









-
Climate change-associated acute events such as drought, flood and extreme temperatures, pose a threat to the sustainability of global agriculture systems[1, 2]. Perennial crops growers face great challenges as adaptive management approaches such as the adoption of new varieties or the relocation of habitats are economically unfriendly or physically impossible due to the long lifespans and high establishment cost of these crops[3]. For this reason, weather-associated adversity is a primary factor that constrains the expansion of perennial crop production, such as grapevine[4, 5]. Heat waves in summer significantly impact fruit and wine quality due to disruption of the biosynthesis for secondary metabolites such as phenolic and aromatic compounds[6, 7]. Cold damage can be far more disastrous since late frost in spring and extreme low temperatures in winter can kill young shoots and threaten the survival of the whole vine, respectively[8, 9].
Grapevines, like other perennial plants, overwinter and resume growth through a dormancy cycle along with gaining and losing of cold hardiness[10]. The dormancy cycle is described as two phases of change between three types of dormancy: a transition from paradormancy (apical dominance induced growth inhibition) to endodormancy (endogenous unknown molecular lock induced growth inhibition), and a transition from endodormancy to ecodormancy (environmental factor induced growth inhibition)[11]. The transition from paradormancy to endodormancy usually occurs in late summer along with cold acclimation. Cold acclimation is triggered by short-day photoperiod and progressively induced by non-freezing and freezing temperatures[12, 13]. During cold acclimation, plants initiate a flush of metabolite activities, which results in the altering of photosynthesis efficiency in leaves[14], the modification of plasma membrane composition[15], accumulation of functional metabolites in buds[16] and enhancement of cold hardiness of all overwintering tissues[17]. As a component of establishing cold hardiness, grapevine buds progressively isolate from the cane vascular tissue[18, 19]. Previous multi-omics analysis revealed that the sugar biosynthesis, including monosaccharides, disaccharides and raffinose family oligosaccharides (RFOs) were upregulated at both transcriptomic and metabolomic level during cold acclimation, indicating the importance of sugars in grapevine cold hardiness[20]. However, comprehensive examination of the process of cold acclimation in dormant grapevine tissues remains understudied.
The transition from endodormancy to ecodormancy occurs in correlation with the accumulation of chilling hours[21]. For grapevine, many different chilling hour models can be used, but most assume temperatures between 7.2 °C to 0 °C effectively contribute to chilling fulfillment[22]. However, the quantity of chilling requirement needed for budbreak exhibits enormous variation across Vitis species[23]. The loss of cold hardiness when warm temperatures occur, termed deacclimation, is enhanced once buds have reached ecodormancy[24, 25]. RNA-seq analysis of the deacclimation process during ecodormancy revealed dozens of biological pathways were differently regulated in four grapevine genotypes[26]. These pathways included critical regulatory pathways (e.g. plant hormone metabolism, hormone signaling and transcription factors), metabolism pathways (e.g. fatty acid metabolism, galactose metabolism, starch and sucrose metabolism and phenylpropanoid metabolism) and basal pathways (e.g. cell cycle, circadian rhythm and photosynthesis).
Overall, the dormancy cycle is a complex mechanism that merges the onsets or offsets of numerous biological pathways, cascades, and networks. However, abscisic acid (ABA), a plant phytohormone, was noted for its concomitant rhythm with cold acclimation, chilling accumulation and deacclimation[12]. ABA serves as a critical regulator in response to abiotic stresses through the signaling mediated by ABA responsive genes and ABA responsive element binding proteins (AREBs)[27]. For grapevine, endogenous ABA content increases during acclimation and decreases during deacclimation, correlating with the gain and loss of bud cold hardiness[28, 29]. Foliar ABA application on grapevines in late fall (early cold acclimation) enhanced bud cold hardiness in winter[29, 30], and ABA application on ecodormant grapevine buds delayed budburst in spring[26, 28]. These findings not only emphasize an important role for ABA in grapevine cold hardiness, but also suggest that exogenous ABA application might serve as a tool for the mitigation of cold-related injury in grapevines. More precise investigations, such as exogenous ABA's impact on key pathways (e.g. sugar metabolism, protein processing or hormonal signaling) would help understand the underlying mechanism and in turn justify its utilization as a culture practice in vineyards.
ABA-mediated genetic mechanisms of grapevine cold acclimation or deacclimation have been suggested in recent studies. These include ABA's regulation on RFOs biosynthesis[31], synergy with low temperature on the expression of C-repeat (CRT)-binding factor/dehydration-responsive element (DRE) binding protein 1 (CBF/DREB1)[32], promotion of starch biosynthesis[33], repression of cell cycle genes[34], interaction with transcriptome factors[35] and impact on the biosynthesis of other plant phytohormones[36]. However, most of these studies investigated individual genes or pathways through targeted analytical tools such as qPCR, overlooking the importance of co-regulation networks or signaling cascades. In the era of bioinformatics, tools like RNA-seq coupled with specialized machine learning tools would not only eliminate the biases associated with targeted approaches, but also help generate new knowledge to identify unknown underlying mechanisms.
This two-part study leverages transcriptomic sequencing (RNA-seq), unsupervised bioinformatic tools, such as principal component analysis (PCA) and weighed gene co-expression network analysis (WGCNA) along with gene set enrichment analysis (GSEA) and 3D modeling to investigate the genetic functionality of ABA during grapevine cold acclimation and deacclimation. The first objective was to identify the functionality of ABA on gene co-expression networks during cold acclimation and deacclimation. The second objective was to evaluate the feasibility of ABA application as a tool for the mitigation of grapevine cold injury and identify transcriptomic targets for future mitigation studies.
-
Two experiments were conducted, (1) during grapevine acclimation (the acclimation experiment) and (2) deacclimation (the deacclimation experiment), to investigate the impact of ABA application on the grapevine transcriptome. Leaves and buds were collected periodically within hours or days after ABA application for RNA-seq in the acclimation and the deacclimation experiment, respectively. RNA-seq samples were collected as triplicates, which generated 40 libraries and 34 libraries in the acclimation experiment and the deacclimation experiment, respectively.
All libraries in both experiments were examined and passed quality assessment using FastQC to assess read quality (www.bioinformatics.babraham.ac.uk/projects/fastqc). In the acclimation experiment, the mean read length per sequence was 77 bp after trimming of barcode adaptors. Reads per library averaged at 3.8 million, and uniquely mapped rate per library after alignment in STAR[37] averaged at 89.2%. Gene count matrix was generated accordingly using all 42,413 genes in VCost.V3 annotation[38]. After low count filtering, a total of 17,056 genes remained in the gene count matrix, and these genes were examined with DESeq2[39]. Variance stabilization transformation (VST) count of genes was used for hierarchical clustering, which demonstrated that no sample significantly deviated from the main cluster, indicating no outlier presented in the dataset (Supplemental Fig. S1). In the deacclimation experiment, the mean read length per sequence was 66 bp after barcode adaptor trimming. The reads per library averaged at 2.9 million, and the uniquely mapped rate per library after alignment in STAR was 91.0%. After low count filtering, a total of 18,906 genes were left in the gene count matrix and used for downstream analysis. Hierarchical clustering of VST data did not identify any outliers in the dataset (Supplemental Fig. S2).
WGCNA and identification of differentially expressed genes (DEGs)
-
WGCNA[40] was implemented to detect gene co-expression modules in response to ABA application during the course of both experiments.
