






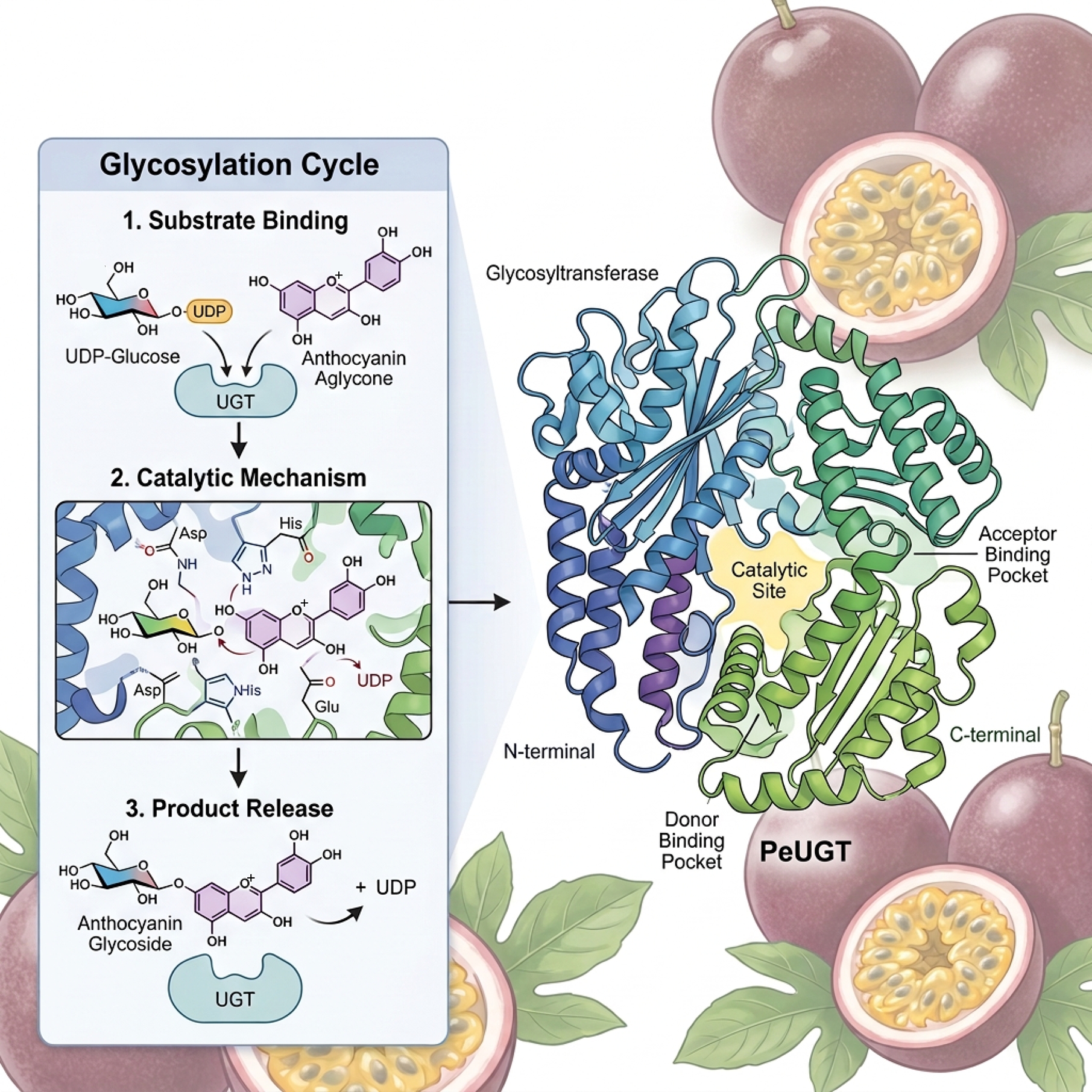



-
Glycosylation is a critical post-synthetic modification of plant secondary metabolites, significantly enhancing their solubility, stability, transportability, and structural diversity[1]. This modification plays a pivotal role in plant growth, development, and responses to both biotic and abiotic stresses[2]. Flavonoids—including flavonols, isoflavones, flavones, and anthocyanins—represent a major class of plant secondary metabolites that predominantly exist as glycoside conjugates. These glycosylation reactions are catalyzed by UDP-glycosyltransferases (UGTs), which transfer glycosyl moieties from activated donor molecules to specific acceptor substrates such as sugars and lipids[3,4].
The UGT family constitutes the predominant group within the CAZy glycosyltransferase family GT1 across the plant kingdom[5]. Structurally, plant UGTs exhibit a complex N-terminal domain responsible for substrate recognition and binding, enabling the accommodation of diverse substrates[6]. The C-terminal region harbors a highly conserved PSPG-box motif (Plant Secondary Product Glycosyltransferase box), which is essential for glycosyltransferase catalytic activity[7]. This 44-amino acid signature sequence is invariant across all characterized plant UGTs[8]. Owing to this conserved structural architecture, UGT gene families have been systematically identified and annotated in numerous plant species, including Arabidopsis thaliana[9], rice (Oryza sativa)[10], apple (Malus domestica)[11], potato (Solanum tuberosum)[12], and tea (Camellia sinensis)[13].
Furthermore, emerging evidence indicates that numerous glycosyltransferase genes are implicated in fruit growth, development, and responses to abiotic stress. During sweet cherry (Prunus avium) fruit development, PavUGT48 mediates the glycosylation of cyanidin and other anthocyanidins, thereby promoting anthocyanin accumulation and fruit coloration—playing a pivotal role in fruit ripening and quality formation[14]. In citrus, CitUGT72AZ4 participates in the regulation of flavonoid metabolism by catalyzing the 4-O-glycosylation of flavonols, thereby modulating the biosynthesis of citrus flavonoid glycosides during fruit development[15]. Additionally, UGT genes contribute to enhanced plant stress tolerance through the accumulation of protective glycosides. For instance, apple MdUGT88F1 regulates phloridzin biosynthesis and is involved in disease resistance[16], while tea plant CsUGT78A14 and CsUGT91Q2 modulate cold tolerance by regulating the synthesis of flavonoid and terpenoid glycosides[17]. Nevertheless, functional characterization of UGT genes remains predominantly confined to model plants and a limited number of economically important crops, leaving the biological functions of UGTs in many non-model plants largely unexplored.
Passiflora edulis Sims is a perennial evergreen climbing vine indigenous to tropical regions of South America and represents the most extensively cultivated species within the Passiflora genus[18]. Passion fruit juice is nutritionally rich, containing substantial amounts of sugars, organic acids, amino acids, vitamins, and flavonoids[19]. Notably, flavonoids exhibit potent biological activities, including anti-carcinogenic, antioxidant, and anti-inflammatory properties, conferring significant health benefits. In recent years, the release of the chromosome-scale passion fruit genome has catalyzed increasing research interest in the biosynthetic pathways and genetic underpinnings of its specialized metabolites[20]. Previous studies have demonstrated that heterologous overexpression of PeMYB114 in tobacco enhances flavonoid accumulation[21]; PeLOX4 expression elevates lipoxygenase activity, thereby promoting volatile ester biosynthesis and intensifying fruit aroma[22]; and PeCWINV5 overexpression augments soluble sugar accumulation[23], PeWRKY20 regulates the production of malic acid and volatile compounds by inhibiting the expression of PeMDH1[24]. However, systematic investigation of the UGT gene family in passion fruit remains scarce. Therefore, comprehensive characterization of the PeUGT family holds substantial significance for elucidating glycosylation-mediated metabolic regulation in this economically important fruit crop.
In this study, we conducted a genome-wide analysis of the UGT gene family in Passiflora edulis based on the published chromosome-scale genome, identifying a total of 149 UGT genes. We systematically characterized these genes through comprehensive analyses of phylogenetic relationships, gene structure, conserved motifs, chromosomal distribution, gene duplication events, promoter cis-acting elements, KEGG and GO functional annotations, and expression patterns across fruit developmental stages. This genome-wide investigation provides a valuable foundation for the functional characterization of UGT-mediated glycosylation in passion fruit secondary metabolism.
