









-
Stephania japonica is a vine plant of the family Menispermaceae[1]. Which is widely used in traditional medicine and ornamental gardening[1,2]. The COVID-19 outbreak in 2019 led to a global disaster, and scientists are working together to find effective drugs[3]. This brings S. japonica back into the view of scientists. Among study results of effective drugs, cepharanthine (CEP) stands out because of its high coronavirus suppression effect in vitro according to the cell experiment of Fan et al.[4] and and Rogosnitzky et al.[5]. CEP is a bisbenzylisoquinoline alkaloid that has been shown to have effects like antiviral, anti-inflammatory and antitumor[6]. It is a secondary metabolite of S. japonica, and extraction from plants is still one of the main ways of its production[7]. CEP supply remains depends on the extraction from the root of S. japonica, however, the current production mode unable sustain the demand of high-quality CEP required by the market.
CEP biosynthesis mainly begins with dopamine and 4-hydroxyphenylacetaldehyde via norclaurane synthesis (NCS) to yield norclaurane[8], norclaurane under the catalysis of 6-O-methyltransferase (6OMT) to produce coclaurine[9], coclaurine under the catalysis of coclaurine N-methyltransferase (CNMT) to produce methycoclaurine[10], and methycoclaurine is the precursor of the synthesis of CEP, it was catalyzed by O-methyltransferase (OMT) and cytochrome P450 (CYP450) (Supplemental Fig. S1). With the increased clinical demand for cepharanthine, it is important to study the biosynthesis of cepharanthine and how transcription factors (TFs) and microRNA regulate its biosynthesis. Previous studies indicated that microRNAs and TF play an important role in the biosynthesis of alkaloids[11,12]. For example, CrERF5 positively regulate the biosynthesis of indole alkaloids and their precursors in Catharanthus roseus[13,14]. The overexpression of NtORC1/ERF221 significantly increased the accumulation of nicotine alkaloid in tobacco plants[15]. Overexpression of CjWRKY1 could increase benzylisoquinoline alkaloid in California poppy cells[16]. MicroRNA is a common regulatory factor that can affect alkaloids biosynthesis by regulating transcription factors[17]. An Arabidopsis miR5021 homologue have a regulatory effect on the key enzymes involved in alkaloid biosynthesis in C. roseus[18]. Three microRNAs in Opium poppy are related to the biosynthesis of alkaloids[11]. miR164 targets the NTNAC-R1 gene to increase nicotine content in tobacco[19]. Study on the regulation of microRNA on transcription factors can play a guiding role in the production of plant secondary metabolites.
MicroRNA is a class of non-coding RNA with a length of 18 - 24 nt, which is ubiquitous in living organisms and play a role in regulating gene expression[20]. MicroRNA has emerged as important regulators in plants, partcipating in plant biological and metabolic process, such as plant development[21−22], and various environmental response[23−24]. Interestingly, increasing evidence has shown that the biosynthesis and accumulation of secondary metabolites in plants were meditated by microRNAs. microRNA-mediated silencing targeted the repressor class of MYBs to promote anthocyanin biosynthesis in grape lines with high anthocyanins[25]. microRNAs may regulate the biosynthesis of flavor compounds in different tissues of tea plant, for instance, the content of gallate catechins was related with the expression of miR156, miR166 and miR172, and the increased content of non-gallated catechins was shown to be positively correlated with the expression of miR169a, miR169l and miR319h[26]. In addition, the biosynthesis of terpenoid and alkaloid has also been meditated by microRNAs. The research suggested that the microRNAs including pmi-miR6173, pmi-miR6300, pmi-Nov_13, miR396a, miR398f/g and pmi-Nov12 post-transcriptionally regulate terpenoid biosynthesis in Persicaria minor (kesum)[27]. The investigation uncovered unknown microRNA families including miR396, miR319, miR399, miR858, miR5083, miR6111 in the Artemisia annua and these microRNAs may influence artemisinin (ART) accumulation[28]. As the depth study of microRNA increases, microRNAs are already being regarded as future gene therapy drugs to be developed[29,30]. MicroRNAs and binding sites of their target genes are evolutionarily conserved in different species[31,32]. Based on their conservation, microRNAs in other species can be predicted by computational methods[33]. The public database such as miRbase[34], PmiREN[35], PMRD[36] makes it easier to carry out research on more species.
