data analysis, short reads are assembled to restore the sequence information of microbial DNAs, which usually obtain a set of long sequences named contigs instead of complete genomes[3,4]. The follow-up taxonomic and functional quantification is generally achieved by mapping short reads in turn to the assembled contigs or predicted coding genes[5,6].
A series of algorithmic tools have been developed to accomplish an efficient and accurate short read mapping, among which the most prevalent choices are Bowtie and BWA, both with more than 40,000 citations in total[7−9]. Bowtie and BWA both were implemented based on the alignment-based Burrows-Wheeler Transform (BWT) method, while another alignment-free tool named Salmon has recently been developed using a dual-phase parallel inference algorithm[10]. Although Salmon was originally designed and tested for the mapping of RNA-seq reads, it has also been recommended in metagenomic analysis due to its ultra-fast speed, and was widely used to deal with the increasingly large sequencing data[6,11−13]. In the latest version, Salmon provides a '--meta' option for metagenomic analysis. The above tools were commonly vaguely applied, without an explicit description of relevant parameters, to estimate the sequencing depth and abundance of contigs and/or genes. However, unlike contigs and the transcripts in RNA-seq which themselves are directly assembled from short reads, the coding genes in metagenomic analysis are just predicted open reading frames (ORFs) on contigs[14]. This would imply the existence of abundant reads with only a subset of their sequences (on the right or the left side) mapping to the gene sequences, thereby producing local alignments that affect the abundance estimation.
In our previous practices of metagenomic analysis, some short genes obtained remarkably low abundance values (or even 0%) through direct mapping of reads using Bowtie 2 or Salmon, likely owing to the removal of local alignments. Some studies suggested a filtration of short genes with lengths less than those of reads[15,16]. Nevertheless, it is proposed that such a problem should intrinsically influence the abundance calculation of most genes with different effect sizes depending on gene lengths. In other words, the abundance values of a gene might partly be determined by its biological nature (i.e., length). A local-alignment mode of reads mapping is supported by Bowtie 2, as well as the BWA-MEM algorithm[9,17]. Moreover, it would also be better to calculate gene abundance values based on the mapping of reads to contigs, which theoretically does not produce local alignments. However, no assessment has been conducted to date to evaluate the accuracy of these methods in regard to the aforementioned issue. Therefore, in this study six methodologies of read mapping in calculating the gene abundance values of short-read sequencing datasets were tested and compared, considering the importance of gene quantification for downstream analyses.
-
A total of 43 samples of cyanobacterial enrichment cultures which were provided by the Freshwater Algae Culture Collection of the Institute of Hydrobiology (FACHB) at Wuhan, China, were used in this study. In addition, two samples of surface seawater were collected at a depth of ~1 m from Daya Bay, China. Microbial DNA was extracted as described in our previous studies[18,19]. The extracted DNA samples were then randomly fragmented to an average length of ~350 bp using an ultrasonicator (Covaris M220). Paired-end sequencing libraries were prepared and sequenced using different strategies, i.e., different read lengths and different platforms (Supplementary Table S1 & S2). For metagenomic analysis, raw reads were firstly trimmed to clean reads using Trimmomatic v0.36 to remove adapters and low-quality bases (Q < 20). Trimmed reads with lengths < 50 bp or having more than five unidentified nucleotides (N) were discarded using custom Perl scripts. All filtered reads were then assembled using the SPAdes v3.13.0 pipeline[20].