-
The Passiflora edulis genome sequence was retrieved from previously published studies. A total of 73 Arabidopsis thaliana UGT protein sequences were downloaded from the TAIR database (
https://arabidopsis.org/ ). These sequences were employed as queries for BLASTP searches against the passion fruit genome, with stringent filtering criteria of E-value < 1e-10 and sequence identity > 50%[25]. Concurrently, the UDP-glucosyltransferase domain (PF00201) was retrieved from the Pfam database, and Hidden Markov Model (HMM) searches were performed using default parameters to identify putative UGT proteins in P. edulis. Candidate genes identified by both BLASTP and HMM approaches were merged as the preliminary PeUGT family members. Subsequently, the Conserved Domain Database (CDD;https://ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi ) was employed to verify the presence of the characteristic UGT domain, thereby determining the final set of PeUGT family members.Phylogenetic analysis of the PeUGT gene family
-
Phylogenetic analysis was performed using PeUGT and selected UGT amino acid sequences from Arabidopsis thaliana and other representative plant species. A neighbor-joining (NJ) phylogenetic tree was constructed using MEGA 12 software with 1,000 bootstrap replicates. Subsequently, the resulting tree was visualized using iTOL (
https://itol.embl.de )[26].Chromosome distribution and collinearity analysis of the PeUGT gene family
-
The chromosomal locations of PeUGT genes were determined based on genome annotation files and visualized using MapChart[27]. Intraspecific collinearity analysis of the PeUGT gene family and interspecific synteny analysis between Passiflora edulis, tomato (Solanum lycopersicum)[28], and Arabidopsis thaliana were performed using MCScanX[29], with the results visualized using TBtools[30].
Gene structure and conserved motif analysis of the PeUGT gene family
-
Gene structure analysis of PeUGT genes was performed using the GSDS online tool (
https://gsds.gao-lab.org ) based on genome annotation data[31]. Additionally, conserved motifs in PeUGT protein sequences were predicted using MEME (Multiple Em for Motif Elicitation) with a maximum motif number of 10 and default parameters for all other settings.Prediction of cis-elements in PeUGT gene family promoters
-
For each PeUGT gene, the 2,000 bp sequence upstream of the translation start codon was extracted from the Passiflora edulis genome as the putative promoter region. These sequences were submitted to PlantCARE (
https://bioinformatics.psb.ugent.be/webtools/plantcare/html ) for cis-acting regulatory element identification[32]. The predicted elements were subsequently filtered, statistically analyzed, and visualized.GO annotation and KEGG enrichment analysis of PeUGT gene family
-
To perform Gene Ontology (GO) annotation and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis of the PeUGT gene family, functional annotation of all Passion fruit protein-coding genes was first conducted using eggNOG-mapper[33]. Subsequently, GO term mapping and KEGG pathway enrichment analysis specific to PeUGT genes were performed using TBtools[30], with the results visualized accordingly.
Analysis of expression levels during fruit development and maturation of the PeUGT gene family
-
Passion fruit peel samples were collected at four developmental stages: 18 d after flowering (S1), 28 d after flowering (S2), 41 d after flowering (S3), and 58 d after flowering (S4). S1 (18 d after flowering, the young fruit stage) is mainly characterized by cell division; S2 (28 d after flowering, the fruit expansion stage) features a significant accumulation of water and sugar; S3 (41 d after flowering, the coloring stage) is marked by the degradation of chlorophyll, the synthesis of anthocyanins and carotenoids, and a substantial accumulation of organic acids (primarily citric acid); S4 (58 d after flowering, the mature stage) sees the content of soluble sugar reach its peak and a large release of volatile aroma substances. All samples were immediately frozen in liquid nitrogen and stored at −80 °C prior to RNA extraction. Total RNA isolation and transcriptome sequencing were performed by Haikou Heliyuan Biotechnology Co., Ltd. (Haikou, China). Three biological replicates were collected for each developmental stage.
Construction of the transcriptional regulatory network of the PeUGT gene
-
To construct the putative transcriptional regulatory network, the FIMO (Find Individual Motif Occurrences) program was first used to scan the promoter sequences of candidate genes to identify potential transcription factors that could bind to the promoter region of the PeUGT gene[34]. Subsequently, the prediction results were integrated with the weighted gene co-expression network analysis (WGCNA) based on the transcriptome data of four ripening stages of passion fruit using TBtools[30]. Finally, the transcription factors that could bind to the promoter region of the PeUGT gene and were in the same co-expression module as the PeUGT gene in the WGCNA were screened as candidate regulatory factors.
-
BLASTP alignment against AtUGT sequences combined with HMM domain searches identified 149 PeUGT-encoding genes in the passion fruit genome. These genes were designated PeUGT1 to PeUGT149 based on chromosomal order. The encoded proteins exhibited substantial physicochemical diversity, with lengths ranging from 177 amino acids (PeUGT122) to 931 amino acids (PeUGT56), molecular masses of 20.2 kDa (PeUGT122) to 105.3 kDa (PeUGT56), theoretical isoelectric points (pI) spanning 4.71 (PeUGT98) to 8.93 (PeUGT42), instability indices of 27.76 (PeUGT22) to 54.20 (PeUGT16), aliphatic indices of 72.15 (PeUGT122) to 114.89 (PeUGT35), and grand averages of hydropathicity (GRAVY) from −0.442 (PeUGT122) to 0.352 (PeUGT35) (Supplementary Table S1), indicating varying degrees of hydrophilicity. These parameters collectively highlight significant hydrophilicity variations across the 149 UGT proteins in passion fruit.
PeUGT phylogenetic analysis
-
To elucidate evolutionary relationships among PeUGT family members, full-length protein sequences from 73 Arabidopsis thaliana AtUGTs and 149 passion fruit PeUGTs were analyzed using BLASTP and multiple sequence alignment tools. Neighbor-Joining phylogenetic reconstruction generated a robust tree (Fig. 1) demonstrating that PeUGT members clustered into 11 distinct subfamilies, exhibiting significant size heterogeneity. Comparative analysis revealed that intra-subfamily sequences shared high pairwise identity between Arabidopsis and passion fruit, suggesting strong purifying selection and functional conservation. Notably, subfamilies IV, X, and XI contain only passion fruit UGT proteins. Subfamily V has also undergone significant expansion in passion fruit. These findings establish that while core UGT functions are evolutionarily conserved, subfamily-specific diversification reflects adaptation to distinct ecological niches.
