









-
Thanks to its small size, diploidy, rapid life cycle, and the availability of genetic tools, woodland strawberry (Fragaria vesca) is the preeminent model system for the cultivated strawberry Fragaria × ananassa, which is allooctoploid. It also serves as a model for the Rosaceae family of fruit crops, which include apple, pear, peach, raspberry, and others. Several accessions of F. vesca have been inbred seven times and developed as models; they are Hawaii 4 (H4), Yellow Wonder 5AF7 (YW5AF7), and Rügen[1−3]. The genome of Hawaii 4 was first published in 2011[4] and later in 2018 with a much improved quality in both assembly and annotation[5−7]. Recently, a new genome was published of F. vesca accession CFRA2339[2], a red-fruited, runnerless variety that is less commonly used in research. However, at this time, the 4th Hawaii 4 annotation and assembly serves as the standard reference genome for genomic studies in strawberry.
Hawaii 4 (H4) is just one of the inbred F. vesca accessions and may not capture the diversity of F. vesca[1−3]. Most importantly, Yellow Wonder 5AF7 (YW5AF7) has been a preferred research model in many laboratories because of several of its characteristics. First, YW5AF7 plants do not form runners, allowing for convenient growth and maintenance in high-density settings like a lab growth chamber (Fig. 1a, b). This feature also makes it easier in mutagenesis screens without contamination of different mutant individuals by runners. Second, the yellow fruit, the product of a recessive mutation in the MYB10 gene[1], provides a convenient visual marker in distinguishing hybrid F1 from self-fertilized progeny in genetic crosses between YW5AF7 female and red-fruited WT accessions (Fig. 1c, d), and for transient expression assays to study fruit pigment genes and ripening processes[1,8]. Third, YW5AF7 is ever-flowering, making it convenient to perform genetic crosses and study the development of flower, seed, and fruit in all seasons[3]. Finally, YW5AF7 was heavily inbred, leading to an improved genetic homozygosity[1].

Figure 1.
Plant architecture and fruit phenotype of three F. vesca accessions Yellow Wonder (YW), Hawaii 4 (H4), and Rügen (Rü). (a)−(c) Photographs of YW, H4, and Rü plants. Note the absence of runners in YW and Rü, but prolific runners (arrows) in H4. (d) Both YW and H4 develop yellow color fruits as shown, but Rü develops red color fruit. Plants are pictured in 4 in × 4 in pots.
A survey of the literature illustrates the widespread use of YW5AF7 in studies ranging from mutagenesis screens and mutant characterization[8,9], genome editing[10], transcriptome profiling[11,12], and gene functional characterization[1,13]. However, YW5AF7 does not currently have its own reference genome as most studies in F. vesca rely on the H4 reference genome. The lack of a true YW5AF7 reference increases the difficulties in primer design, gene cloning, genome editing and analysis in YW5AF7 due to SNPs and structural variations between YW5AF7 and the H4 genomes. Therefore, a high-quality genome of the Yellow Wonder 5AF7 accession is highly desirable.
We have assembled, annotated and released the YW5AF7 genome as FvYW Version 1.0 (FvYW1.0). The genome was assembled de novo using Oxford Nanopore long reads, and polished with Illumina reads. To annotate the genome, we lifted over the annotation from H4 v4.0.a2[7]. FvYW1.0 exhibits high quality, with a BUSCO score of 97% − reflecting the identification of the vast majority of a conserved set of plant genes − and an N50 of 34MB, indicating chromosome-scale assembly. We further examined and confirmed the molecular lesions in the loci determining the loss of runnering, ever-flowering, and yellow fruit color. This genome assembly will further improve YW5AF7 as a desirable model and greatly facilitate genetic and genomic research in F. vesca and commercial strawberry at large.