In the acclimation experiment, 37 distinct gene co-expression modules with a number of genes ranging from 40 (modules 'yellowgreen' and 'skyblue3') to 2,123 (module 'turquoise') were detected. The genes being independent from any co-expression modules (n = 2,663) were categorized into module 'grey' and excluded for any downstream analysis. PCA analysis of the remaining 14,393 genes explained 36.9% of variance among all samples in the first three components (Supplemental Fig. S3A & S3B). No obvious clustering or separation of samples could be identified in two-dimensional profiling with principal component 1 (PC1) and PC2 (Supplemental Fig. S3A). The treatment effect was partially reflected with PC2 and PC3. Although ABA samples at 2 h post-treatment were not clearly separated from control samples, ABA samples at 4 h, 24 h and 48 h post-treatment clustered towards the lower right section (positive PC2 and negative PC3) of the coordinate system, while control samples clustered towards the upper left section (negative PC2 and positive PC3) of the system (Supplemental Fig. S3B).
Module eigengenes (ME, PC1 of all genes in the module) of 37 modules were correlated with ABA treatment and time post-treatment and visualized across all timepoints in the experiment (Supplemental Fig. S4). Numerous MEs (e.g. 'tan' and 'green') showed very significant correlations with time post-treatment, and other MEs (e.g. 'red' and 'royalblue') showed very significant correlations with ABA treatment (Supplemental Fig. S4A). Based on MEs visualization, most MEs remained unchanged in response to ABA at 2 h post-treatment, indicating that exogenous ABA's impact on grapevine transcriptome might not be as significant within such small window (Supplemental Fig. S4B). Numerous MEs showed significant responses to ABA treatment from 4 h to 48 h post-treatment (Supplemental Fig. S4B), indicating a weak grouping of ABA-impacted genes in WGCNA. Thus, no target module was specifically selected for the experiment. Instead, a contrast of 'all ABA samples after 2 h post-treatment (n = 12) vs. all control samples after 2 h post-treatment (n = 12)' was applied to all 14,393 genes. Eventually, 273 and 61 genes were found significantly upregulated and downregulated, respectively, by ABA treatment after filtering using p-adj (FDR) < 0.05 and log2 fold change (LFC) > |1| of the contrast. These 334 genes were identified as target DEGs for any downstream analysis in the acclimation experiment.
In the deacclimation experiment, 17 distinct gene co-expression modules with a number of genes ranging from 66 (modules 'grey60') to 4,357 (module 'turquoise') were detected. As above, 2,091 genes were categorized into module 'grey' and were excluded for any downstream analysis. PCA analysis of the remaining 16,815 genes explained 55.2% of variance among all samples in the first three components (Supplemental Fig. S3C & S3D). ABA samples and control samples were clearly separated between '0' on PC1 (Supplemental Fig. S3C). Time post-treatment effect was revealed by an obvious trend in PC2 and PC3: earlier samples located at the lower right section, and later samples located at the upper left section (Supplemental Fig. S3D).
Module eigengenes (ME) of 17 modules were correlated with ABA treatment and time post-treatments and visualized across all timepoints in the experiment (Supplemental Fig. S5). Numerous MEs (e.g. 'grey60' and 'blue') showed very significant correlations with time post-treatment, and other MEs (e.g. 'turquoise', 'green' and 'yellow) showed very significant correlations with ABA treatment (Supplemental Fig. S5A). It was also noted that the p-values of the correlations for these three MEs and ABA treatment were much lower than that of the others (Supplemental Fig. S5A). Thus, modules 'turquoise', 'green' and 'yellow' were identified as target modules for this experiment. Based on MEs visualization, the MEs of target modules showed significant and consistent response to ABA treatment, indicating that exogenous ABA's impact on the genes in these modules might be significant during the entire experiment (Supplemental Fig. S5B). A contrast of 'all ABA samples (n = 15) vs. all control samples (n = 15)' was conducted to the genes in target modules. After filtering using FDR < 0.05 and LFC > |1| of the contrast, 1,814 and 523 genes were found significantly upregulated and downregulated, respectively, by ABA treatment. These 2,337 genes were identified as target DEGs for any downstream analysis in this experiment.
GSEA and identification of enriched pathways
-
To clarify functional roles of genes, the target DEGs (V3 annotation) identified from WGCNA modules were matched with their corresponding CRIBI V1 annotations (http://genomes.cribi.unipd.it/grape) for better functional annotations to facilitate GSEA. V3 genes without matching V1 annotations (15 genes in the acclimation experiment and 83 genes in the deacclimation experiment) were excluded for downstream analysis. Pre-ranked GSEA was separately conducted using target DEGs in the acclimation experiment, target DEGs in the deacclimation experiment and shared target DEGs in both experiments. Within each DEGs list, GSEA was further separately conducted according to response category (all, downregulated by ABA or upregulated by ABA). For the GSEA of target DEGs in the acclimation and the deacclimation experiments, the ranking criterium was the FDR of the specific contrast created for the experiment per se. For the GSEA of shared target DEGs, the ranking criterium was the mean FDR of two contrasts for two experiments. Only the pathways with NOM p-value < 0.1 are shown in GSEA summary (Fig. 1).
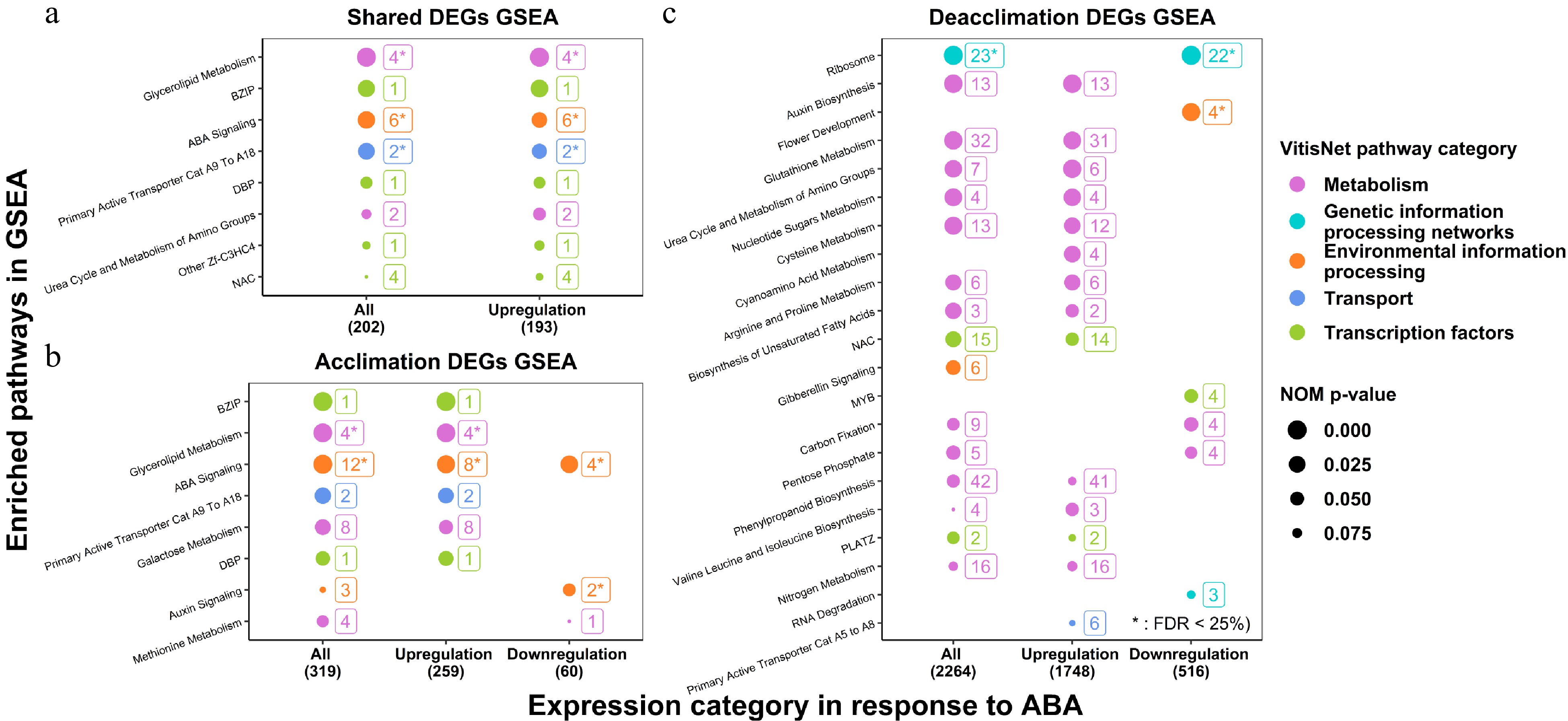
Figure 1. GSEA of ABA-induced DEGs in the acclimation and the deacclimation experiments. (a) GSEA of shared DEGs in two experiments; (b) GSEA of DEGs in the acclimation experiment; (c) GSEA of DEGs in the deacclimation experiment. Only the pathways with NOM p-value < 0.1 are shown.