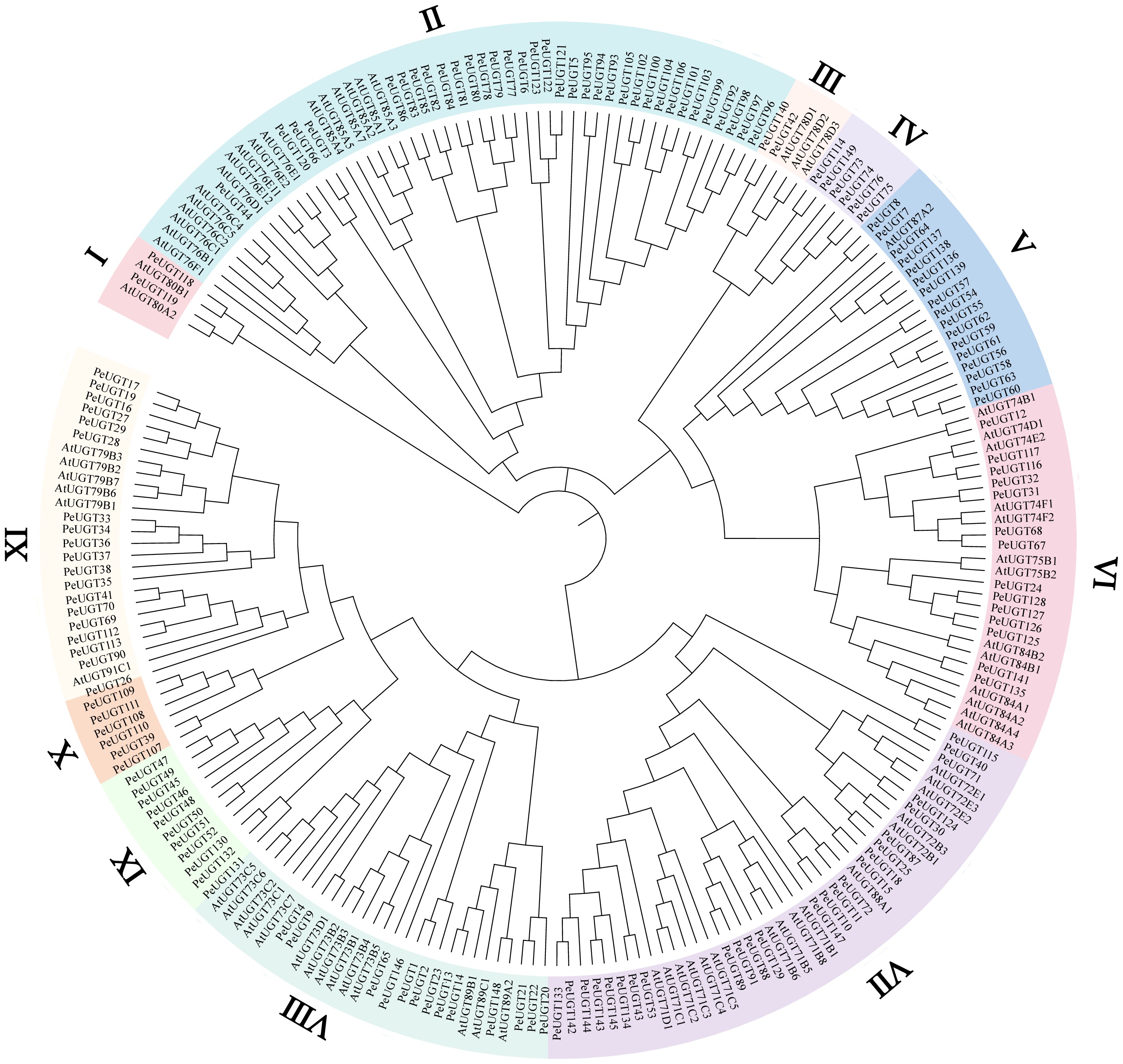
Figure 1.
Phylogenetic analysis of UGT proteins in Arabidopsis and passion fruit. Using the neighbor-joining (NJ) method implemented in MEGA12 software, a phylogenetic tree was constructed using 73 AtUGT proteins from Arabidopsis and 149 PeUGT proteins from passion fruit. Different subfamilies are represented by different colors.
Chromosome localization and collinearity analysis of the PeUGT gene
-
To elucidate the genomic distribution of PeUGT genes, chromosomal mapping was performed. Among the 149 identified PeUGT genes, 147 were mapped to nine passion fruit chromosomes, while two remained on unassembled scaffolds (Fig. 2). Chromosomal density analysis revealed significant variation, with chromosome 1 harboring the highest gene density (39 PeUGTs), followed by chromosomes 2 (26), 3 (21), and 4 (20). Subsequent chromosomes exhibited progressively lower densities, ranging from one to 16 genes per chromosome (chromosomes 5–9).
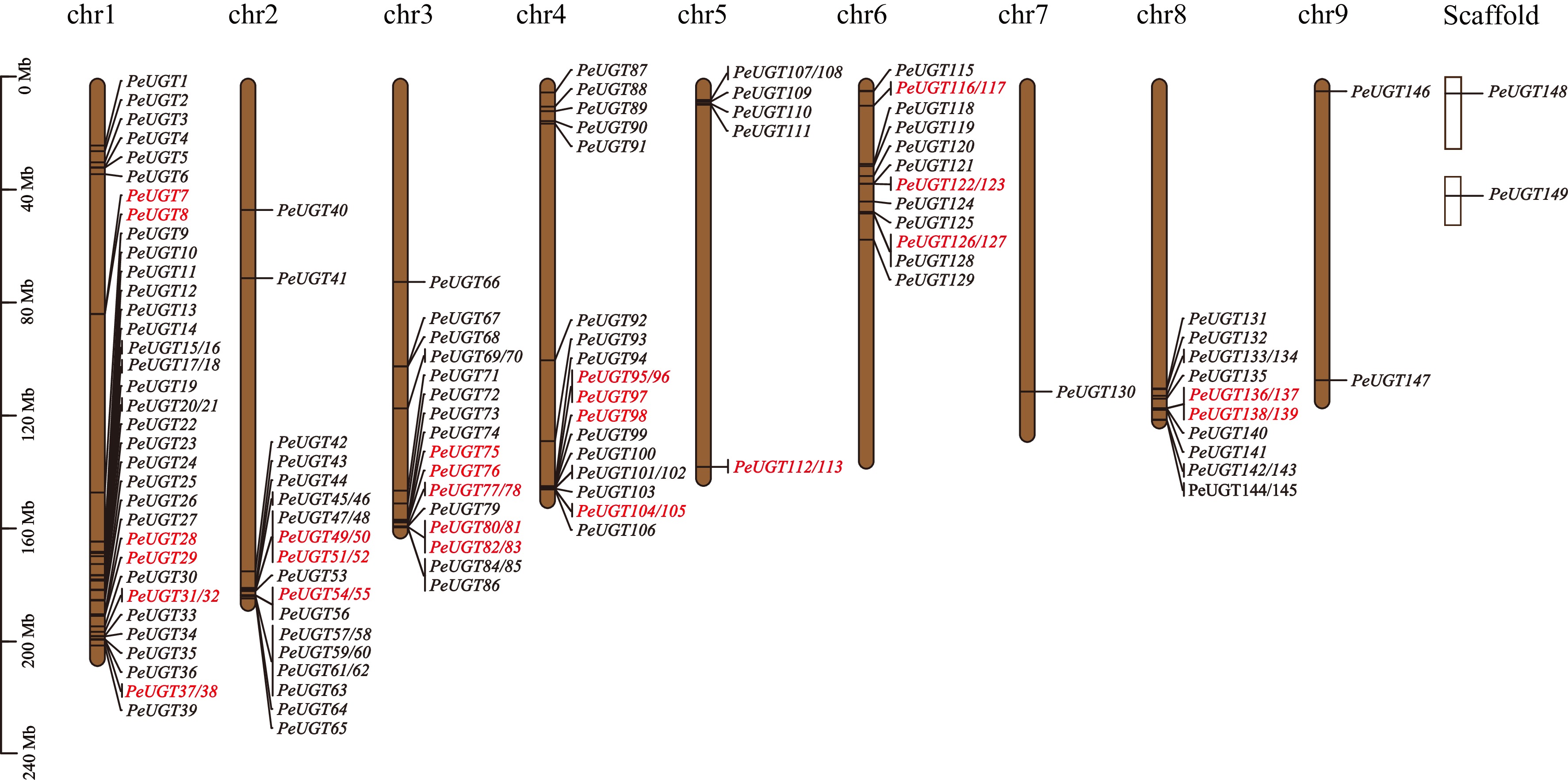
Figure 2.
Chromosome distribution and tandem repeat analysis of the PeUGT gene. Each line represents a chromosome, with duplicated gene pairs highlighted in red. The approximate location of each PeUGT gene on each chromosome is indicated.
Syntenic analysis identified 20 tandem and 11 segmental duplication events within the PeUGT family, suggesting both intrachromosomal and interchromosomal expansion mechanisms (Fig. 3a). Comparative genomics revealed 21 PeUGT genes with collinear relationships to AtUGT genes and 26 with Solanum lycopersicum SlUGTs (Fig. 3b). Notably, 12 PeUGT genes demonstrated dual synteny with both AtUGT and SlUGT orthologs, indicating functional conservation across evolutionarily divergent species.