Comparison of different mapping methodologies
-
The ORFs on contigs were predicted as protein-coding genes using Prodigal v2.6.3[14]. Mapping of short reads to contigs and genes were first performed using Bowtie 2 v2.5.1 in both the global mode (i.e., with default parameters) and the local-alignment mode (with the parameter '--local')[9]. Read mappings were also performed with the help of BWA v0.7.17, using the BWA-MEM program which tacitly support the local-alignment mode[17]. The SAM files produced by Bowtie 2 and BWA-MEM were converted to BAM files, which were then sorted, both using SAMtools[21]. The mean coverage depth (i.e., the mean number of times a nucleotide was sequenced) were calculated using custom Perl scripts. The abundance values of protein-coding genes were calculated using the following equation:
$ A=\dfrac{{M}_{d}}{{N}_{r}}\times {10}^{7} $ (i) where A is the abundance, Md is the mean coverage depth, and Nr is the total number of paired clean reads. Mapped reads and TPM were further calculated for genes using the alignment-free Salmon with the methodology called selective alignment and the parameter '--meta' as recommended by the authors[10,22]. Moreover, Salmon was run both without decoys and with the contigs as decoy sequences. In the 'contig mapping' method presented in this study, the coverage depth of each site on a contig was first calculated by the command 'samtools depth'. Values of Md for genes were calculated based on the above results of contig depths and the GFF files generated by Prodigal, which record the location information of genes. In other words, the Md of a gene was calculated as the mean of the coverage depths of nucleotides between the start and end site of the gene on the contig. A custom Python script was developed to output the Md and abundance of genes ( more details at
https://github.com/biotengwk/Meta_pipeline ). Statistical analysis and visualization were mainly performed using R v4.0.3.All coding genes were annotated using the Hidden Markov Models (HMM) database of KEGG Orthologs (KOfam, downloaded from
ftp://ftp.genome.jp/pub/db/kofam/ on 18 April 2023) implemented in HMMER v3.1b2 with the parameters: e-value ≤ 10−5; alignment coverage ≥ 0.5[23]. Considering the non-normal distribution of abundance data[24], non-parametric Wilcoxon signed-rank tests (wilcox.test in R) was conducted to analyze the intergroup differences of KEGG Orthologs (KOs) and all estimated p-values were adjusted using the 'Bonferroni' method (p.adjust in R). Comparison of the results using six different mapping methodologies was shown by the UpSet plot with the help of UpSetR package. A circular diagram of the chromosome sequence was generated using Circos v 0.69 as described previously[25]. -
To examine the technical variability probably induced by mapping algorithms, the abundance values of protein-coding genes on a single contig were calculated with the help of alignment-based tools Bowtie 2 and BWA-MEM. As part of our previous study[18], an enrichment culture of Anabaena sp. FACHB-83 was subjected to shotgun sequencing on an Illumina HiSeq platform which generates 150 bp pair-end reads (i.e., PE150). After quality control processing, a total of 6,500,346 paired clean reads were obtained with a mean length of 145 bp. Metagenomic assembly yielded a long contig of 2.9 Mb in length, which formed a circular chromosome of a bacterium belonging to the Armatimonadota phylum as assigned by the Genome Taxonomy Database Toolkit (GTDB-Tk)[26]. The abundance values of both the chromosome and the predicted 2,737 protein-coding genes on it were subsequently calculated.
In theory, the abundance value of the chromosome (175.79 by Bowtie 2 and 176.35 by BWA-MEM, both with default parameters) should be the mathematical expectation of gene abundance values. However, the results for coding genes were rather unexpected. By using Bowtie 2 with default parameters, the abundance values of genes ranged from near 0 to more than 200 with a mean of 147.86 which was much less than the chromosome abundance, and surprisingly gradually decreased with the diminution of gene length (Fig. 1b). It was speculated that the elimination of local alignments in a global mode should account for this variability. To assess this effect size, it was assumed that there were adequate reads of equal length (d) which could map to each site of the contig with length l totally at random. As l >> d, it could be further assumed that there was an equal number (n) of reads on average mapped to the contig and started at each site around a specific gene. Therefore, there would be n(l-d+1) reads forming global alignments with the gene, and 2n(d−1) reads forming local alignments with the gene. Because in local alignments only half of a read on average was mapped to the gene, the global alignments and local alignments contributed nd(l-d+1) and nd(d−1) bases, respectively, to the coverage depth of the gene. In other words, when calculating the coverage depth and abundance values of the gene, a total of ndl reads, instead of only nd(l-d+1) in a global mode, should be included. Thus, the equation relating the gene abundance (Agene) generated using Bowtie 2 and the contig abundance (Acontig), which served as a proxy for the theoretical gene abundance, could be inferred as shown in Fig. 1a. Excitingly, this relationship model (the red line shown in Fig. 1b) fits the gene abundances well, supporting the conjecture that the elimination of local alignments leads to such variability.