MicroRNAs are important pilots of secondary metabolites, and research on regulatory effects of microRNA in S. japonica are more and more necessary. None of the microRNA-related studies have been performed so far in S. japonica, and none are listed in the miRBase. This research used genomic data to explore potential microRNA in S. japonica and analyzed their regulatory effects to related TFs. Then generated the microRNA and respective target profile of S. japonica and performed a comparative microRNA expression study between vegetative and reproductive tissues (leaves, roots, stems, and buds), which may help to understand the microRNA-mediated regulation of transcription factors and further regulate the biosynthesis of CEP in S. japonica.
-
The S. japonica genome data was obtained by our research group (unpublished data)[37,38]. Known microRNAs downloaded from the miRBase database (
www.mirbase.org ) used as a reference microRNA set for predicting the conserved microRNAs[39]. To identify the potential microRNA genes in the S. japonica genome, mature microRNAs were filtered out and aligned with S. japonica genome sequences by the bowtie method (params: -f -m 20 -v 1 -a)[40]. In order to obtain the relatively conservative and maximum possible microRNA, the bowtie process allows only one mismatch, and microRNAs identified within other plants were retained and kept for analysis.Secondary structure analysis of microRNA
-
To check the reliability of 31 putative microRNAs, the 200 bp upstream and downstream of putative microRNAs were predicted secondary structures using MFOLD (
http://unafold.rna.albany.edu/?q=mfold )[41] and the UNAFold website (www.unafold.org/mfold/applications/rna-folding-form.php )[42]. To further improve credibility of candidate microRNA, the filtering standards described by Paul et al.[43] were used to screen for microRNAs with stem-loop structure.Prediction of microRNA target transcription factor family
-
All gene models in S. japonica genome were assigned to TFs families based on PlantTFDB classification. The TF database was obtained from PlantTFDB (http://planttfdb.gao-lab.org/
)[44]. In this study, Targetfinder ( https://github.com/carringtonlab/TargetFinder )[45] was used with default parameters to identify the potential microRNA targets. Potential target genes, including transcription factors were subsequently analyzed.Chromosome distribution and expression pattern analysis
-
The physical location distribution of 47 transcription factors was obtained from the genome data of S. japonica. The TBtools was used to visualize the relationship between 47 transcription factors and four microRNA in S. japonica. RNA-Sequencing (RNA-Seq) data of various S. japonica tissues were obtained by our research group (unpublished data), including leaf, root, stem, and bud. In our study, the expression patterns of 47 transcription factors were analyzed using TBtools software based on the FPKM values.
RNA-seq and qRT-PCR analysis
-
S. japonica plants were obtained from Hubei province, China. Stem, leaf, root, and bud were collected respectively. The samples from six individual plants were pooled as one biological replicate used for RNA-seq analysis and qRT-PCR analysis. The qualified RNAs were used for cDNA library preparation. Agilent 2100 Bioanalyzer was used to detect the quality of the library and DNBSEQ was used for RNA sequencing. The raw data of low-quality, irrelevant sequences were filtered out using fastq1 (v.0.21.0. option: default). Clean sequences were aligned to the reference genome using Star3. StringTie4 was used to assemble the transcripts, and use the merge function of the software to merge the transcript of each sample. The combined transcripts were compared with the known transcripts in gffcompare5 (v. 0.12.1. option: -R-C-K) to obtain new transcripts and new genes. The gene expression statistics were generated using RSEM6 to obtain the number of reads in the transcript of the sample and convert to Fragments Per Kilobase per Million bases (FPKM). The co-expression network was visualized using Cytoscape.