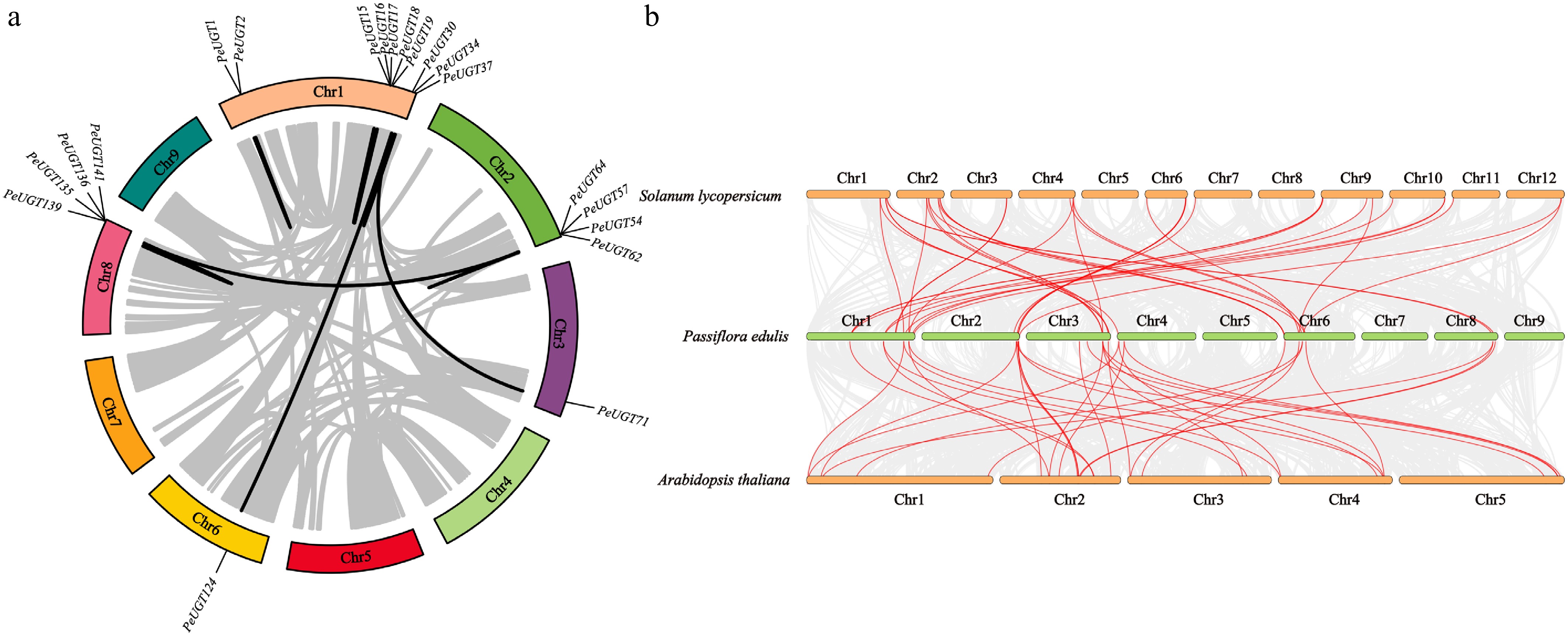
Figure 3.
Analysis of intraspecific and interspecific collinearity of UGT genes. (a) Intraspecific collinearity analysis of the PeUGT gene in passion fruit. (b) Collinearity analysis of UGT genes in passion fruit, Arabidopsis thaliana, and tomato.
Conservative motif and gene structure analysis of the PeUGT gene
-
MEME analysis identified ten conserved motifs across the PeUGT proteins, exhibiting high conservation among members. Motif 7 was exclusively present in subfamily II, while a core set of motifs (4, 9, 10, 2, 8, 1, 3, and 5) was distributed across nearly all PeUGT members. Notably, motifs 1 and 3 corresponded to the canonical PSPG-box signature. Members containing PSPG-box exhibit strict conservation in the C-terminus region, but show significant differences in the N-terminus region, consistent with observations from other plant species such as apples[11] and blueberries[35] (Fig. 4). In addition, other typical domains—including GTB (motifs 2, 5, and 9), YjiC (motif 4), and GT1 (motifs 3, 6, and 8)—were identified across all PeUGT members, despite each being annotated in only one member. These differential domain distributions likely contribute to functional diversification within the PeUGT family.
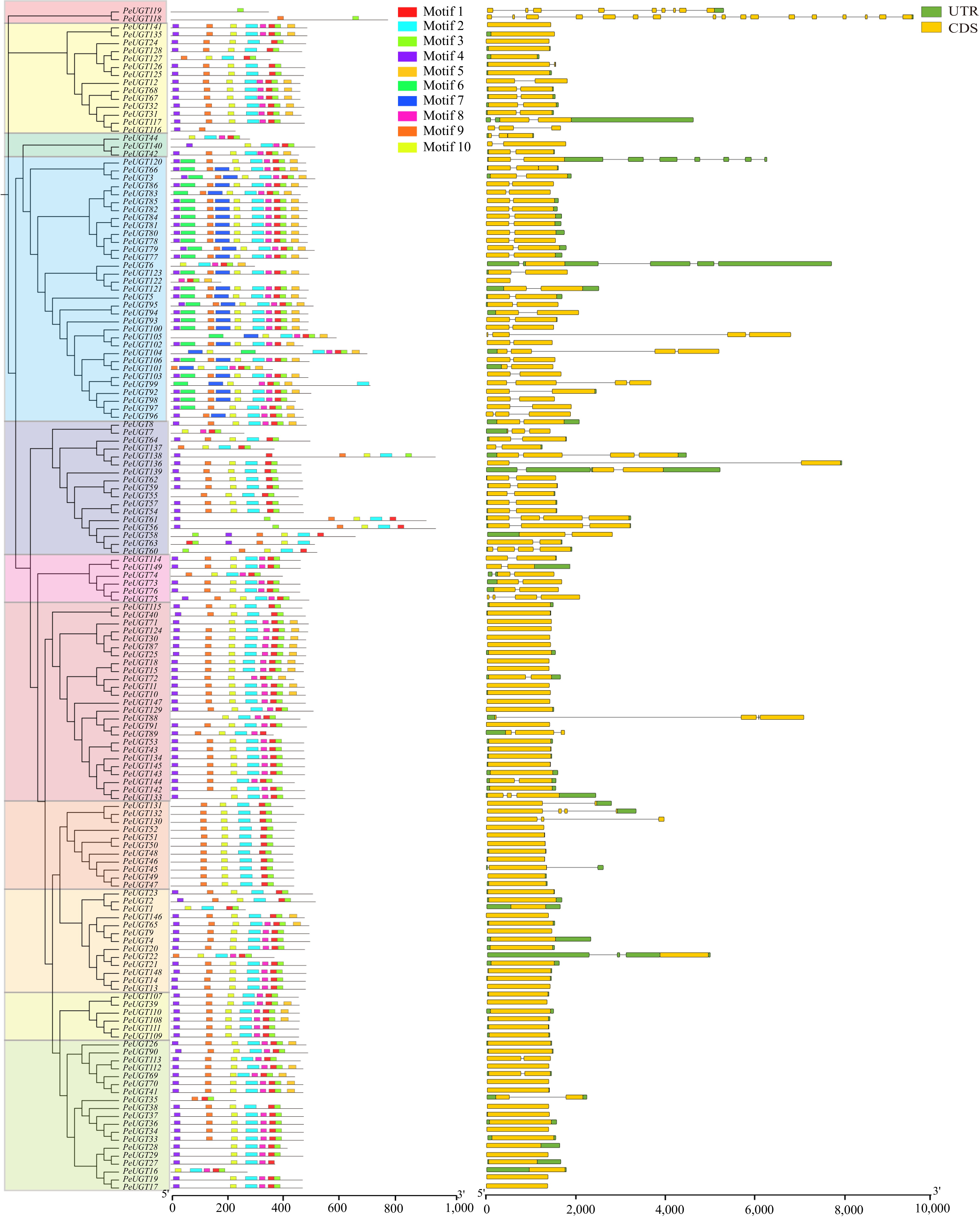
Figure 4.
Analysis of PeUGT conserved motifs and gene structure. PeUGT conservative motifs are sorted according to different subfamilies, with different colors representing different subfamilies in the evolutionary tree. There are a total of ten motifs, each represented by a colored box, and non-conservative regions represented by black lines. In the PeUGT gene structure analysis, CDS is represented by yellow boxes, UTR is represented by green boxes, and introns are represented by black lines.