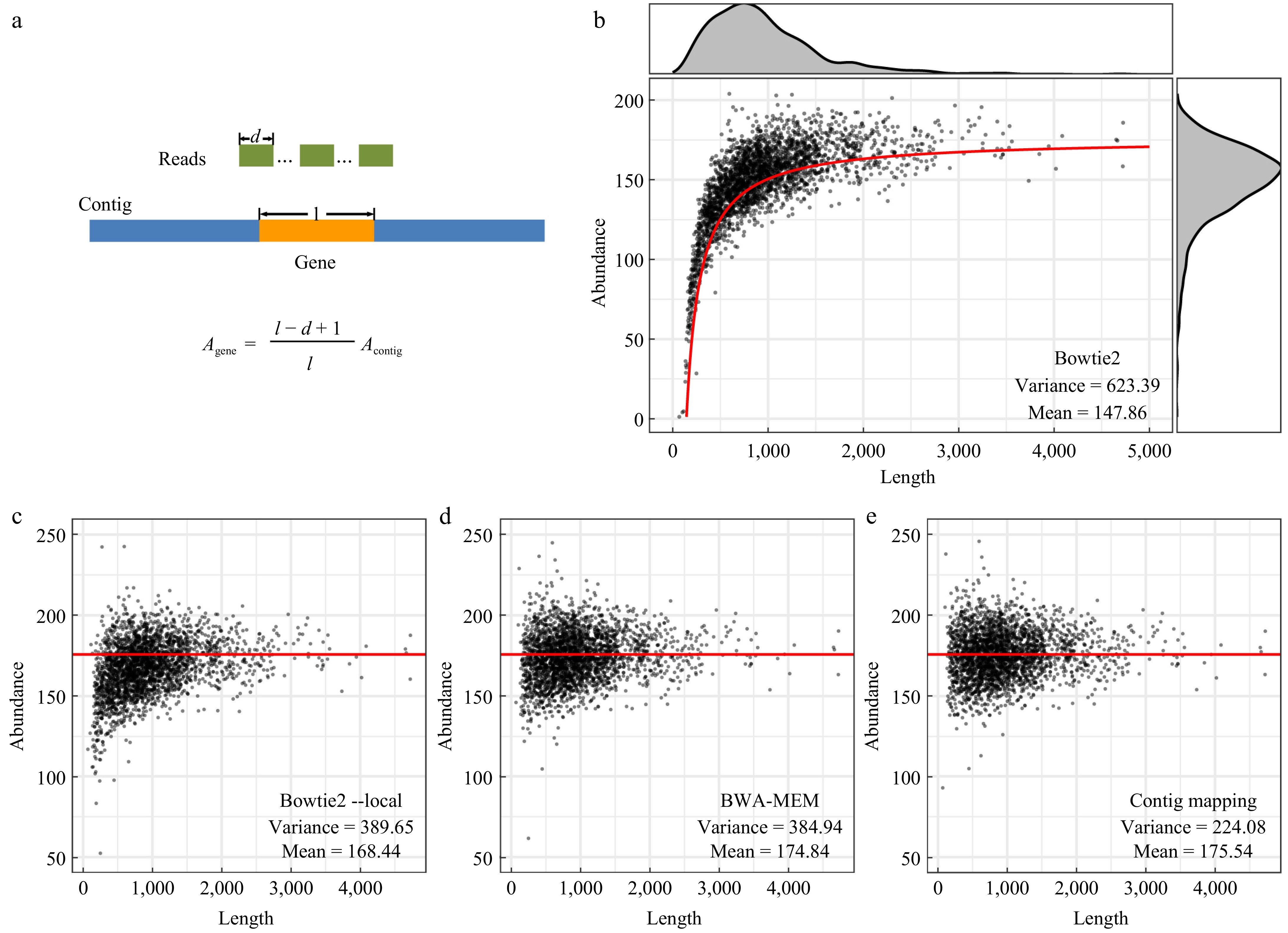
Figure 1.
Gene abundance values calculated using the alignment-based algorithms. (a) Schematic diagram showing the generation of local alignments in read mapping of genes and the equation relating the abundance of genes and that of the chromosome. (b) Gene abundance values calculated using Bowtie 2 with default parameters. The red line indicates the abundance value of genes calculated based on the equation shown in (a). (c) Abundance values calculated using Bowtie 2 with the parameter '--local'. (d) Abundance values calculated using BWA-MEM with default parameters. (e) Gene abundance values calculated based on gene position and the coverage depth of the chromosome sequence. The red lines in (c)−(e) indicate the abundance value of the chromosome calculated using Bowtie 2 with default parameters.