-
To assemble the YW5AF7 genome, we combined long-read Oxford Nanopore sequencing and high coverage short read Illumina sequencing. About 2.3 million Nanopore reads, totaling 26 gigabytes (GB) and ~125x coverage of the genome, were used in assembling the genome following a pipeline described in Fig. 2. First, the Canu assembler[14] was used to trim and correct the raw Nanopore reads and then assemble a preliminary genome. This initial assembly yielded 163 contigs, with an N50 of 5.6 MB. RagTag was then used to correct and scaffold misassemblies, using homology-based alignments to realign contigs against existing F. vesca reference genomes and merge scaffolds into pseudomolecules[2,15]. We compared the RagTag result of aligning the YW assembly to the H4 (v4.0) reference genome with that of aligning to the CFRA2339 genome (Supplemental Table S1); scaffolding to the CRFA2339 genome yielded superior statistics in terms of continuity (7 kb gap sequence) but slightly reduced N50 (32.8 MB vs 33.3 MB). Hence, we selected the CFRA2339 assembly as the final RagTag reference. POLCA, a subprogram of the MASURCA genome assembly software[16], was used to polish the assembly over three rounds with 11.6 GB of previously-published Illumina DNA-seq data of YW5AF7[1]. The final genome assembly of YW5AF7 spanned 219.5 MB across 99 contigs with an N50 length of 33.6 MB (Fig. 2; Table 1). Fragaria vesca has seven chromosomes. In the final assembly, 216 MB of the 219.5 MB total sequence are contained in seven YW5AF7 pseudomolecules.
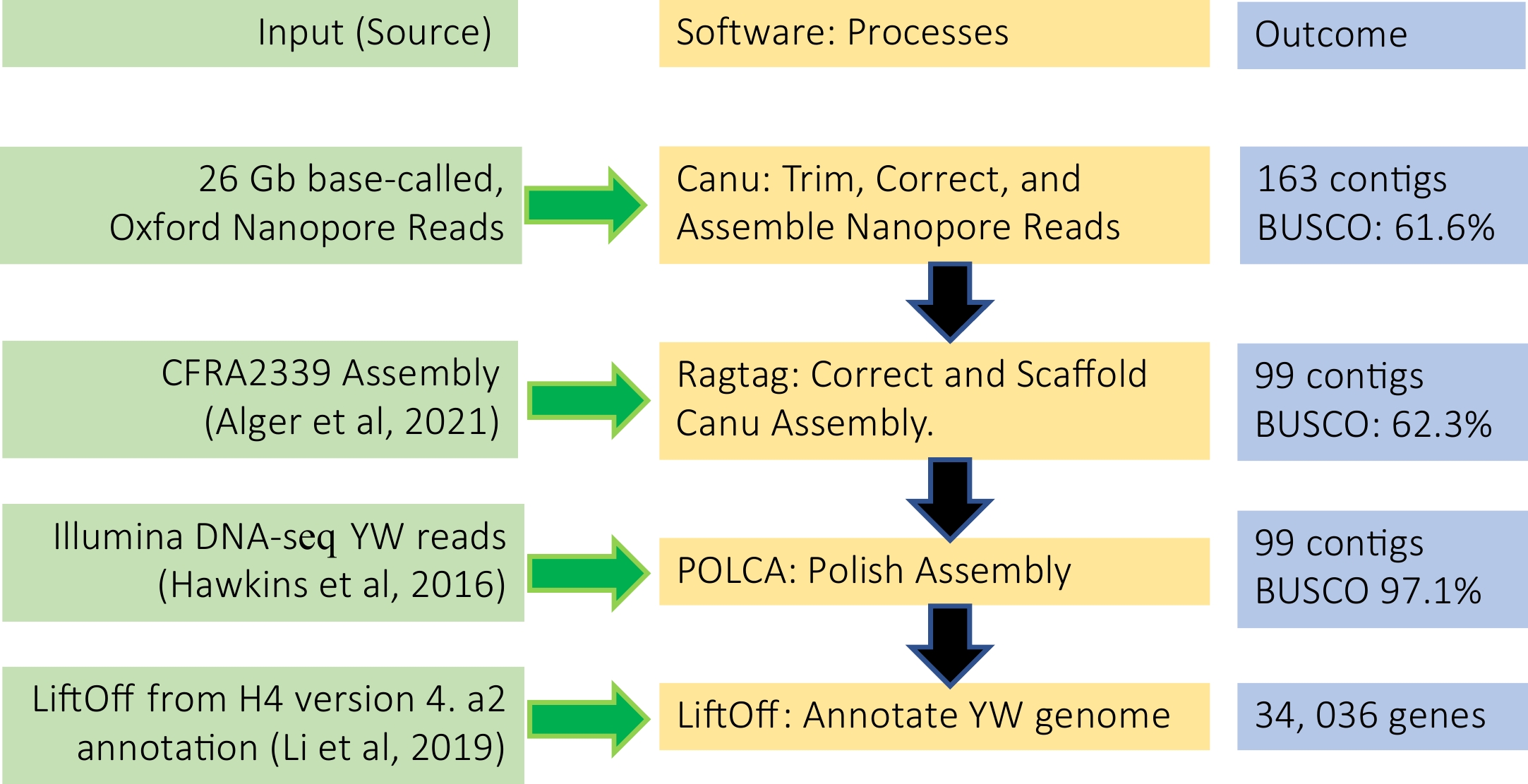
Figure 2.