Investigation of intron-exon architectures revealed PeUGT genes ranged from 1 to 17 exons (median: nine exons) and 0 to 16 introns (median: five introns). Subfamily I exhibited the highest structural complexity (9–17 exons, 8–16 introns), whereas subfamilies II–V predominantly contained three or more introns, whereas the remaining subfamilies displayed less intron content, potentially attributable to shorter sequence lengths or evolutionary loss of intron-rich domains (Fig. 4).
Analysis of cis acting-elements of the PeUGT gene
-
Analysis of the 2 kb upstream promoter regions of PeUGT genes identified 4,271 predicted cis-regulatory elements, categorized into three functional classes: phytohormone responsiveness, plant growth and development, and stress response. Stress-responsive elements demonstrated significant enrichment, including 325 anaerobic induction elements, 64 defense and stress-responsive elements, 163 drought-inducible elements, 339 light-responsive elements, and 165 low-temperature-responsive elements. This pronounced bias toward stress-related motifs aligns with the proposed role of UGTs in abiotic stress mitigation, suggesting their evolutionary adaptation to environmental perturbations.
A total of 1,793 cis-regulatory elements associated with six phytohormone response pathways were identified, including 726 abscisic acid-responsive elements, 130 auxin-responsive elements, 102 gibberellin-responsive elements, 515 methyl jasmonate-responsive elements, and 156 salicylic acid-responsive elements. This substantial enrichment of hormone-responsive elements indicates that phytohormones play pivotal roles in regulating PeUGT gene expression (Fig. 5).
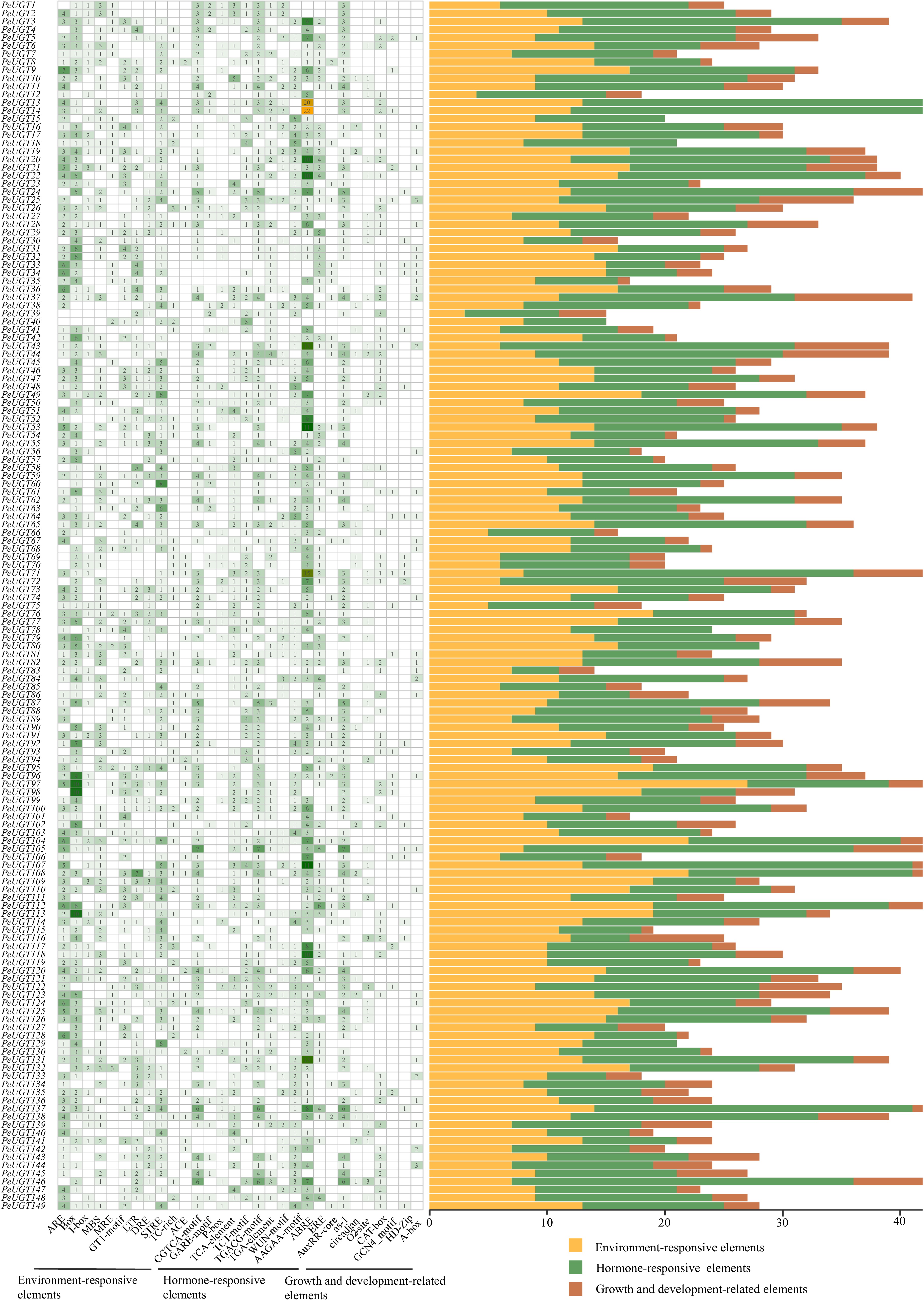
Figure 5.
Identification of cis-regulatory elements in the PeUGT gene promoter. The left figure shows the number of each type of cis-regulatory element present in each gene promoter region, while the right figure shows the number of elements belonging to plant hormone response, plant growth and development, and stress responsiveness within each genome promoter.
Additionally, the PeUGT promoter regions contained 532 cis-acting elements associated with growth and developmental processes, including 30 circadian rhythm control elements, 25 endosperm expression elements, and 56 zein metabolism regulatory elements. The diversity of these regulatory elements suggests that PeUGT genes participate in a broad spectrum of physiological processes during passion fruit development.
PeUGT gene GO and KEGG enrichment analysis
-
To elucidate the functional roles of PeUGT genes in passion fruit developmental biology, Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses were performed. PeUGT proteins demonstrated predominantly catalytic activity, with primary functional annotations in metabolic processes, biological regulation, and developmental processes. The most significantly enriched biological process categories included cellular metabolite biosynthesis, anthocyanin-containing compound metabolism, and secondary metabolic pathways. Notably, the majority of PeUGT members were annotated to specific metabolic pathways, including biosynthesis of secondary metabolites, and terpenoid and polyketide metabolism (Fig. 6). These findings suggest that PeUGT genes likely participate in biotic and abiotic stress responses, as well as plant growth and development, through the regulation of secondary metabolite glycosylation.