When using Bowtie 2 in the local-alignment mode (i.e., with the parameter '--local'), such length-decay of gene abundance was largely mitigated, with the total variance reduced from 623.39 to 389.65 and the mean value increased to 168.44 (Fig. 1b & c). Most genes however still showed abundance values lower than that of the chromosome, especially those with short lengths. Slightly better performance was achieved using BWA-MEM, suggesting that read mapping in local-alignment mode indeed reduced the technical variability (Fig. 1d). Encouragingly, by using the 'contig mapping' method (i.e., calculating the abundance values of a gene based on its position and the coverage depth of each site on the contig, see Materials and methods), the best performance was accomplished with the minimum variance of 224.08 and a mean abundance value of 175.54 which was very close to that of the chromosome (Fig. 1e). Potential sources of the remaining variability may be complex and derived from systematic differences in sequencing depth between genome regions resulted from genome organization, GC-content, and so on[5,27]. Particularly, extraction and sequencing of genomic DNAs undergoing replication will lead to higher sequencing depths of sequences around the replication origin oriC[28]. In line with this interpretation, a symmetric pattern of coverage depth was observed on the chromosome similar to that of the GC-skew (Supplementary Fig. S1), reflecting a theta mode of chromosome replication which is prevalent in bacteria[29]. The GC-skew transition corresponds with the oriC or replication terminus in general[29]. Thus, the approximate location of oriC can be inferred according to GC-skew and sequence depth (Supplementary Fig. S1), although without a reference in the latest DoriC database[30].
Length-dependent gene abundances produced by alignment-free methods
-
By using Salmon, it is nevertheless striking to find that the calculated transcripts per million (i.e., TPM) increased rapidly with the diminution of gene length (Fig. 2a). Especially, for genes with lengths less than 1,000 bp, the TPM could range from 217.76 to more than 1,000, only except for three genes that were shorter than reads and had TPM values near 0 (Fig. 2a). The variance of the TPM values increased by about two orders of magnitude (up to 38,451.37). More dramatically, when the chromosome was used as a decoy sequence to help for the filtration of mismapped reads, the TPM values diverged more and more during the diminution of gene length (Fig. 2b). By analyzing the intermediate result files of Salmon, it was found that an excessive restriction in the value of 'EffectiveLength' likely accounted for such dramatic results (Fig. 2c). This measure, as described by the authors of Salmon, was employed considering the probability of sampling fragments from a specific transcript, which reflects the nature of RNA-seq (