GSEA analysis demonstrated enriched pathways for shared (8) (Fig. 1a), acclimation (8) (Fig. 1b) and deacclimation (21) (Fig. 1c). These pathways varied by experiment but included specific pathways of all five major categories of gene function: Transcription factors, Metabolism, Environmental Information Processing, Genetic Information Processing, and Transport. Specific subcategory pathway designations are shown in Fig. 1, but it should be noted that pathways related to ABA signaling were enriched in shared target DEGs, additional ABA signaling genes were enriched in the acclimation experiment target DEGs, and many more genes related to Metabolism and Genetic Information Processing were observed in the deacclimation experiment target DEGs. The details of these specific pathways are discussed below.
Critical pathways in detail
-
Several critical pathways were examined in detail for their significance in the analysis (FDR of GSEA < 25% and NOW p-value of GSEA < 0.05 as suggested in by GSEA guideline[41]) or their known functionality in grapevine acclimation and deacclimation. Metabolite acronyms are defined in the figure captions.
The ABA signaling pathway and ABA biosynthesis reactions were defined in detail in Fig. 2 as this pathway was significantly enriched in the GSEA of shared target DEGs and target DEGs in the acclimation experiment (Fig. 1a & b). Briefly, VDE functions as a negative regulator in ABA biosynthesis by catalyzing the transition of violaxanthin (ABA precursor) to zeaxanthin, and NCED serves as a positive regulator by catalyzing violaxanthin to xanthoxin. In ABA signaling, PYL/RCARs bind to PP2C upon the reception of ABA, which forms a ternary complex (PYL-ABA-PP2C). PP2C is deactivated in the ternary complex, facilitating the autophosphorylation of SnRK2 members which in turn phosphorylates transcription factors such as AREB/ABFs. In the nucleus, activated AREB/ABFs stimulate the expression of ABA responsive genes by binding at upstream promoter or enhancer regions. In ABA biosynthesis, two genes encoding NCED were upregulated by ABA in both experiments, and one gene encoding VDE was downregulated in the deacclimation experiment (Fig. 2). In ABA signaling, two genes encoding RCAR10, a member of PYL/RCAR family, were downregulated in deacclimation (Fig. 2). Three genes encoding ABI1, AHG3 and DBPPP2C, members of group A PP2C family, were upregulated in both experiments (Fig. 2). A gene encoding SRK2H, a member of SnRK2 family, was upregulated in the acclimation experiment (Fig. 2). A gene encoding AREB2, a member of AREB/ABF family, was upregulated in both experiments (Fig. 2).
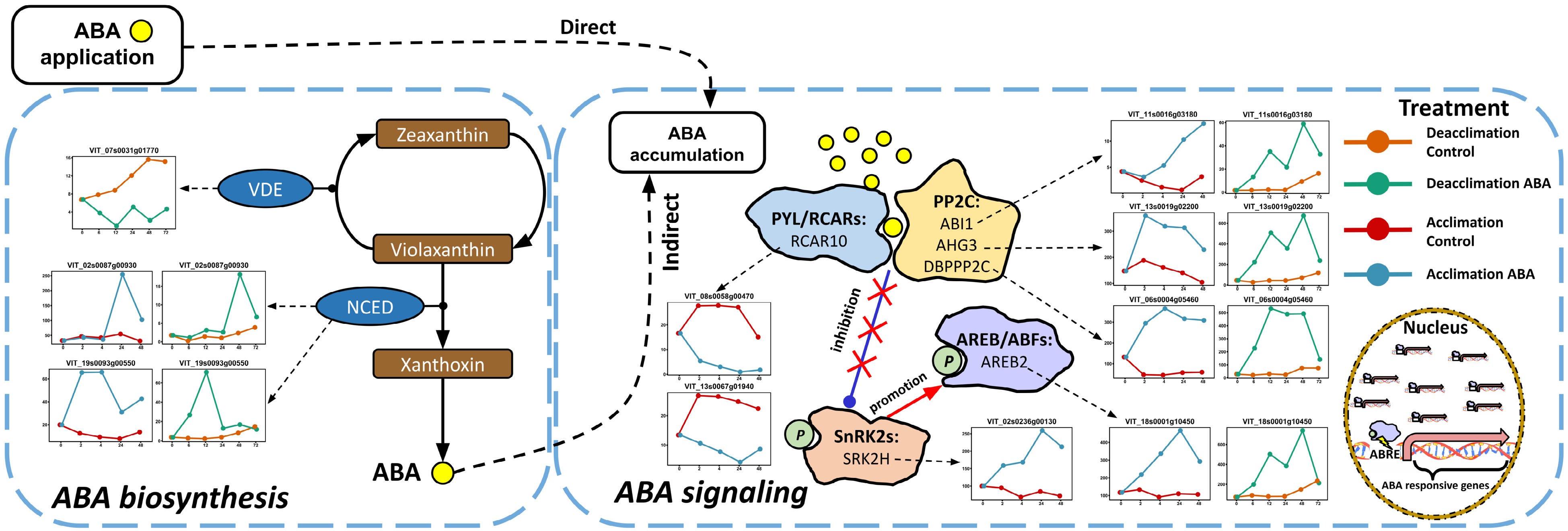
Figure 2. Impact of ABA application on ABA biosynthesis and signaling pathways in the acclimation and the deacclimation experiments. The pathways were reduced based on the full pathways in VitisNet. The expression value of genes was normalized by DESeq2. Normalized expressions of the DEGs observed in the deacclimation experiment for control and ABA treatment are orange and green, respectively. Normalized expressions of the DEGs observed in the acclimation experiment for control and ABA treatment are red and blue respectively. All shared and unshared ABA-induced DEGs are shown. Abbreviations: ABRE, ABA responsive element; AREB/ABF, ABRE-binding protein/ABRE-binding factor; NCED, 9-cis-epoxycarotenoid dioxygenase; PP2C, group A protein phosphatase 2C; PYL/RCAR, pyrabactin resistance 1-like protein/regulatory component of ABA receptor; SnRK2, sucrose non-fermenting 1-related protein kinase 2; VDE, violaxanthin de-epoxidase.
Genes encoding multiple key components in Auxin/IAA biosynthesis pathway and Auxin/IAA conjugation reactions were significantly upregulated by ABA in the deacclimation experiment (Fig. 3). In IAA biosynthesis, upregulated genes included four genes encoding nitrilase, an enzyme catalyzing the transition from IAN to IAA, and two genes encoding AOP, an enzyme catalyzing the transition from TAM to IAA (Fig. 3). In IAA conjugation, upregulated genes included four genes encoding IaaGlu, an enzyme catalyzing the conjugation of IAA and glucose, two genes encoding GH3-1, an enzyme catalyzing irreversible conjugation of IAA and amino acids, and one gene encoding ILR/ILL, an enzyme catalyzing reversible conjugation of IAA and amino acids (Fig. 3).