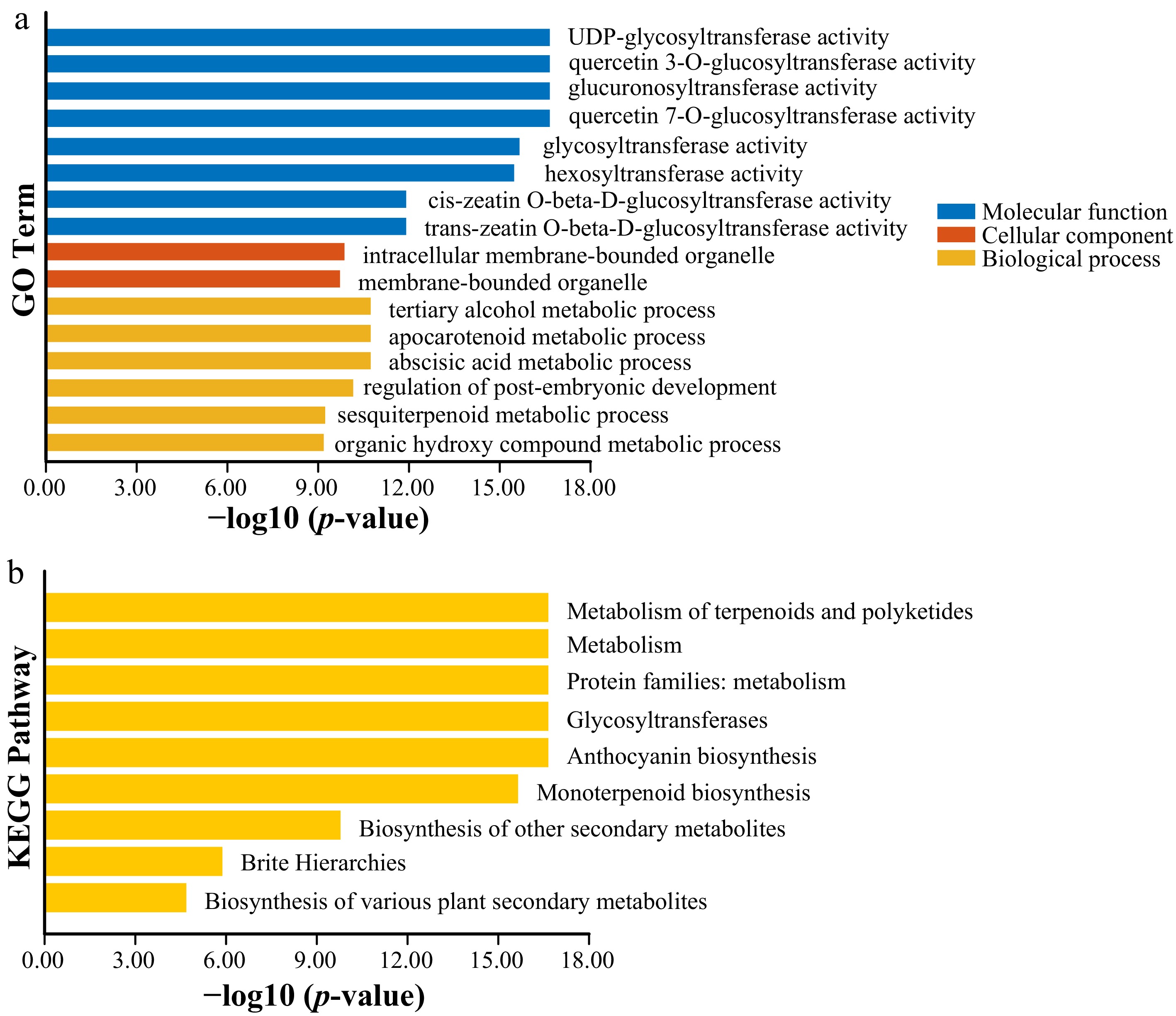
Figure 6.
GO and KEGG enrichment analysis of the PeUGT gene. (a) GO analysis of the PeUGT gene. GO classification of cellular components, molecular functions, and biological processes has been determined. (b) Annotation of the KEGG pathway of the PeUGT gene.
Analysis of expression patterns of PeUGT gene during developmental stages
-
RNA sequencing (RNA-seq) analysis was performed to elucidate the spatiotemporal expression dynamics of PeUGT genes in passion fruit peel across four post-flowering developmental stages: S1−S4. Quantitative transcriptomic profiling revealed distinct temporal expression patterns, with 36 PeUGTs exhibiting peak expression at S1, 47 at S2, 27 at S3, and 24 at S4. Notably, the highest aggregate expression level occurred during S2, whereas the lowest was observed at S4, suggesting a developmental phase-dependent regulatory mechanism (Fig. 7).
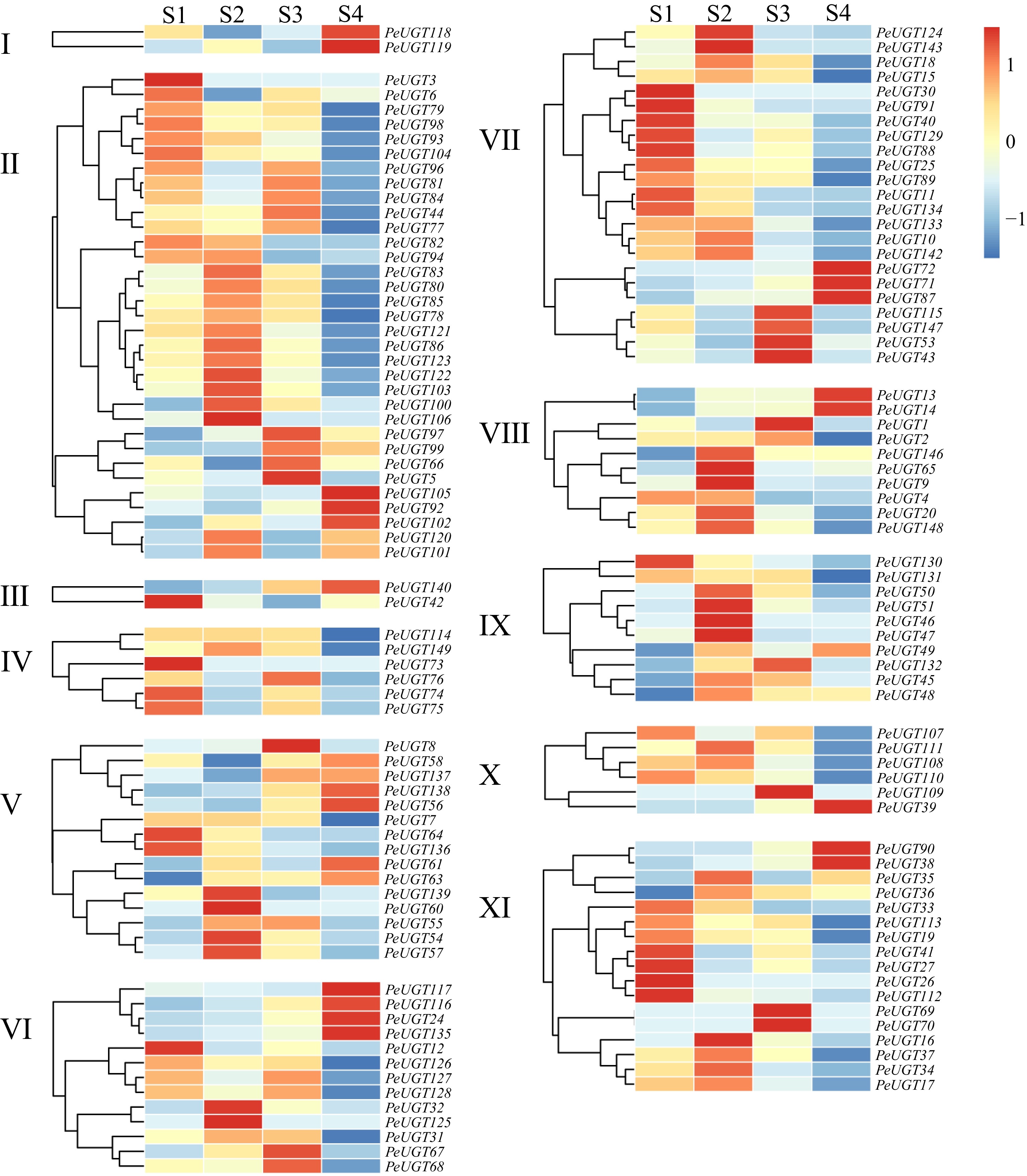
Figure 7.
Analysis of PeUGT gene expression pattern during the developmental stage of passion fruit. Different subfamilies of PeUGT are clustered separately, with S1 representing 18 d after flowering, S2 representing 28 d after flowering, S3 representing 41 d after flowering, and S4 representing 58 d after flowering. Gene expression levels are represented as TPM (Transcripts Per Million) values and subjected to Z-score normalization (row-wise scaling). The color gradient indicates the deviation from the mean expression level: red denotes higher-than-average expression (Z-score > 0), while blue denotes lower-than-average expression (Z-score < 0).
Members of the tandemly duplicated gene family exhibit highly coordinated expression patterns across four developmental stages. Specifically, PeUGT75/76 shows significantly high expression in stages S1 and S3, but is significantly downregulated in stages S2 and S4; PeUGT122/123 expression gradually increases with development, reaching a peak in stage S2 and then showing a decreasing trend. These expression profile characteristics suggest that tandemly duplicated genes may have functional redundancy and cooperatively regulate specific biological processes throughout the entire developmental cycle (Fig. 7).