https://salmon.readthedocs.io/en/latest/salmon.html ). However, there is no need to take into consideration such an effect for predicted coding genes in metagenomic sequencing. Thus, the TPM values were recalculated using the number of mapped reads (i.e., the 'NumReads' generated by Salmon) and real gene lengths instead. As anticipated, this analysis revealed similar results to those of Bowtie 2 (Fig. 1b & 2d). Because reads best aligned to the decoy sequence were discarded, there were lower values of TPM to different extents in the decoy-aware mode (Fig. 2b & e). Interestingly, it was also found that genes with short (less than 145 bp) upstream and downstream noncoding regions decreased to the greatest extent as indicated by the TPM results (Fig. 2b), implying that the arrangement of genes on the chromosome affected the assignment of a paired-end read regarding whether it constituted a valid mapping to the genes.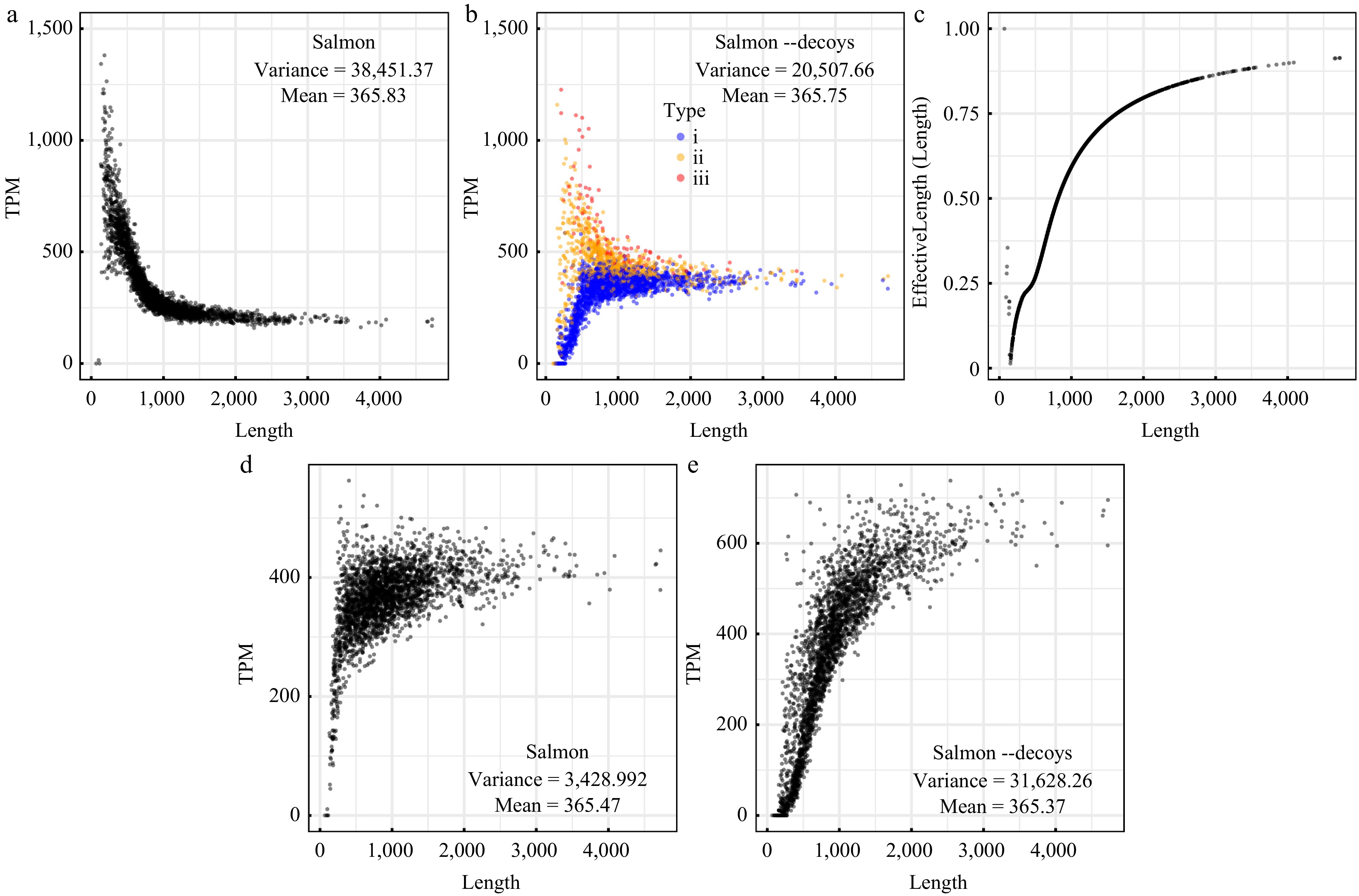
Figure 2.
Gene abundance values calculated using the alignment-free algorithms. (a) Values of the TPM (transcripts per million) measure calculated using Salmon with the parameters '--meta --validateMappings'. (b) Values of the TPM measure calculated using Salmon with the chromosome as a decoy sequence. Dots with different colors correspond to three types of genes: (i) genes with upstream and downstream noncoding regions less than 145 bp, (ii) genes with upstream or downstream noncoding regions less than 145 bp, and (iii) genes with upstream and downstream noncoding regions more than 145 bp. (c) Variation in the EffectiveLength/Length ratio as a function of gene length. (d) Values of TPM calculated using the numbers of mapped reads generated by Salmon and real gene lengths. (e) Values of TPM calculated using the numbers of mapped reads generated by Salmon with the chromosome as a decoy sequence and real gene lengths.
Length-dependent gene abundances influence the functional quantification
-
In theory, length-dependent gene abundance resulted from inappropriate application of mapping algorithms are ubiquitous in all metagenomic practices using the NGS platforms. To support this, three additional datasets were adopted, including the metagenome of another enrichment cyanobacterial culture (Calothrix sp. FACHB-168) using Illumina HiSeq PE125, and two marine metagenomes from our previous study[19], DAYAB-A02 and DAYAB-A35, using the DNBseq PE150 and DNBseq PE100 (MGI Tech, China) platform, respectively (Supplementary Table S1). For each dataset, the longest contig with coverage depth > 30 was selected to calculate the abundance values of coding genes using six different mapping methodologies as described above. As anticipated, length-dependent gene abundance values were observed in all four datasets using Bowtie 2 or Salmon (Supplementary Figs S2−S5). An unexpected observation was that mapping methodologies based on local alignment generated high variances even exceeding those generated using Bowtie 2 with a global-alignment mode (Fig. 3a). This was largely due to multiple outliers with extremely high abundance values (Supplementary Figs S2−S5). The 'meta' mode of Prodigal (vs the 'single' mode used above), which was designed to apply to metagenomes[14], probably account for these outliers. For example, Prodigal with the 'meta' mode predicted more genes (2,743 vs 2,737) for the chromosome from FACHB-83 and the gene with the highest abundance value resided in a non-coding region as predicted with the 'single' mode. Anyway, the 'contig mapping' method proposed here still had the best performance, showing the minimum variance of gene abundance values regardless of sequencing strategies (Fig. 3a).