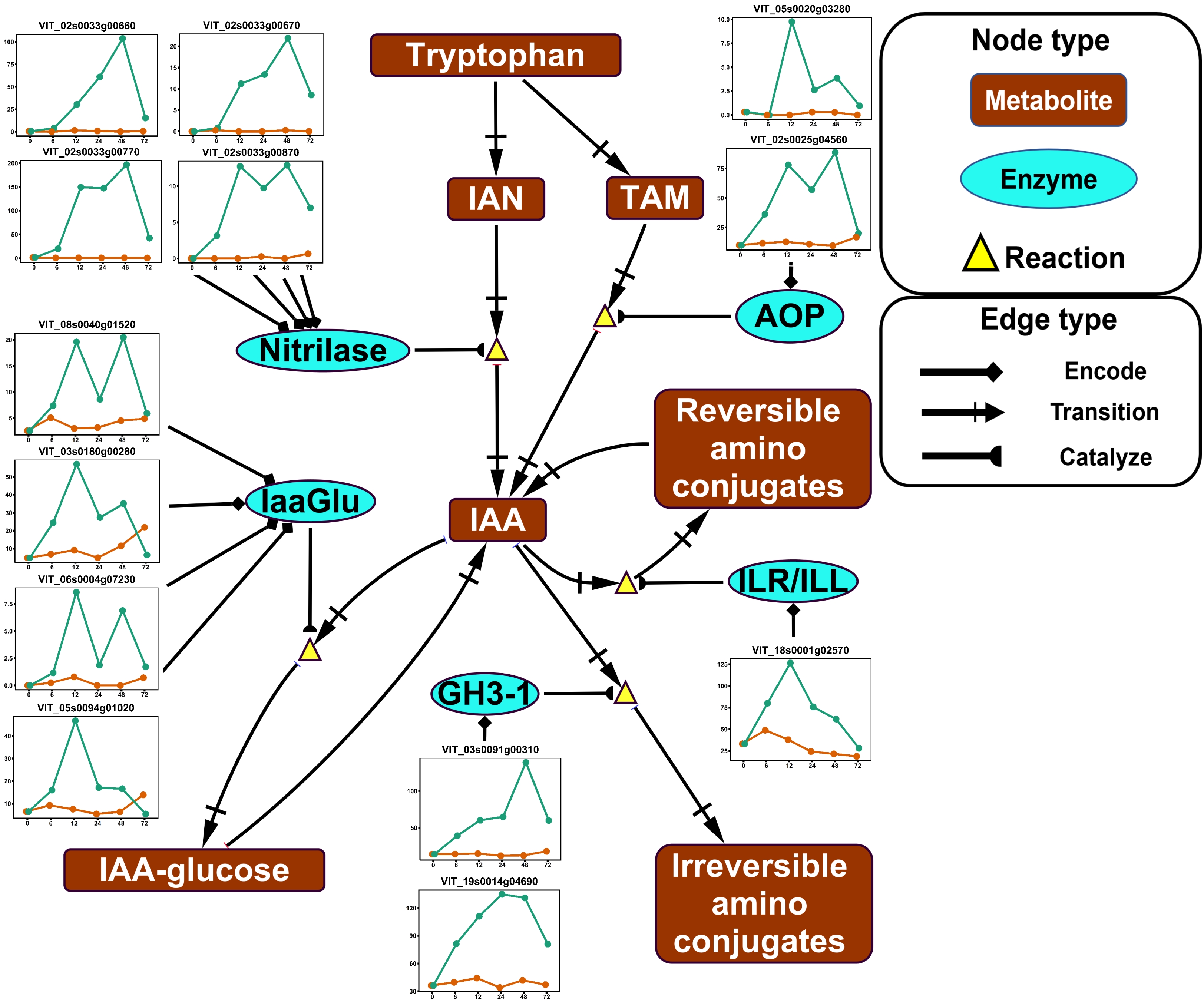
Figure 3. Impact of ABA application on auxin (IAA) metabolism in the deacclimation experiment. The pathway was reduced based on the full pathway in VitisNet. The expression value of genes was normalized by DESeq2. Normalized expressions for control and ABA treatment groups are shown by orange and green lines, respectively. Abbreviations: AOP, amine ocidase flavin containing; GH3, GRETCHEN HAGEN 3; IAA, indoleacetic acid; IaaGlu, indoleacetic acid glucosyltransferase; IAN, indole-3-acetonitrile; ILR/ILL, IAA-Leu resistant/ILR-like; TAM, tryptamine. Refer to Fig. 2 for keys of treatments in gene expression plots.
Eight genes encoding key enzymes in the galactose metabolism were significantly upregulated by ABA in the acclimation experiment (Fig. 4). In RFO biosynthesis, upregulated genes included four genes encoding GolS, an enzyme catalyzing the synthesis of galactinol, one gene encoding RafS, an enzyme catalyzing the transition of galactinol to raffinose, and one gene encoding StaS, an enzyme catalyzing the transition of raffinose to stachyose (Fig. 4). In RFO degradation, two genes encoding α-GAL, an enzyme catalyzing the degradation of RFOs, were upregulated (Fig. 4).
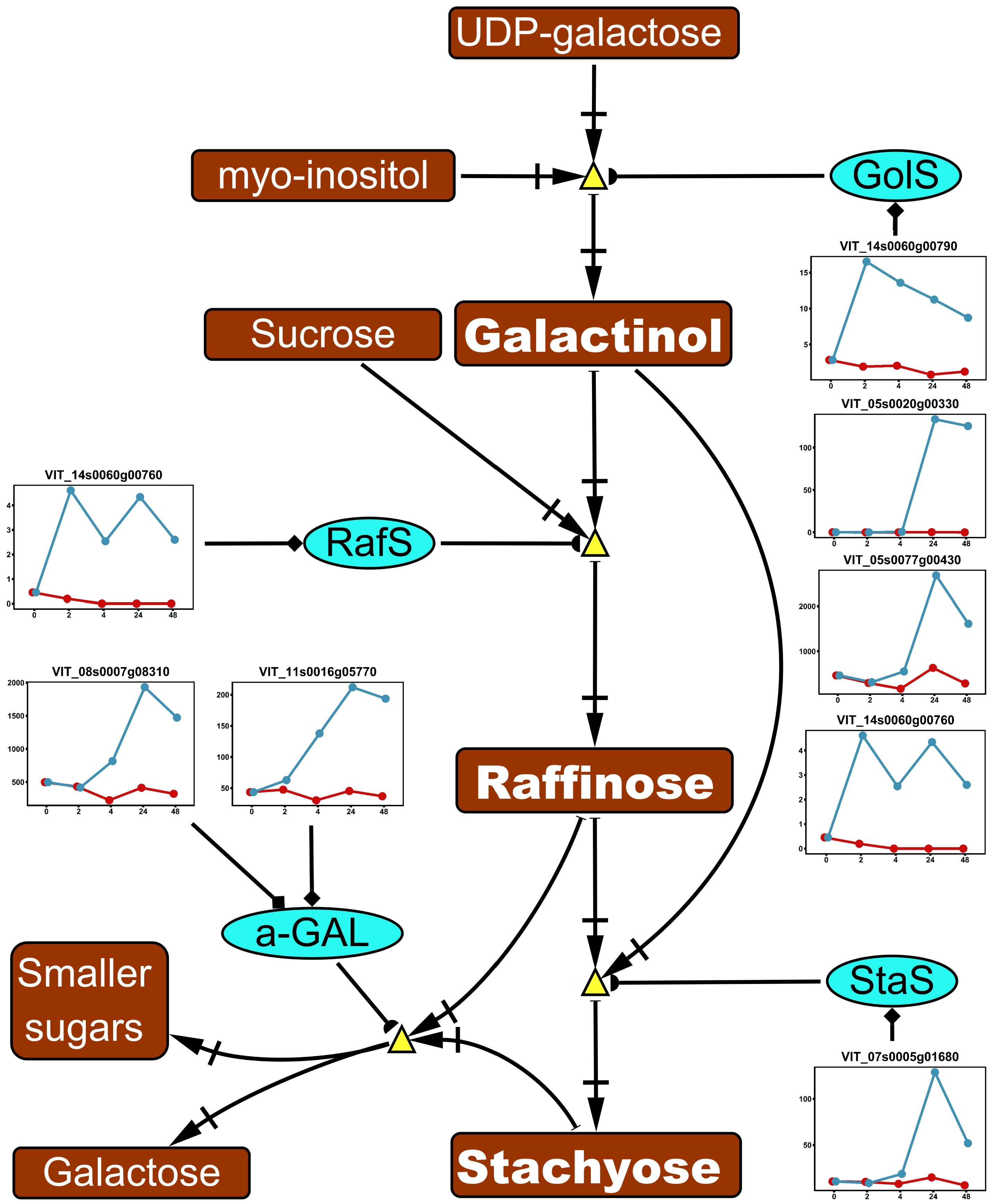
Figure 4. Impact of ABA application on raffinose family oligosacchrides (RFOs) metabolism in the acclimation experiment. The pathway was reduced based on the full pathway in VitisNet. The expression value of genes was normalized by DESeq2. Normalized expressions for control and ABA treatment groups are shown by red and blue lines respectively. Abbreviations: α-GAL, alpha-galactosidase; GolS, galactinol synthase; RafS, raffinose synthase; StaS: stachyose synthase. Refer to Fig. 2 and Fig. 3 for figure keys.