Construction of transcriptional regulatory network for the PeUGT gene
-
Our integrated analysis of functional enrichment and developmental expression dynamics revealed that PeUGT genes associated with anthocyanin biosynthesis exhibited distinct stage-specific expression patterns. PeUGT10 and PeUGT17 displayed significant upregulation during early development stages, peaking at S2, followed by progressive downregulation. Conversely, PeUGT38 exhibited sustained upregulation across all developmental stages, achieving maximum at S4 (Fig. 7).
To elucidate the transcriptional regulatory architecture, we performed comprehensive co-expression network analysis coupled with FIMO-based motif enrichment. This identified 28, 2, and 20 candidate transcription factors (TFs) regulating PeUGT10, PeUGT17, and PeUGT38, respectively. The candidate TF families for PeUGT10 included ARF, bHLH, bZIP, C2H2, Dof, EIL, ERF, G2-like, GATA, HD-ZIP, HSF, MIKC_MADS, MYB, WOX, and WRKY; PeUGT17 candidate regulators comprised G2-like and HD-ZIP family members; while PeUGT38 candidate TFs encompassed ARF, BBR-BPC, bZIP, C2H2, Dof, G2-like, MIKC_MADS, MYB, NAC, and WRKY (Fig. 8). These findings establish key regulatory targets for investigating the transcriptional control of anthocyanin-related PeUGT genes during passion fruit ripening.
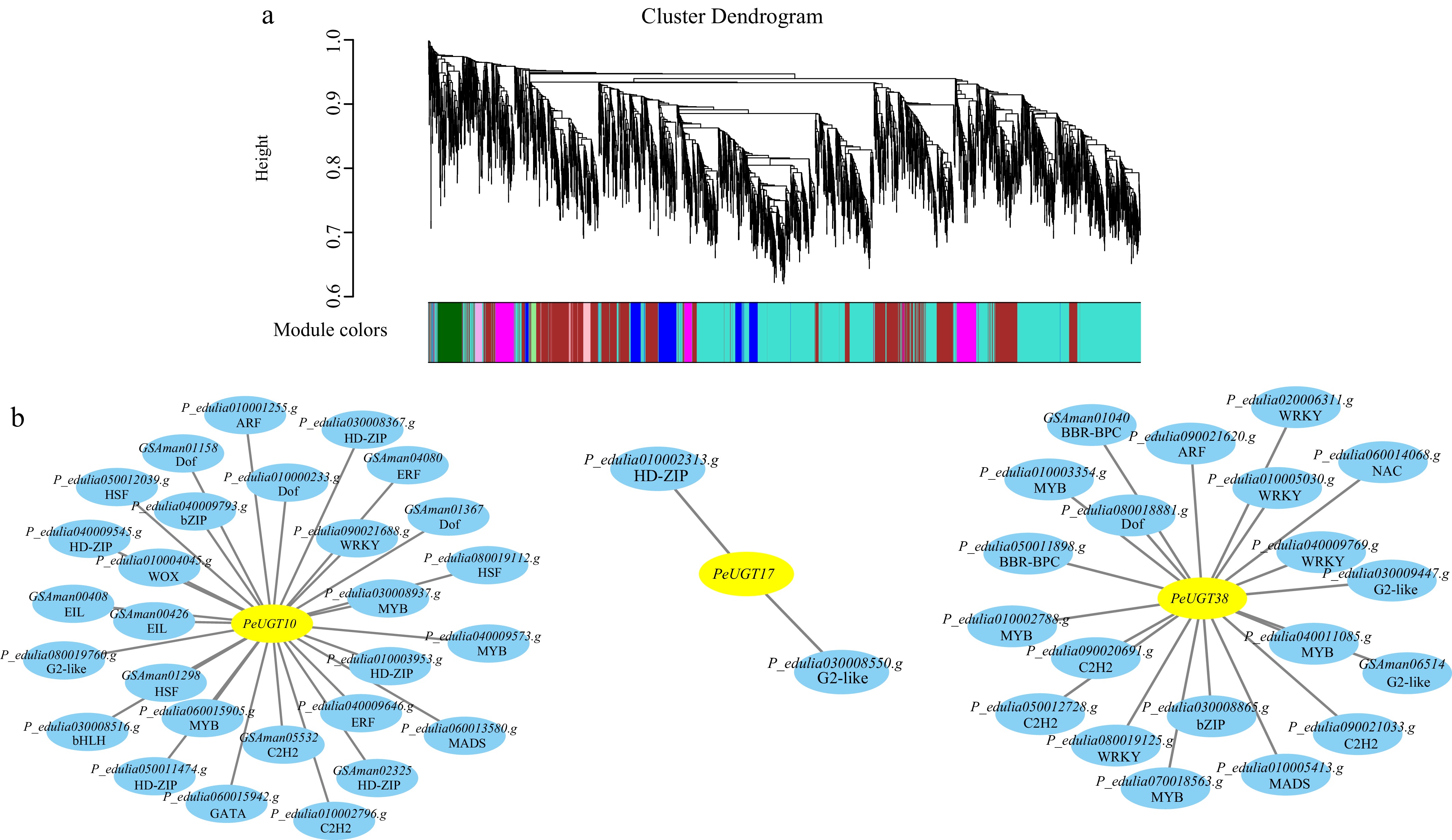
Figure 8.
Potential transcriptional regulatory networks of PeUGT10, PeUGT17, and PeUGT38. (a) Identify gene co-expression modules through WGCNA. (b) Identifying transcription factors with potential regulatory relationships with PeUGT10, PeUGT17, and PeUGT38 through integration of co-expression and FIMO analysis. The first row in the circle in the figure is the gene number, and the second row is the corresponding transcription factor family.
-
In this study, we conducted a comprehensive genome-wide identification and characterization of the UGT gene family in Passiflora edulis, including phylogenetic relationships, gene structure, conserved motifs, chromosomal distribution, duplication events, intraspecific and interspecific synteny, promoter cis-regulatory element analysis, developmental expression patterns, and functional enrichment analyses (GO and KEGG). A total of 149 PeUGT genes were identified and clustered into 11 subfamilies. Comparative analysis revealed that the PeUGT family size was comparable to that of other plant species, including Arabidopsis (120 members)[9], maize (147)[36], wheat (179)[6], citrus (145)[37], and upland cotton (174)[38].
Phylogenetic and structural analyses demonstrated substantial conservation within PeUGT subfamilies, with members of the same clade exhibiting identical or highly similar exon-intron architectures. This pattern is consistent with previous findings in tomato[28] and blueberry[35], underscoring the evolutionary conservation of UGT family organization across diverse plant taxa. Conserved motif analysis confirmed that motifs 1 and 3 constitute the characteristic PSPG-box signature present in all PeUGT members. Notably, genes containing the PSPG-box exhibited high conservation in the C-terminal region and significant variability in the N-terminal region, the latter being responsible for substrate specificity. These findings align with established evidence that the PSPG-box directly interacts with sugar donors, thereby modulating glycosylation activity and substrate specificity[39]. Analysis of the 2 kb upstream region of the PeUGT gene identified three categories of cis-regulatory elements associated with plant hormone responses, growth and development, and stress responses. Notably, hormone-responsive elements were found to be particularly critical. In rice, hormones such as strigolactones (SLs) can directly regulate the biosynthesis of flavonoid and terpenoid phytoalexins through the WRKY45 transcription factor, thereby coordinating the balance between plant growth and defense metabolism. This suggests that PeUGT may participate in flavonoid glycosylation modifications under hormone-mediated regulatory signaling[40].