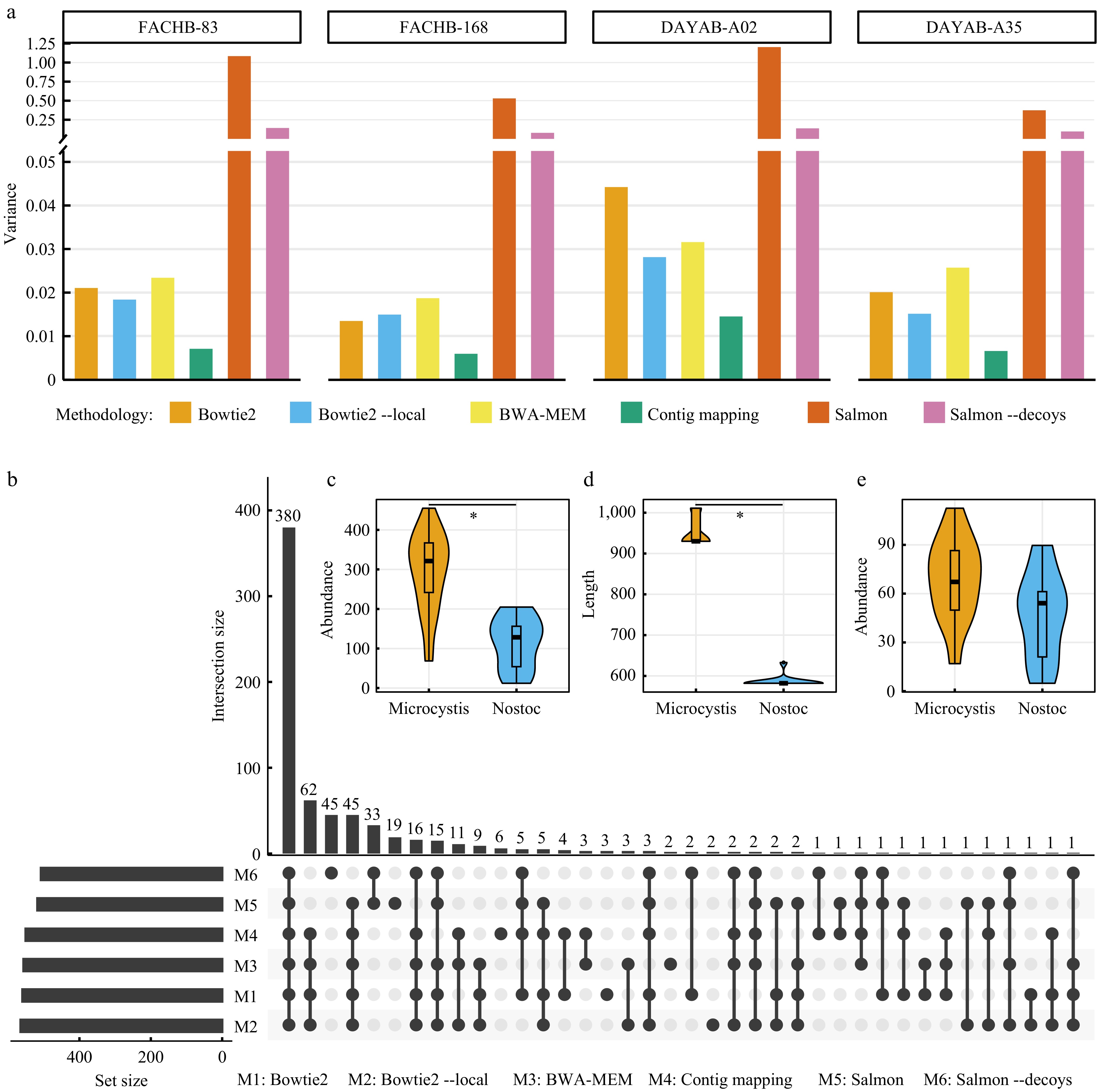
Figure 3.
Length-dependent gene abundances pervasively influence the quantification of microbial function. (a) Variances of calculated abundance values of genes from four different datasets for each mapping methodology. To facilitate the comparison, the original gene abundance values of each dataset are normalized by dividing by the average abundance of respective long (> 3,000 bp) genes. (b) Identification of KEGG Orthologs (KOs) with significant differences (adjusted p-value < 0.05) in abundance between two different cyanobacterial cultures (Microcystis sp. and Nostoc sp.) using six mapping methodologies. In this plot, the combination matrix at the bottom shows the intersections of six sets of identified KOs and the bars at the left and top shows the sizes of sets and intersections, respectively. (c) Comparison of the abundance values of K08480 in two different cyanobacterial cultures using the 'contig mapping' method proposed in this study. (d) Comparison of the lengths of genes annotated to K08480 in two different cyanobacterial cultures. (e) Comparison of the abundance values of K08480 in two cyanobacterial cultures using Salmon. * in (c) and (d) indicate adjusted p-value < 0.05.