De novo genome assembly and annotation pipeline used in this study.
Table 1. Quality metrics for FvYW_v1.0 and previously published F. vesca genomes.
Fragaria vesca
accessionsHawaii-4
(FvH4_v4.0)CFRA2339 Yellow Wonder
(FvYW_v1.0)Assembly length (MB) 220 244 220 Total contigs 29 402 96 auN (MB) 32 31 32 N50 (MB) 34 31 34 L90 (MB) 7 7 7 Number of annotated genes 34,008 30,349 34,007 BUSCO 0.947 0.967 0.961 The Benchmarking Universal Single-Copy Orthologs (BUSCO) with the plantae database was used to estimate the quality of the assembly and annotation[17]. The YW5AF7 genome was found to have 96% of the core genes in the BUSCO plantae dataset (Table 1), supporting a high-quality genome assembly and annotation, with 22 duplicated, 10 fragmented and 22 missing BUSCOs of the 956 searched. Table 1 summarizes the key characteristics of the YW genome in comparison to the two previously published F. vesca genomes, H4 and CFRA2339. These key indicators such as total contig number, N50, and BUSCO score indicate that FvYWv1.0 is of a quality similar to the previously published FvH4_v4.0 and CFRA2339 genomes.
LiftOff[18] was used to port the high-quality H4 genome annotation (v4.0.a2)[7] to the YW5AF7 genome assembly that resulted in a final annotation that annotates 34,007 genes. The number of YW5AF7 genes is very similar to H4 with 34,008 annotated genes (Table 1). All but 62 genes were localized to the seven chromosomes, the remaining 62 genes are located in the assembly’s remaining unanchored 92 contigs. Of the 10 most overrepresented biological process gene ontologies (GOs) related to those 62 genes, six of them contribute to ribosome assembly and peptide translation (Supplemental Table S2). This result likely reflects the repeated nature of ribosomal genes, which causes difficulty in assigning them to unique loci.
In the annotations, YW5AF7 gene names are derived from their LiftOff-identified H4 syntenic homologs. Thus, in the FvYWv1.0.a1 annotation, the MYB10 gene has a gene ID of FvYW_1g22020, which correlates with its homolog in H4, FvH4_1g22020 from the FvH4 annotation v4.0.a2.
As mentioned earlier, YW exhibits certain plant architecture and fruit characteristics due to DNA variants in specific genes. We examined the new assembly to confirm that these molecular variations underlie the phenotypes of YW (Supplemental Fig. S1). Specifically, in the YW5AF7 assembly, MYB10 (FvYW_1g22020) is found to possess the G-to-C SNP that underlies the tryptophan to serine change in the gene’s 12th amino acid that leads to production of yellow colored fruit (Fig. 1d)[1]. Second, the YW5AF7 assembly harbors a 9-bp deletion in the GA20OX4 gene (FvYW_2g35050), which underlies YW’s runnerless phenotype (Fig. 1b)[19]. Finally, the TFL1 (FvYW_3g24700) aligns with 100 percent sequence identity to FvH4_3g24700, indicating that the gene of YW5AF7 harbors the same 2-bp deletion responsible for the perpetual flowering phenotype found in H4 and other previously-identified perpetual flowering varieties[20,21]. The recapitulation of these DNA variations in FvYWv1.0 strengthens previous findings and confirm the quality of this assembly.
Genome comparison between YW5AF7 and previously published F. vesca accessions
-
The YW5AF7 genome is relatively distinct from previously published F. vesca genomes of H4 and CFRA2339 accessions. We used Sourmash to calculate the Jaccard similarity coefficients across the three F. vesca genomes, using Fragaria iinumae as an outgroup (Fig. 3a). This comparison reveals that the CRFA2339 and H4 genomes are more similar to each other than they are to the YW5AF7 genome, although the three F. vesca genomes cluster strongly from the outgroup. This phylogeny also is supported by synteny maps, which show that the YW5AF7 accession contains major inversions in three loci in comparison to the genomes of Hawaii 4 or CFRA2339 (Fig. 3b). The inversions are distal to the centromere in chromosomes 1 and 3 and near the midpoint of the length of chromosome 3. One such inversion (midpoint of chromosome 3) contains the TFL1 (FvYW_3g24700), showing that the SNP polymorphism responsible for the perpetual flowering phenotype is robust to a genetic inversion. As such, synteny and similarity comparisons demonstrate that YW5AF7, at a genetic level, is more distantly related to Hawaii 4 than CFRA2339 (Fig. 3b). Together, the genome described here for F. vesca YW5AF7 will be a valuable new resource for the strawberry research community.