Twenty-two genes encoding ribosomal proteins were downregulated by ABA in the deacclimation experiment (Supplemental Fig. S6). The grape ribosome was remodeled based on the 3D model of Arabidopsis thaliana mitochondrial ribosome[42] with only homologous proteins shared by Vitis and Arabidopsis presented (Supplemental Fig. S6). Three grapevine ribosomal proteins (L12, L29 and L34) encoded by target DEGs in the deacclimation experiment were not present in the current mito-ribosome model in Arabidopsis. Compared to the modeled structure of the ribosome under permissive growing conditions (Supplemental Fig. S6A & S6B), the biosynthesis of 13 and 5 ribosomal proteins on large subunit (LSU) and small subunit (SSU), respectively, appeared to be downregulated in grapevine in response to ABA treatment (Supplemental Fig. S6C & S6D).
-
Deciphering the regulation of ABA during cold acclimation and deacclimation is critical for the sustainability of viticulture under climate change. In this study, we applied exogenous ABA during cold acclimation and deacclimation and precisely investigated the impact of exogenous ABA on the grapevine transcriptome. We followed a data-driven approach in the analysis of RNA-seq data, which facilitated the construction of a gene co-expression network and the identification of target DEGs. In our discussion, we dive into the pathways that exhibited most intensive response to exogenous ABA, correlate these pathways and conclude with a proposed model of how exogenous ABA impacted grapevine transcriptome and ultimately led to altered phenotypes. The coexistence of ABA-induced transcriptomic response and the balancing systems not only enriches the knowledge of grapevine regulation of phytohormone, but also suggests that ABA application might not have any long-term effect on grapevine biology. Implication of this finding may justify the application of exogenous ABA as a cultural practice in vineyards to acquire desired phenotypes to counter cold damage in winter and spring.
Identification of DEG is a major challenge for the analysis of RNA-seq data, especially in factorial design experiments composed by time courses and treatments. Standard approaches such as pairwise comparison at individual time points or contrasting with basal condition (e.g. pre-treatment) may successfully identify DEGs, nevertheless, ignore the nature of gene co-expression networks[43]. The analysis through a posterior approaches (e.g. only evaluating gene expression on certain pathways) may facilitate the investigation at pathway/cascade level but may also constrain the ability to identify unknown mechanisms. Therefore, network approaches such as WGCNA have gained popularity in RNA-seq data analysis[44]. WGCNA can be conducted through a supervised or an unsupervised approach, but can result in an overwhelming number of genes in large modules, which exacerbates the power of any downstream analysis such as pathway enrichment analysis or the identification of hub players[44].
To resolve these complications, we followed an analysis pipeline incorporating both standard and network analysis. This pipeline used unsupervised WGCNA to assess expression patterns followed by a target DEGs filtering based on FDR and LFC. We then leveraged the detailed gene functional annotation in VitisNet[45] and conducted pre-ranked GSEA on a ranked DEG list with contrast FDR as ranking criterium. Through this approach, the genes revealing most significant treatment impact on their expressions were assigned with more weight in the pathway enrichment analysis, thus making the analysis more robust by incorporating quantitative gene expression information. While our study was designed to characterize the major changes in gene expression which correlate with ABA treatments, we cannot exclude the possibility that important differences in low expressed genes also occur in response to ABA. Future studies are underway to explore more deeply the pathways uncovered in this study to identify critical candidate genes for gene knockout and functional characterization studies
One difference of note between the two experiments is the number of modules generated by WGCNA: 37 modules in the acclimation experiment and 17 modules in the deacclimation experiment. Two facts might account for these differences: (1) different transcriptomic activities in different sequenced tissue; (2) different experimental environments. The sequenced tissues in the acclimation experiment were actively growing leaf tissue, while the deacclimation experiment utilized ecodormant buds. Research has demonstrated that most basal pathways for plant growth with large number of genes (e.g. ribosome, spliceosome and photosynthesis) are more active during active growth than during dormancy[46]. We speculate that these active pathways along with the genes in these pathways complicated the co-expression network, which was detected by WGCNA and resulted in increased number of modules in the acclimation experiment. In addition, the acclimation experiment was conducted in a greenhouse with a semi-controlled environment, while the deacclimation experiment was conducted in a growth chamber. Compared to more controlled environments, such as a growth chamber, semi-controlled environments usually lead to an increase of within-replicate variation (error) due to uncontrolled diurnal dynamic and spatial variation[47]. Our results from hierarchical clustering and PCA agreed with this statement as the replicates in the acclimation experiment are less tightly clustered compared with the deacclimation experiment (Supplemental Figs. S1−S3). These factors might also account for the fact that the impact of ABA on the transcriptome was less intense regarding the number of DEGs and the magnitude of response (LFC of ABA vs. control) in the acclimation experiment.
GSEA indicated that ABA signaling pathway was significantly enriched in the shared target DEGs in two experiments (Fig. 1a) In the detailed examination of the pathway, we identified that two genes encoding NCED, a key enzyme in ABA biosynthesis, were upregulated by exogenous ABA in both experiments, however, the upregulation was either transient or unstable (Fig. 2). The increase of NCED transcript abundance in response to exogenous ABA has also been reported in other plants[48, 49], suggesting the existence of a positive feedback for ABA biosynthesis by ABA itself. This positive feedback is balanced by another ABA-dependent negative feedback system mediated by AREB/NAC protein complex[50]. AREBs and NACs are the transcription factors that mediate ABA signaling and induce the expression of ABA responsive genes[27] yet the complex formed by these two proteins can also bind to the promoter region of NCEDs and constrain its transcription in response to excessive ABA[50]. Interestingly, numerous genes coding for AREB or NAC were significantly upregulated in our experiments (Figs 1 & 2), supporting a potential dual role of AREB/NAC also exists in grapevine. The interaction of AREB, NAC and NCED may be critical to maintaining ABA homeostasis in grapevine.