GO and KEGG enrichment analyses revealed that ten PeUGT genes were associated with anthocyanin biosynthesis pathways, consistent with established roles of UGTs in flavonoid glycosylation across plant species. In Arabidopsis, UGT79B subfamily members (UGT79B1, UGT79B2, and UGT79B3) catalyze anthocyanin modifications, with distinct substrate specificities: UGT79B1 preferentially recognizes 3-O-glucosylated anthocyanins and flavonols, whereas UGT79B2 and UGT79B3 favor anthocyanidin aglycones over 3-O-glucosylated substrates. Their expression is directly regulated by CBF1[41]. The first plant UGT gene identified, maize (Zea mays) UDP-glucose: flavonoid 3-O-glucosyltransferase BRONZE1, is endosperm-specific and responsible for seed pigmentation through flavonoid 3-O-glycosylation[42]. Additionally, quercetin glucosyltransferase activity has been extensively characterized: Arabidopsis UGT78D subfamily members (UGT78D1, UGT78D2, and UGT78D3) glycosylate flavonoids with varying specificities - UGT78D2 catalyzes glucose transfer to cyanidin[43], kaempferol, and quercetin, while UGT78D1 and UGT78D3 specifically modify flavonol glycosides[44]. The second maize flavonol glycosyltransferase, UFGT2, exhibits high catalytic efficiency toward kaempferol and quercetin and enhances abiotic stress tolerance[45]. These findings suggest that passion fruit PeUGT genes enriched in anthocyanin-related pathways may fulfill analogous biochemical functions, providing a foundation for subsequent functional characterization.
Furthermore, we investigated developmental expression dynamics across fruit ripening stages. Fourteen PeUGT genes exhibited progressively increasing expression, including PeUGT13, 14, 24, 38, 39, 63, 71, 72, 87, 90, 116, 135, 138 and 140; conversely, 23 genes displayed declining expression patterns, comprising PeUGT3, 11, 19, 25, 30, 33, 40, 64, 73, 82, 89, 91, 93, 94, 104, 110, 112, 114, 130, 131, 133, 134, and 136. Notably, three anthocyanin-associated genes—PeUGT10, PeUGT17, and PeUGT38—exhibited contrasting trajectories (decreasing vs increasing), prompting transcriptional regulatory network analysis. This identified 28, 2, and 20 candidate transcription factors (TFs) potentially regulating PeUGT10, 17, and 38, respectively. PeUGT10 candidates encompassed ARF, bHLH, bZIP, C2H2, Dof, EIL, ERF, G2-like, GATA, HD-ZIP, HSF, MIKC_MADS, MYB, WOX, and WRKY families; PeUGT17 candidates comprised G2-like and HD-ZIP; while PeUGT38 candidates included ARF, BBR-BPC, bZIP, C2H2, Dof, G2-like, MIKC_MADS, MYB, NAC, and WRKY.
Strikingly, all three gene sets shared G2-like TFs, members of the GARP superfamily implicated in chloroplast development, chlorophyll biosynthesis, and abiotic stress responses. Arabidopsis GLK2 positively regulates high-light-induced anthocyanin accumulation, establishing a molecular link between light signaling and anthocyanin biosynthesis[46]. In cotton, GhBLH2 antagonizes GhGLK1-mediated salt tolerance by interacting with its activation domain[47]. Metabolites may also vary among different varieties[48], In passion fruit, differential FLS enzyme activities between YPF-p and PPF-p may alter the competitive dynamics with F3'H, DFR, and F3'5'H, consequently leading to distinct anthocyanin synthesis patterns between the two varieties[49]. We hypothesize that passion fruit G2-like TFs may directly activate anthocyanin glycosyltransferase genes (PeUGT10, PeUGT17, and PeUGT38) to promote pigment synthesis and stability, or alternatively, interact with environmental signaling pathways (light, temperature) to coordinate fruit development.
-
In this study, we identified 149 PeUGT genes in Passiflora edulis and systematically characterized them through integrated phylogenetic, structural, chromosomal, syntenic, and functional analyses. Three candidate genes, PeUGT10, PeUGT17, and PeUGT38, were prioritized as potential regulators of anthocyanin biosynthesis during fruit development based on their phylogenetic affiliations, conserved domain architectures, chromosomal locations, duplication histories, promoter cis-regulatory element profiles, developmental expression patterns, and pathway enrichment signatures. Co-expression analysis and candidate transcription factor screening revealed distinct yet overlapping regulatory networks for these genes, with G2-like TFs emerging as shared hub regulators. This comprehensive characterization of the PeUGT family establishes a transcriptional regulatory framework for key anthocyanin glycosylation genes, implicating G2-like transcription factors as central nodes integrating developmental and environmental signals. These findings advance the mechanistic understanding of anthocyanin biosynthesis in passion fruit and provide candidate gene targets and regulatory hubs for marker-assisted breeding and genome editing-directed improvement of fruit color traits.
-
During the preparation of this work, the author used Stork (version: 2026, date of use: March 12, 2026) to enhance the images of the graphical abstract. The author reviewed and edited all content generated with the assistance of these tools, verified its accuracy, and takes full responsibility for the integrity and originality of the final manuscript. This work represents the author's own intellectual contribution, and no artificial intelligence tool is regarded as an author.
-
The authors confirm contribution to the paper as follows: study conception and design: Fang C; data collection: Wang L, Peng J, Luo L; analysis and interpretation of results: Wang L, Luo L; draft manuscript preparation: Fang C, Wang L, Luo L. All authors reviewed the results and approved the final version of the manuscript.
-
Data will be made available on request. RNA sequence data that support the findings of this study have been deposited under SRA Bio-Project accession number CRA038695.
-
The authors are grateful to the Passion Fruit Science and Technology Institute in Baisha for providing the plant materials used in this study. This research was supported by the Key R & D Project of Baoting Li and Miao Autonomous County (Grant No. BTZDYF2025001) and the Project of Sanya Yazhou Bay Science and Technology City (Grant No. SKJC-JYRC-2024-29).
-
The authors declare that they have no conflict of interest.
-
accompanies this paper online at: https://doi.org/10.48130/tp-0026-0009.
-
Received 9 February 2026; Accepted 13 March 2026; Published online 27 March 2026
- Supplementary Table S1 Physicochemical properties of PeUGT transporter in passion fruit.
- Copyright: © 2026 by the author(s). Published by Maximum Academic Press on behalf of Hainan University. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Wang L, Luo L, Peng J, Fang C. 2026. Genome-wide identification and characterization of the UDP-glycosyltransferase gene family in passion fruit (Passiflora edulis). Tropical Plants 5: e010 doi: 10.48130/tp-0026-0009 

Genome-wide identification and characterization of the UDP-glycosyltransferase gene family in passion fruit (Passiflora edulis)
- Received: 09 February 2026
- Revised: 10 March 2026
- Accepted: 13 March 2026
- Published online: 27 March 2026
Abstract: Glycosylation, catalyzed by UDP-glycosyltransferases (UGTs), is a pivotal modification enhancing the solubility, stability, and diversity of plant metabolites, influencing growth, development, and stress responses. Passion fruit (Passiflora edulis) is an economically important fruit crop renowned for its rich flavor and health-promoting metabolites, particularly flavonoids. However, a comprehensive understanding of the UGT gene family underlying its metabolic diversity remains limited. In this study, we performed a genome-wide analysis and identified 149 PeUGT genes in the passion fruit genome. Phylogenetic analysis clustered these genes into 11 distinct subfamilies, revealing high evolutionary conservation. Structural analyses demonstrated conserved exon-intron architectures and motifs within subfamilies, with the PSPG-box signature present in all members. Chromosomal mapping revealed an uneven distribution across nine chromosomes, and duplication event analysis suggested that tandem and segmental duplications were the primary drivers of family expansion. Promoter cis-element analysis identified an abundance of motifs related to stress, hormone signaling, and development. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment highlighted associations with flavonoid biosynthesis, particularly anthocyanin metabolism. Expression profiling across four fruit developmental stages revealed dynamic patterns, with 14 genes up-regulated and 23 down-regulated during ripening. Notably, three key anthocyanin-related genes—PeUGT10, PeUGT17, and PeUGT38—showed contrasting expression trajectories. Subsequent co-expression network analysis identified potential transcription factors (e.g., G2-like, MYB, WRKY) regulating these genes. Our findings provide a foundational resource for the functional characterization of PeUGT-mediated glycosylation in passion fruit and offer prime candidate targets for molecular breeding aimed at improving fruit quality traits, such as color and nutritional value.
-
Key words:
- Passion fruit /
- UGT gene /
- Fruit development /
- Anthocyanins