Total RNA of leaves, roots, stems, and buds in S. japonica was extracted according to plant total RNA Extraction Kit (Foregene Biotech, Chendu, China, CodeNo. RE-05011) and reverse-transcribed according to the reagent Kit with gDNA Eraser (Foregene Biotech, Chengdu, China, CodeNo. RT-01032). Specific reverse transcription primers were design for the microRNA using the Stem-loop qRT-PCR method[46]. During reverse transcription, microRNAs were reverse transcribed with individual tubes to get cDNA for qPCR. The cDNA was used as a template to measure gene expression. The specific primers and the GAPDH gene (internal control) are listed in Supplemental Table S1. A quantitative real-time polymerase chain reaction (qRT-PCR) was conducted by a realtime PCR system (QuantStudio5) using TB Green® Premix ExT aq™ II (Vazyme Biotech, Beijing, China, CodeNo. Q711-02). The comparative CT method (2−ΔΔCᴛ method) was used to quantify relative gene expression. Correlation analysis is completed using the online analysis website of Cloudtutu (
www.cloudtutu.com ). -
A public data of mature plant miRNAs was downloaded from miRBase database (
http://www.mirbase.org/ ), and possible microRNA sites was found use genome-wide matching by bowtie (removes non-plant microRNAs and repeats). In total, 31 potential microRNAs were predicted from S. japonica using a rigorous filtration method. Most of the identified S. japonica microRNAs were match to Picea abies (20%), Arabidopsis thaliana (13%), Oryza sativa (10%), and Camelina sativa (10%) (Fig. 1a). Thirty one potential microRNAs belong to ten microRNA families (miR1134, miR11418, miR11592, miR1168, miR156, miR415, miR5021, miR529, miR535, and miR6196) (Supplemental Table S1 & Fig. 1b).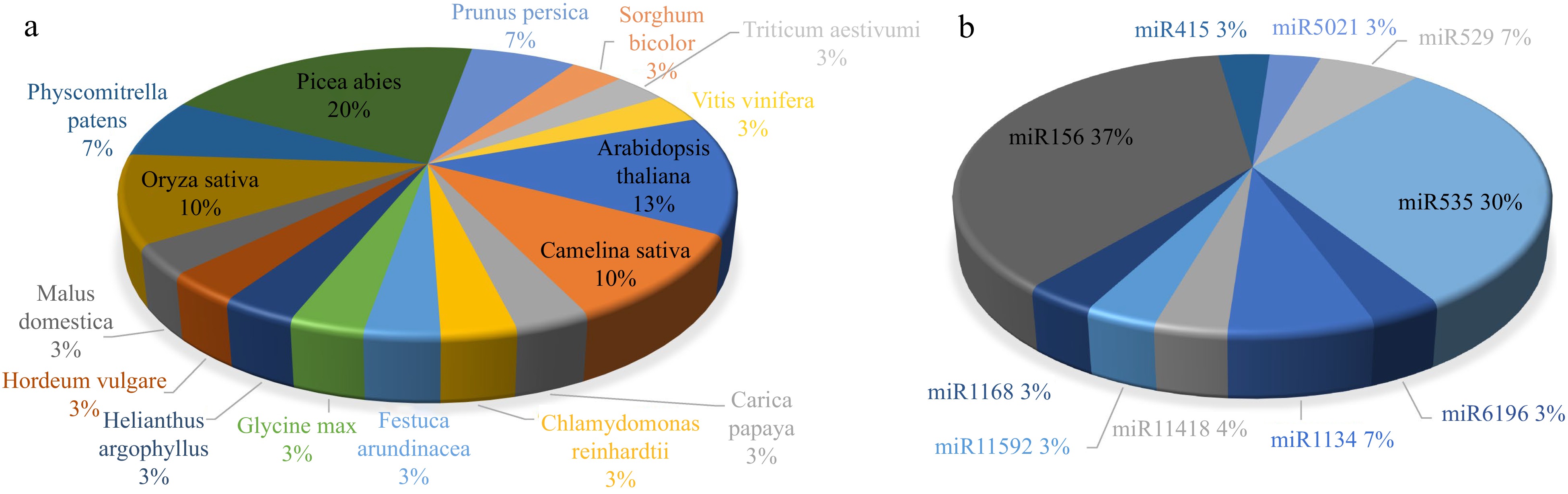
Figure 1.
(a) Potential microRNAs found by bowtie software come from known microRNAs of different species. (b) Potential microRNAs found by bowtie software from known microRNAs families.