We also identified a potential negative feedback mechanism in ABA signaling in response to exogenous ABA. This mechanism appears to be mediated by altered expression of genes encoding key proteins in ABA signaling[27]. In the acclimation experiment and in response to ABA application, genes encoding three members in group A PP2C family, a repressor of ABA signaling, were upregulated, and genes encoding a member in PYL/RCARs, a group of ABA receptors, were downregulated (Fig. 2). In the deacclimation experiment and in response to ABA, the same PP2C family members were upregulated, and the response was more intense (relatively speaking) than in the acclimation experiment (Fig. 2). If grapevine proteomic and metabolomic patterns match these transcriptomic responses, signal transduction in the ABA signaling pathway would be constrained, leading to an insensitivity of ABA in cells and generating a negative feedback in ABA signaling. A recent study demonstrated that AREB protein enhances PP2Cs expression by binding at their promotor region after ABA application[51]. This mechanism might be also pivotal for the negative feedback in ABA signaling in response to ABA application in grapevine.
The enrichment of galactose metabolism pathway and auxin/IAA biosynthesis/metabolism pathway was found in the acclimation and the deacclimation experiment, respectively, suggesting that ABA's regulation of these pathways may depend on the dormancy stage (Figs 3 & 4). However, our data are not sufficient to support this hypothesis due to the complexity of tissue and timing in our experimental design. Among the genes of the galactose metabolism pathway, the metabolism of RFOs have been shown to be important for plant cold hardiness, glass formation, osmotic protection, and hydroxyl radical scavenging[52]. In the acclimation experiment, multiple genes encoding three key enzymes which catalyze the biosynthesis of RFO precursor (galactinol) or RFOs (raffinose or stachyose) were upregulated by ABA application (Fig. 4). Our result agrees with qPCR analysis of the same genes in grapevine buds in response to ABA application[31]. However, it is unreasonable to extrapolate these findings to assess the change of RFOs content in our experiment since two genes encoding a RFO degradation enzyme (α-GAL) were also upregulated during the experiment (Fig. 4). The same scenario was found in auxin/IAA biosynthesis/metabolism pathway in the deacclimation experiment (Fig. 3). Auxin is a plant phytohormone that regulates various plant growth activities including dormancy[53]. It was proposed that the interaction of auxin and ABA along with their signaling pathways might serve as a key regulator in the plant dormancy cycle[54]. Exogenous application of synthetic auxin in grapevine accelerated the removal of callose, which physically facilitates shoot growth after budburst[55]. Gene expression analysis also revealed that IAA biosynthesis was enhanced during dormancy release[36]. As an inference, ABA application might negatively impact IAA biosynthesis and constrain IAA accumulation, thus slowing the process of budburst. This inference is supported by the finding that exogenous ABA inhibited IAA biosynthesis during bud outgrowth in Arabidopsis[56]. However, in our deacclimation experiment, stimulatory effects of ABA application were identified on the pathways contributing to both increase and decrease of free IAA: two tryptophan-dependent IAA biosynthesis pathways were upregulated, and three IAA conjugation/degradation pathways were also upregulated (Fig. 3). These results confound our interpretation of exogenous ABA's impact on IAA metabolism during deacclimation.
The potential mechanism of negative feedback of exogenous ABA on ABA signaling as discussed above may explain these contradictions. On one hand, ABA application increases free ABA content in cell, which enhances ABA signaling and onset downstream ABA responsive genes as a ubiquitous process. This might be the underlying mechanism for the transcriptional enhancement of RFOs biosynthesis in the acclimation experiment and IAA conjugation/degradation in the deacclimation experiment. One the other hand, when excessive ABA is present in the cell, an unknown scavenging system is activated to balance the overwhelming responses by reversely regulating ABA-induced responses. It is reasonable to infer that the pathways regulated by the scavenging system might intensivelyoverlap with ABA responsive pathways. The activation of this scavenging system might explain ABA-induced constraining of ABA signaling in both experiments, upregulation of RFOs degradation in the acclimation experiment and upregulation of IAA biosynthesis in the deacclimation experiment. The activation of this scavenging system may depend on the concentration of exogenous ABA. This proposed mechanism might be supported, in part, by the fact that the application of exogenous ABA at different concentrations led to distinguished phenotypes of root growth[57].
ABA application was seen to potentially impact many ribosomal protein genes in the deacclimation experiment. Twenty-two genes, encoding 21 ribosome proteins were downregulated in response to ABA application (Supplemental Fig. S6). Examining the spatial distribution of these proteins using the 3D model of the Arabidopsis mito-ribosome suggests that a large portion of the LSU and SSU monosomes may be impacted by exogenous ABA (Supplemental Fig. S6). Plant ribosomal heterogeneity conferred by different ribosomal components plays crucial roles in plant development by selectively translating specific mRNAs[58]. Arabidopsis ribosomal protein mutants showed distinguished developmental phenotypes, including delayed growth[59]. This finding suggests that different ribosomal proteins might specifically function at different developmental stages or in response to different stresses. Although the transcript abundance of individual ribosomal protein genes does not necessarily correlate with translation efficiency[60], a systematic downregulation of numerous ribosomal protein genes might impact global translation. Guo et al.[61] reported that exogenous ABA inhibited global protein translation in Arabidopsis, and the inhibition might be mediated by ABA-induced downregulation of Receptor for Activated C Protein Kinase1 genes (RACK1). We identified a Vitis homolog (VIT_17s0000g02750) of a Arabidopsis RACK1 gene (AT1G18080)[62], which was significantly downregulated in the deacclimation experiment (Supplemental Fig. S7A), indicating the existence of similar mechanism in Vitis. The same response was not identified in the acclimation experiment (Supplemental Fig. S7B) which suggests that this response may be specific to grapevine deacclimation.
A schematic representation of a proposed model based on our result and reasoning is shown in Fig. 5. The application of exogenous ABA during grapevine cold acclimation and deacclimation increases free ABA content in cells through direct penetration or indirect positive feedback to endogenous ABA biosynthesis mediated by upregulating NCEDs. Accumulationof free ABA triggers ABA signaling and stimulates downstream ABA responsive metabolism pathways (e.g. RFO metabolism or IAA metabolism) or basal pathways (e.g. ribosomal biogenesis) as a ubiquitous process. The stimulation of the pathways may be selective according to developmental stage, thus making some metabolomic responses specific to cold acclimation or deacclimation. In the meantime, multiple balancing systems are activated to maintain homeostasis of metabolism. These might include inhibition of NCED expression by increased AREB/NAC protein complex, insensitivity of ABA by constrained ABA signaling, and a putative scavenging system that reversely regulates various ABA responsive metabolism pathways. The altered phenotype in response to ABA application, such as deeper dormancy or delayed budburst[26, 31], is likely a consequence of a lingering effect from a short-term disequilibrium of these mechanisms. The existence of the scavenging system also suggests that the application of exogenous ABA would likely induce short-term favored phenotypes without generating long-term negative effects. This finding further justifies the utilization of ABA as a culture practice in vineyards. The onset of the proposed scavenging system is yet to be deciphered and might be of interest for developing other practical tools to acquire more desired phenotypes. Further investigation should be conducted using metabolomics techniques to complement this research with validations at the metabolite level.