The ubiquity of length-dependent gene abundance in analyzing short-read sequencing data potentially compromises the quantitative profiling of metagenomic datasets. In a comparison of functional genes between the enrichment cultures of Microcystis sp. and Nostoc sp. (Supplementary Table S2, data from our previous study[18]), the above six different mapping methodologies resulted in different sets of KOs with significant differences in abundance values (Fig. 3b). Particularly, only 55% (380 of 694) of KOs were identified by six approaches simultaneously. Further, the largest difference was found between alignment-based and alignment-free algorithms (Fig. 3b). Except for the above 380 KOs, four alignment-based methods had an intersection of 62 KOs while two alignment-free approaches using Salmon identified 97 distinct KOs in total. Overall, the results demonstrate that inappropriate application of mapping algorithms likely decreases the accuracy of downstream analyses in metagenomic analysis. One representative example is K08480, an important circadian clock gene named kaiA[31]. When using the 'contig mapping' method, K08480 showed a significant difference in abundance between two groups of enrichment cultures with adjusted p-value < 0.05 (Fig. 3c). However, due to significantly smaller lengths of the genes annotated to K08480 in Nostoc sp., the abundance values were elevated using Salmon and resulted in no significant difference (Fig. 3d & e). It is difficult to categorically declare which approaches produce the estimates closest to the ground truth. Nevertheless, as demonstrated above, the 'contig mapping' method minimizes the technical variability and is at least more plausible than other approaches.
-
Metagenomic analysis workflows employ a large and diverse array of tools[3,6]. Subtle differences among different tools of the same class as well as different settings of the same tool potentially bring multiple biases influencing the quantification of microbial community and function[5,32]. In this study, multiple mapping algorithms were applied to calculate gene abundance values for short-read sequencing datasets. Results indicated that mapping reads directly to predicted genes intrinsically induce length-dependent gene abundance values regardless of whether an alignment-based or an alignment-free algorithm was applied. Such interference primarily caused by the elimination of partly mapped reads had a serious implication especially for the quantification of genes less than 1000 bp in length (Fig. 1b & 3), and further influenced downstream analysis (Fig. 3b). According to our previous data[33], more than 60% of the total genes from 11,502 representative prokaryotic genomes are less than 1000 bp in length. Moreover, genes of the same function can be very variable in length due to different evolutionary histories as well as errors from sequencing and metagenomic assembly, which in turn perplexes the comparison between datasets (Fig. 3c−e). Local-alignment methods (Bowtie 2 with '--local' and BWA-MEM) performed a little better than those based on global-alignment, but were still inferior to the 'contig mapping' method. Therefore, we recommend calculating gene abundances based on gene coordinates and coverage depths of contigs, and developed a publicly available Python script to accomplish this task, which was suitable for all short-read sequencing datasets from NGS platforms. In addition to the best performance, this also reduce the computational cost because reads mapping to contigs is necessary for metagenomic binning[34].