Secondary structure of microRNA
-
To further improve credibility of candidate microRNA, secondary structures of 31 potential pre-microRNA were predicted using RNAfold and mfold. The possible precursor (pre-miRNA) sequences of approximately 400 nucleotides (nt) (200 nt upstream and 200 nt downstream to the BLAST hit region) were mined. Seven out of 31 potential pre-microRNA have been screened out with stem-loop structure, which belonging to three microRNA families (miR156, miR535, miR8528) (Supplemental Fig. S2, Table 1). In the current study, the maximum number of microRNA members was found to be present in the miR156 family. The GC content of S. japonica microRNAs had an average of 52.14%. The MFE values ranged from −36.3 to −48.6 with an average of −45.06, while the MFEI values oscillated between 0.86 and 1.25 with an average of 1.16, excluding the possibility of being other microRNAs.
Table 1. Summary of the identified microRNAs from S. japonica.
Name LM
(nt)Query
microRNAChromosome
positionmicroRNA
sequencesAccession Loc LP
(nt)GC
(%)MFEs
(ΔG)MFEI Sja-miR156a 20 ath-miR156h chr10_4372847_4373267 UGACAGAAGAAAGAGAGCAC MIMAT0001013 5' 85 44 −45 1.2 Sja-miR156b 20 ath-miR156h chr11_610113_610533 UGACAGAAGAAAGAGAGCAC MIMAT0001013 5' 84 39 −42.2 1.3 Sja-miR156c 19 cas-miR156g chr4_50504862_50505281 GACAGAAGAGAGUGAGCAC MIMAT0045276 5' 81 48 −48.6 1.3 Sa-jmiR156d 19 cas-miR156g chr5_16609851_16610270 GACAGAAGAGAGUGAGCAC MIMAT0045276 5' 81 48 −48.6 1.3 Sja-miR156e 19 cas-miR156g chr8_1964637_1965056 GACAGAAGAGAGUGAGCAC MIMAT0045276 5' 84 50 −36.3 0.9 Sja-miR535 20 ppt-miR535a chr3_56476014_56476435 GGCGGCGGCGAGGCGGGGG MIMAT0003139 5' 81 56 −46.5 1.0 Sja-miR8528 18 pxy-miR-8528a chr3_55723033_55723451 GGGCGGCGGCGAGCGGGA MIMAT0033748 5' 49 80 −48.2 1.2 Prediction potential targets of microRNAs in S. japonica