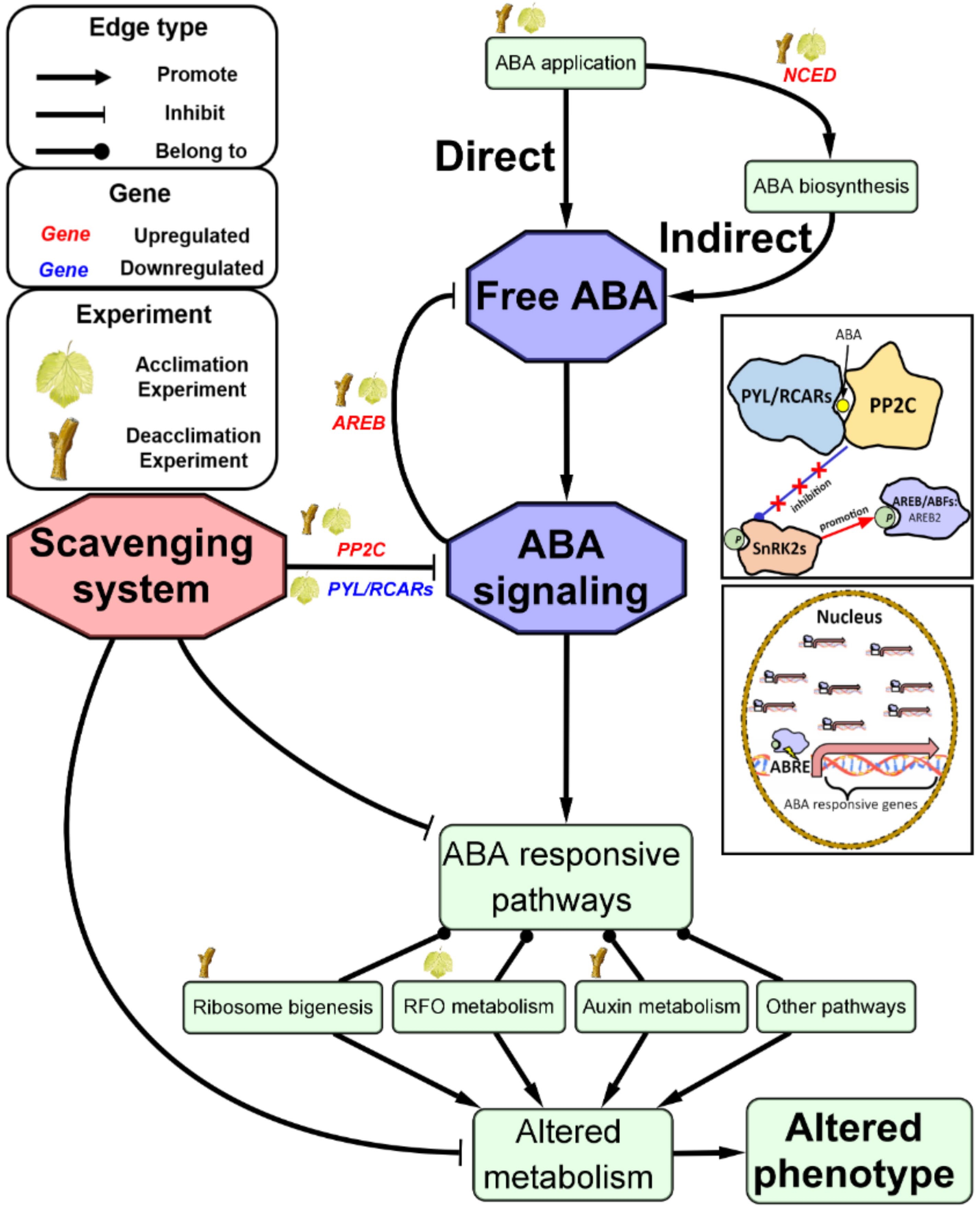
Figure 5. Schematic representation of a proposed model for exogenous ABA’s regulation on gene expression in grapevine cold acclimation and deacclimation.
-
The V. vinifera cultivar 'Cabernet Franc' was used in the experiment, in part for its economic importance in the Eastern U.S. The experiment was conducted in a greenhouse at the Ohio Agricultural Research and Development Center (OARDC, Wooster, OH, USA) using two-year-old, self-rooted grapevines cultivated in 7.6 L pots. Greenhouse environmental conditions, growth medium, grapevine training and other managements associated with grapevine growth are as described in previous study[31]. The experiment involved two treatments (control and ABA) in quadruplicate using randomized complete block design. For control treatment, the solution was composed of deionized water with 0.05% (v/v) and Tween-20 (Acros Organic, Hampton, NH, USA) as surfactant. For ABA treatment, the solution was composed of 500 mg L−1 (1.9 mM) S-ABA diluted from ProTone® SG (Valent BioSciences Corporation, Libertyville, IL, USA) and 0.05% (v/v) and Tween-20 as surfactant. This concentration was determined to be effective and safe regarding inducing proper physiological response and generating minimum phytotoxicity, respectively[63, 64]. Treatments were applied at noon of 18 Aug. 2018 when the leaf age of base node was 95 d. This timing corresponds to the start of early cold acclimation in nature in Eastern U.S.[10]. Both treatments were sprayed on all grapevine leaves until runoff to ensure a full coverage.
Deacclimation experiment
-
Field-grown 'Cabernet Franc' grapevines were used in this experiment. Grapevines were grafted on 3309C rootstocks and commercially cultivated at Ravine's Wine Cellars in Geneva, NY and subjected to standard vineyard management practices during the growing season. Dormant canes were harvested in March of 2017, after vines had been exposed to > 1,200 chilling hour (NC model https://products.climate.ncsu.edu/ag/chill-models) and chopped into single node cuttings. At collection, the cuttings were at 'winter buds' stage, corresponding to Eichhorn Lorenz Stage 1 (EL1)[65] and had fully transitioned to ecodormancy. Cuttings were randomized and divided into six groups, corresponding to six timepoints for pre and post treatment sample collections. Grouped cuttings were incubated with cut ends in cups of water under permissive growing conditions (22 °C and 16/8 h light/dark) in a growth room. When single bud cuttings developed to 'woolly buds' stage, corresponding to EL3[65], treatments were applied on each group with a hand sprayer to runoff. Control treatment was deionized water, and ABA treatment was 1,322 mg L−1 (5mM) S-ABA diluted from ProTone® SG (Valent BioSciences Corporation, Libertyville, IL, USA). This concentration was reported to be effective to delay budburst on woolly buds[26].
RNA-seq library preparation and data processing
-
In the cold acclimation experiment, leaf samples from the ABA and water (control) treatments were collected at pre-treatment, 2 h, 4 h, 24 h and 48 h post-treatment. Four biological replicates, each consisted of five leaves collected from node number three to 10, were used for RNA-seq. Eight replicates were collected at pre-treatment. In the deacclimation experiment, bud samples treated with ABA and water (control) were collected at pre-treatment, 6 h, 12 h, 24 h, 48 h and 72 h post-treatment. Three biological replicates, each consisting of three buds excised from single-node cuttings, were used for RNA-seq. Four replicates were collected at 48 h post-treatment. Samples were flash-frozen in liquid nitrogen immediately after collection and stored at −80 °C until extraction.
Total RNA was extracted using SpectrumTM Plant Total RNA Kit (Sigma Aldrich, St Louis, MO, USA) following the protocol suggested by the manufacturer. Libraries were constructed using Lexogen QuantSeq 3'mRNA-Seq Prep Kit (Lexogen, Greenland, NH, USA) following standard practices as a service provided by Cornell University Institute of Genomic Diversity (Ithaca, NY, USA). Sequencing of the libraries was accomplished using NextSeq500 (Illumina, Inc., San Diego, CA, USA) with 95 samples per lane at Cornell University Institute of Biotechnology (Ithaca, NY, USA). The raw read length was 85 bp and 75 bp in the acclimation and deacclimation experiment, respectively. For each library, sequencing was conducted in triplicate to justify technical validity. As RNA-seq data preprocessing, FastQC was applied to each library for quality control. Reads from each library were subsequently trimmed using BBDuk (http://jgi.doe.gov/data-and-tools/bb-tools) to remove adaptors and poly-A following the pipeline suggested by the manufacturer (www.lexogen.com/quantseq-data-analysis). Trimmed reads were aligned to Vitis vinifera 12X.v2 genome and VCost.v3 annotation[38] using STAR[37]. Gene level quantification was conducted using '-quantMode GeneCounts' in STAR.