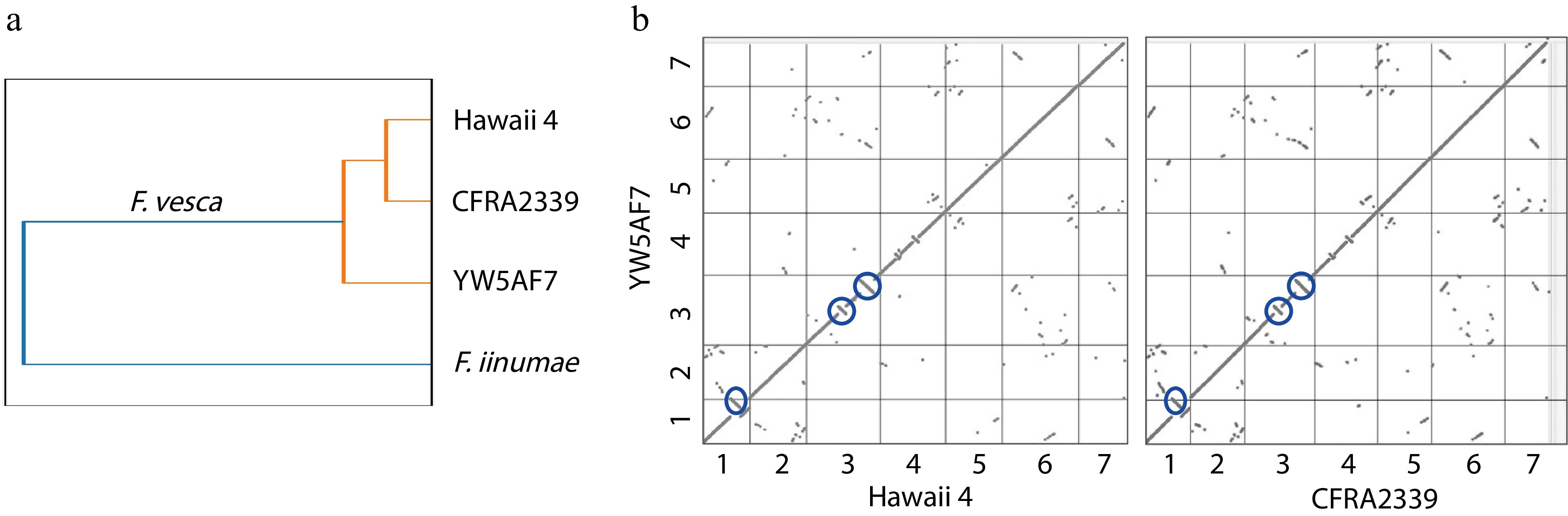
Figure 3.
YW5AF7 genome structure shows consistent differences from previously-published genomes of Fragaria vesca accessions. (a) YW5AF7 genome clusters are distinct from previously published F. vesca genomes, Hawaii 4 and CFRA2339, as well as Fragaria iinumae. This is indicated by Jaccard similarity coefficient calculated by Sourmash. (b) Inversions are observed in Chr 1, 3, and 4 between YW5AF7 and the other two F. vesca accessions (highlighted in blue circles). Y and X axis show seven chromosomes. Synteny plots generated on COGE.
-
Seventh-generation inbred Yellow Wonder accession, 5AF7, of Fragaria vesca was used as previously described[1,3]. Genomic DNA was extracted from young leaves using a method previously described[22]. Samples were sequenced on PromethION (Oxford Nanopore Technologies, Oxford, UK) for 48 h, then raw fast5 data were basecalled with Albacore version 2.1.10 (Oxford Nanopore Technologies, Oxford, UK), yielding 26 GB of raw sequencing data (~125× coverage of the genome).