-
A total of 1125 transcription factors has been predicted and assigned to 56 transcription factor families based on PlantTFDB classification. The transcription factor families with a large number in S. japonica are NAC, bHLH, MYB, ERF, C2H2, and MYB-related transcription factors (Fig. 2a). Fourty seven target sites of microRNA were obtained using Targetfinder. We found that bHLH (7), SBP (5), TCP (4), TALE (3), and ERF (3) account for a large proportion of 47 transcription factors and may be regulated by microRNAs (Fig. 2b & Supplemental Table S2).
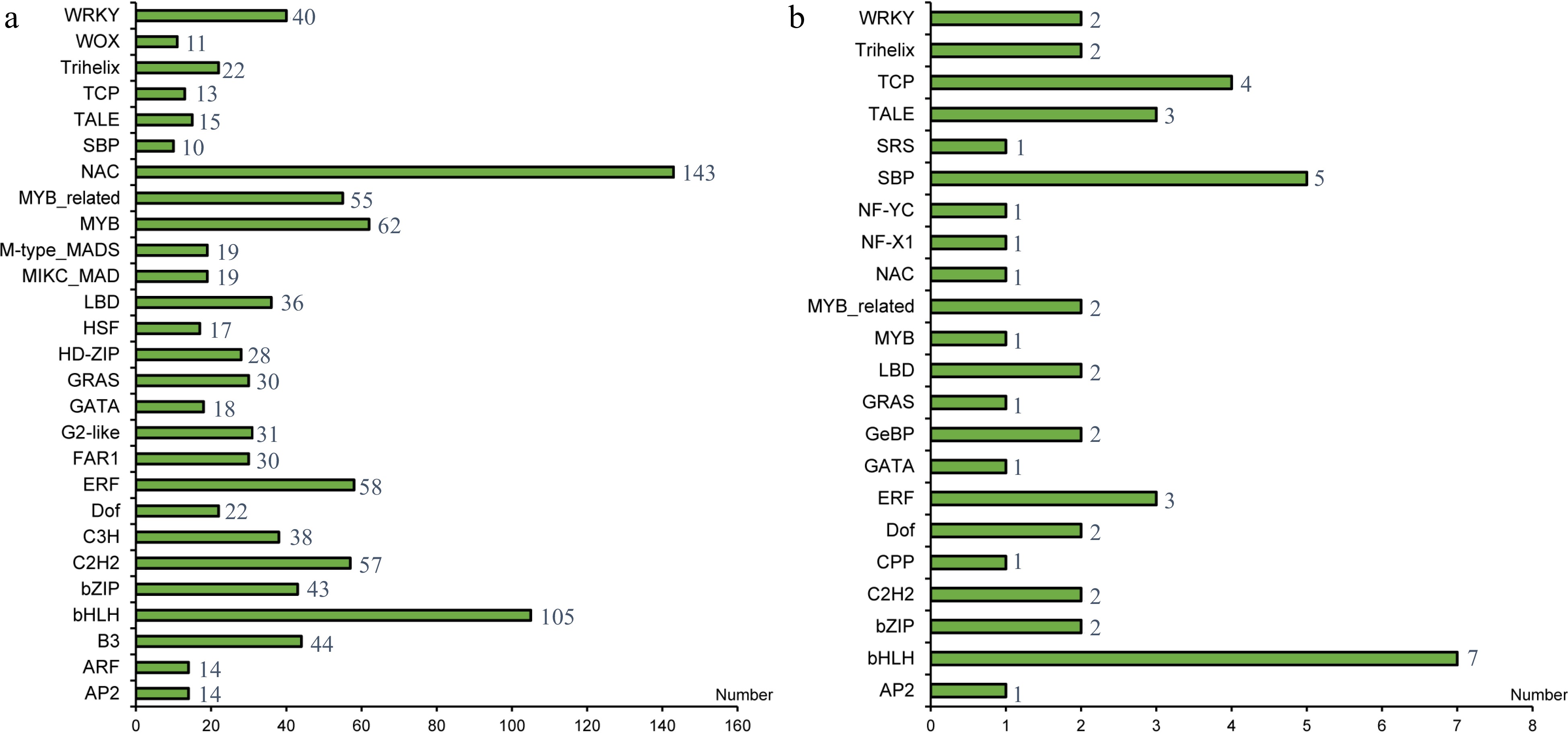
Figure 2.
(a) Number of transcription factor families in S. japonica. (b) Statistical analysis of 47 transcription factors with regulatory relationship with microRNA.
Chromosomal location analysis of microRNA and transcription factors
-
Seven microRNAs and 47 transcription factors were disproportionately distributed on 11 S. japonica chromosomes (Fig. 3). The Sja-miR156b, Sja-miR156c, Sja-miR156d, Sa-jmiR156e, and Sja-miR156f were located in the Chr 4, Chr 5, Chr 8, Chr 10, Chr 11, respectively. The Sja-miR535 and Sja-miR8528 were located in Chr 3. Nine TFs genes on Chr1, four TFs genes on Chr2, two TFs on Chr3, six TFs on Chr4, five TFs on Chr5, two on Chr6, five on Chr7, five on Chr8, four on Chr9, three on Chr 10, and two on Chr11. In addition, a total of 27 potential target proteins of Sja-miR535 were identified, including bHLH (4), TCP (3), ERF (2) and WRKY (2). Sja-miR8528 has target relationship with 11 transcription factors in eight families such as SPB (3). Sja-miR156b has target relationship with five transcription factors, Sja-miR156c has target relationship with four transcription factors, and five target proteins of Sja-miR156d were identified.