The resulting gene count matrices were filtered for low count genes based on total gene count among all samples. The genes with total gene count greater than sample number were considered as expressed genes and subjected to subsequent analysis. The filtered gene count matrix was analyzed for differential expression using DESeq2[39]. The full model of DESeq2 contained time post-treatment as a continuous variable and treatments (ABA or control) as a discrete variable. A normalized gene count matrix was generated by DESeq2 (normalize = TRUE) and was inputted for gene expression visualization in figures. Log transformed gene count matrix was generated by DESeq2 VST and was subjected to downstream analysis.
WGCNA and gene filtering
-
For each experiment, VST count of all genes after low count filtering were used for WGCNA[40] to ensure an unsupervised gene co-expression network construction. A dendrogram of all samples was constructed using hierarchical clustering, and the samples showing substantial distance from the main cluster were removed as outliers. Remaining samples were subjected to one-step signed network construction and module detection using 'blockwiseModules' (power = 12, networkType = "signed", TOMType = "signed", minModuleSize = 50, reassignThreshold = 0, mergeCutHeight = 0.25).
After network construction, genes in module 'grey' were excluded for any further analysis since these genes were recognized as noise genes by WGCNA. The PC1 of all genes in each module, known as module eigengene or ME, was calculated by WGCNA. MEs were correlated with treatments using the Pearson method and visualized across all timepoints to identify the expression pattern that might explain treatment effect per interest of the experiment. Numeric transformation of treatments (ABA as '1', control as '0') was conducted to facilitate the correlation analysis. Contrasts were created based on identified expression patterns and applied on genes in target modules (if the identified expression pattern exists in few modules) or all genes (if the identified expression pattern exists in numerous modules). The genes subjected to contrasting were further filtered based on FDR < 0.05 and LFC > |1| of the specific contrast in DESeq2. Resultant genes were considered as target DEGs for the experiment.
GSEA
-
Identifications of target genes were changed to their corresponding V1 annotations for better functional annotations (http://genomes.cribi.unipd.it/grape). Resulting genes were subsequently ranked using FDR of the contrast as the ranking criterion in a decreasing order. A two-column.rnk file containing the gene list and corresponding ranking list was input to GSEA. The gene set file (.gmt) was generated from predefined pathways in VitisNet database[45]. All pathways in VitisNet were used for GSEA. GSEA was conducted through 'Run GSEAPreranked' in 'weighted' mode using 1000 permutations, and normalization mode was set as 'meandiv'.
3D remodeling of ribosome
-
The cryo-electron microscopy structure of Arabidopsis thaliana mitoribosome (RCSB PDB: 6XYW) was used as template for 3D remodeling to visualize the impact of exogenous ABA on grapevine ribosome during deacclimation[42]. The modeling was accomplished in Chimera 1.15[66]. Only homologous proteins shared by Vitis and Arabidopsis were presented in the model. After splitting the entire model into protein segments using 'split', protein surface was calculated through 'surface' and visualized in 'mesh'.
Data availability
-
All RNA-seq raw data along with processed gene count matrices and sample metadata are available in NCBI-GEO (accession: GSE184114).
- This work was funded in part by the New York Grape and Wine Foundation, by U.S. Department of Agriculture appropriated project 1910-21220-006-00D, by The Ohio State University Research Competitive Grants Program (SEEDS) and by The Ohio State University College of Food, Agricultural, and Environmental Sciences, Department of Horticulture and Crop Science.
- The authors declare that they have no conflict of interest.
- Supplemental Fig. S1 Hierarchical clustering of samples in the acclimation experiment. All genes after low count filtering were used for clustering.
- Supplemental Fig. S2 Hierarchical clustering of samples in the deacclimation experiment. All genes after low count filtering were used for clustering.
- Supplemental Fig. S3 Principal component analysis for the transcriptome of the samples in two experiments. A-B) Two−dimensional profiling of leaf samples in the acclimation experiment using PC1, PC2 and PC3; C-D) Two-dimensional profiling of bud samples in the deacclimation experiment using PC1, PC2 and PC3.
- Supplemental Fig. S4 WGCNA of samples in the acclimation experiment. A) Module-trait relationships; B) ME visualization across all timepoints in the experiment. Input genes are all genes after low count filtering. Error bars represent SE (n = 4). The Module-trait relationships were computed using Pearson method. The first line and second line of each cell represent correlation coefficient and correlation p-value, respectively.
- Supplemental Fig. S5 WGCNA of samples in the deacclimation experiment. A) Module-trait relationships; B) ME visualization across all timepoints in the experiment. Input genes are all genes after low count filtering. Error bars are not shown due to minimum variance among replicates. The Module-trait relationships were computed using Pearson method. The first line and second line of cells represent correlation coefficient and correlation p-value, respectively.
- Supplemental Fig. S6 Remodeling of potential grapevine ribosome in response to ABA application in the deacclimation experiment. Presented ribosome was remodeled in Chimera based on cryo-electron microscopy structure of Arabidopsis thaliana mitochondria ribosome. Only homologous proteins shared between Arabidopsis and Vitis are shown. A-B) Potential ribosome in control samples under permissive growing condition (22 °C) for budburst; C-D) Potential ribosome in ABA-treated samples under same condition. At least one gene encoding each blue-highlighted protein exhibited significantly lower transcript abundance in response to ABA application. Abbreviations: SSU, small subunit; LSU, large subunit.
- Supplemental Fig. S7 Expression of VIT_17s0000g02750, an Arabidopsis RACK1 homolog in Vitis, in two experiments.
- Copyright: © 2022 by the author(s). Exclusive Licensee Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Wang H, Dami IE, Martens H, Londo JP. 2022. Transcriptomic analysis of grapevine in response to ABA application reveals its diverse regulations during cold acclimation and deacclimation. Fruit Research 2: 1 doi: 10.48130/FruRes-2022-0001

Transcriptomic analysis of grapevine in response to ABA application reveals its diverse regulations during cold acclimation and deacclimation
- Received: 26 October 2021
- Accepted: 13 December 2021
- Published online: 24 January 2022
Abstract: Abscisic acid (ABA) plays crucial regulatory roles in cold acclimation and deacclimation of grapevine, making it a potential tool to be utilized in vineyards for the acquisition of preferred phenotypes in winter and spring. To understand the function of ABA, we conducted experiments during cold acclimation and deacclimation and evaluated the impact of exogenous ABA on the grapevine transcriptome. RNA-seq data were collected periodically hours or days after ABA treatment. Transcriptomic data were analyzed using principal component analysis (PCA), hierarchical clustering, unsupervised weighed gene co-expression network analysis (WGCNA), contrast-based differentially expressed genes (DEGs) identification and pre-ranked gene set enrichment analysis (GSEA). Our results suggest that ABA functions differently during cold acclimation and deacclimation by selectively regulating key pathways including auxin/indole acetic acid (IAA) metabolism, galactose metabolism and ribosome biogenesis. We also identified the activation of several apparent negative feedback systems that regulated ABA-induced transcriptomic changes, suggesting the existence of a balancing system in response to excessive ABA. This balancing systems potentially eliminates the long-term negative effect on grapevine growing from using ABA in the field. These findings advance our understanding about the regulation of grapevine physiology during dormancy and supports the potential of applying ABA as a cultural practice to mitigate cold injury in winter and spring.
-
Key words:
- ABA /
- Vitis /
- RNA-seq /
- Cold acclimation /
- Deacclimation /
- Transcriptomics