Canu v1.9[14] was used to correct, trim, and assemble raw Nanopore reads assuming a genome size of 240 MB and under default parameters. We used a conda (version 4.12.0) installation of RagTag[15] in misassembly correction and scaffolding modes to merge the remaining fragmented scaffolds into pseudomolecules, generating a genome guided assembly based on the CRFA2339 reference assembly.
To polished the draft assembly, POLCA, a subprogram of the MASURCA genome assembly software[16], was used in three rounds, using 11.6 GB of 50 bp, single-ended Illumina reads[1] aligned to the draft assembly with minimap2 using the '–ax sr' parameter set[23]. The polished assembly was annotated with Liftoff (2.31.10) in eukaryotic mode using as a reference F. vesca H4 annotation v4.0.a2 downloaded from Genome Database for Rosaceae (GDR), the Rosaceae genomic repository[7,18,24]. Except for the Canu assembly, all computational work was performed using resources provided through the Cyverse iPlant Collaborative[25]. The files of FvYW1.0 genome assembly, annotation, transcripts, proteins, and the index of the assembly are available for download at GDR and as Supplemental Data 1−5; these were generated using gffread options -w and -y, respectively[26].
Gene ontology enrichment analysis was conducted using the BioConductor TopGO software package in R Studio, based on data provided in the F. vesca Hawaii 4 v4.0.a2 annotation[7,27].
Data availability statement
-
The Hawaii 4 and CFRA2339 genome assembly, annotations, and other supporting data are available on the GDR (
www.rosaceae.org )[24] and the CyVerse CoGe[28] platform. The raw nanopore DNA sequencing data of YW5AF7 have been deposited in NCBI Sequence Read Archive under BioProject ID PRJNA871257.The FvYW_v1.0 genome assembly and annotation files have been submitted to GDR (
www.rosaceae.org ) for the community to download. We would like to thank Dr Steve Mount and Dr Muzi Li for their guidance and advice on the genome assembly and annotation. This research was supported by a University of Maryland Faculty-Student Research Grant to DJ and ZL, University of Maryland CMNS Dean's Matching Award to DJ that is associated with the UMD NIH T32 Molecular and Cell Biology Training Grant, a University of Maryland Hockmeyer Fellowship to DJ, and National Science Foundation grant (IOS1444987) to ZL The computational resources were provided by Cyverse (www.cyverse.org), which is supported by the National Science Foundation grants DBI-0735191, DBI-1265383, and DBI-1743442.
-
The authors declare that they have no conflict of interest.
- Supplemental Table S1 Comparison of RagTag results used to scaffold the initial YW5AF7 assembly against two earlier Fragaria vesca assemblies.
- Supplemental Table S2 Gene ontology enrichment analysis of 62 genes not placed on 7 major assembly pseudomolecules shows enrichment for ribosome and peptide assembly.
- Supplemental Fig. S1 Yellow Wonder de novo assembly corroborates with previous phenotype-linked genetic lesions. Blast results showing the alignment in the region of interest. Bold nucleotides highlight the genetic variants underlying specific phenotypes.
- Copyright: 2022 by the author(s). Exclusive Licensee Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Joldersma D, Sadowski N, Timp W, Liu Z. 2022. Assembly and annotation of Fragaria vesca 'Yellow Wonder' genome, a model diploid strawberry for molecular genetic research. Fruit Research 2:13 doi: 10.48130/FruRes-2022-0013 

Assembly and annotation of Fragaria vesca 'Yellow Wonder' genome, a model diploid strawberry for molecular genetic research
- Received: 05 August 2022
- Accepted: 16 August 2022
- Published online: 30 August 2022
Abstract: Fragaria vesca, a wild diploid strawberry, serves as a fundamental research model for cultivated strawberry. The current reference genomes available are limited to two closely-related accessions, Hawaii 4 and CFRA2339. The widely-used model accession 'Yellow Wonder' does not yet have its reference genome. In this study, the genome of a 7th generation inbred 'Yellow Wonder' was assembled using a combination of Oxford Nanopore long reads and Illumina short reads. The de novo chromosome-scale assembly of this 220 megabase genome possesses 34,007 genes which were annotated through lift over from the Hawaii 4 genome annotation. Genome comparisons show that the 'Yellow Wonder' genome is relatively distinct from the two previously published F. vesca accessions, Hawaii 4 and CFRA2339. The availability of a 'Yellow Wonder' reference genome adds another important genomic resource to Fragaria vesca and enables rapid research progress in strawberry.
-
Key words:
- Wild strawberry /
- Yellow Wonder /
- Genome assembly /
- Genome annotation