According to the technical procedures of metagenomic sequencing, a contig sequence represents a real genomic or environmental DNA fragment and in an ideal scenario each site on it has an equal sequencing depth[4,35]. Thus, one may argue that all genes on the same contig can be assigned abundance values equal to that of the contig. This was observed roughly to be the case, as the average value of gene abundances using the 'contig mapping' method was indeed approximately equal to the contig abundance (Fig. 1e), which exists as proof of the accuracy of the present method. Nevertheless, this study was conducted based on the fact that mapping reads directly to genes has been widely applied in previous studies, especially those solely focused on genes related to specific functions, e.g., antibiotic resistance genes[6,12]. Furthermore, genes on the same contig still show variations in their coverage depths, likely indicating important biological processes, including the replication of portions (Supplementary Fig. S1). Another controversy may surround the measure of gene abundance. In the field of RNA sequencing, terms like TPM, reads per kilobase of transcript per million reads mapped (RPKM) and fragments per kilobase of transcript per million reads mapped (FPKM) have been proposed as measures of gene expression[36]. TPM and RPKM, though having been used by many studies in metagenomic analysis workflows, both are calculated using read counts. According to the present results, coverage depths performed much better than the read counts in calculating the abundance of metagenomic genes due to the occurrence of local alignments. The abundance value calculated by the Eqn (1) with the help of our Python script is similar but not identical to RPKM. As the fact that the FPKM measure can easily be converted to TPM by dividing by the sum of all values and multiplying by 106[37], the gene abundance in metagenomic analyses can also be converted to a relative abundance which are similar to the TPM measure using the same method. This is in agreement with the classical thinking in quantitating the abundance of contigs and metagenomic assembled bins (MAGs) in multiple workflows, e.g., CheckM[38]. Because TPM has been considered to be more suitable for inter-group comparisons[37], the above abundance data calculated by using BWA, Bowtie 2, and our 'contig mapping' method were further normalized to relative abundance. As expected, a new comparison revealed similar results, though with a slightly higher proportion of KOs (60%, 399 of 667) which were identified by six approaches simultaneously (Supplementary Fig. S6), indicating again the strong influence of length-dependent gene abundance. In summary, the present results suggest that a cautious application of computer algorithms by taking the biological and experimental nature into account is imperative to improve the accuracy of metagenomic analysis.
This research was supported by grants from the National Natural Science Foundation of China (Grant Nos 92351301 and 32370009), and the Natural Science Foundation of Guangdong Province (Grant No. 2022A1515010464).
-
The authors confirm contribution to the paper as follows: study conception and design: Teng W, Chen M; data collection: Teng W, Chen S; data analysis and interpretation of results: Teng W, Xia T; draft manuscript preparation: Teng W, Xu Y; response to reviewers and manuscript revision: Teng W, Zhou Y; supervision and funding: Shu W, Zhang C. All authors reviewed the results and approved the final version of the manuscript.
-
All data used and generated in this study, including the sequence data, work records, and related scripts are available from the following site: https://doi.org/10.6084/m9.figshare.25807498.
-
The authors declare that they have no conflict of interest.
-
# Authors contributed equally: Wenkai Teng, Mengyun Chen
- Supplementary Table S1 Four metagenomic datasets used in this study.
- Supplementary Table S2 Metagenomic datasets for comparison of functional genes.
- Supplementary Fig. S1 Circular map of the chromosome sequence used in this study.
- Supplementary Fig. S2 Gene abundance values of FACHB-168 calculated using six different mapping methodologies.
- Supplementary Fig. S3 Gene abundance values of FACHB-83 calculated using six different mapping methodologies.
- Supplementary Fig. S4 Gene abundance values of DAYAB-A02 calculated using six different mapping methodologies.
- Supplementary Fig. S5 Gene abundance values of DAYAB-A35 calculated using six different mapping methodologies.
- Supplementary Fig. S6 Length-dependent gene abundances pervasively influence the quantification of microbial function.
- Copyright: © 2024 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Teng W, Chen M, Chen S, Xia T, Zhou Y, et al. 2024. Inappropriate application of mapping algorithms results in length-dependent gene abundances in metagenomic analysis. Genomics Communications 1: e007 doi: 10.48130/gcomm-0024-0007 

Inappropriate application of mapping algorithms results in length-dependent gene abundances in metagenomic analysis
- Received: 23 September 2024
- Revised: 02 December 2024
- Accepted: 04 December 2024
- Published online: 23 December 2024
Abstract: Multiple biases still exist in metagenomic analysis workflows compromising a precise quantification of microbial species and function. In the present study it is illustrated that inappropriate mapping of short reads could result in length-dependent gene abundances and further reduce the accuracy of downstream analyses. Specifically, mapping reads directly to predicted coding genes using alignment-based Bowtie 2 and alignment-free Salmon generated abundance values which dramatically decreased and increased with the diminution of gene length, respectively. This introduced high technical variabilities in gene abundances, which can be reflected by the variances, 623.39 for the abundance values using Bowtie 2 and strikingly 38,451.37 for the transcripts per million (TPM) using Salmon. In contrast, the abundance values calculated using the 'contig mapping' method proposed in this study were not affected by gene lengths with a low variance of 224.08. The universality of this problem was demonstrated by using four short-read datasets from different sequencing strategies. When identifying functional genes with significant differences between groups, only 55% (380 of 694) of KEGG orthologs were not influenced by mapping methodologies. Therefore, the 'contig mapping' method is recommended to minimize technical variabilities.
-
Key words:
- Metagenomic analysis /
- Gene abundance /
- Read mapping /
- Next-generation sequencing