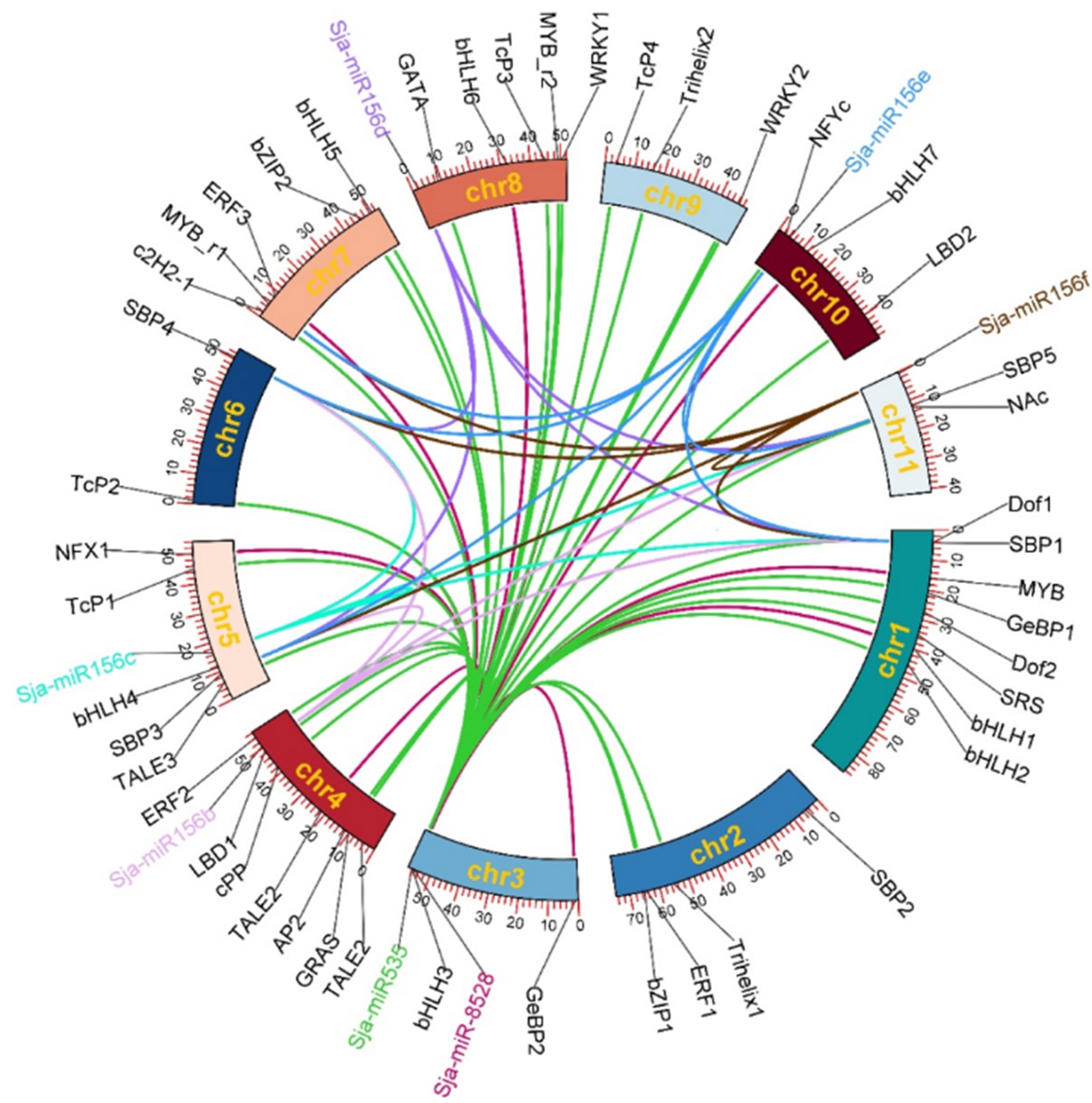
Figure 3.
The chromosome mapping of microRNA and transcription factors.
Tissue expression profiling and co-expression network
-
The expression profiles of 47 transcription factors in different tissues (leaf, root, stem, and bud) of S. japonica were carried out. The heatmap analysis showed that most transcription factors preferentially expressed in roots, and buds. A total of 26 TFs with high expressions were observed in root tissues of S. japonica (Supplemental Fig. S3). Eight TFs with high expressions were observed in bud tissue. Five TFs with high expressions in stem tissue, and six TFs with high expressions in leaf tissue.
The key enzymes involved in CEP biosynthesis pathway have been isolated and identified in other plants (Supplemental Table S3). We identified five NCS, five 6-OMT, and five CNMT genes in S. japonica genome using BLASTP method, which are highly similar with known functional genes at protein level. Heatmap analysis showed that the majority of 47 transcription factors had higher expression level in root, which was consistent with candidate CEP-biosynthetic genes. Co-expression analysis of transcription factors and CEP-biosynthetic genes was constructed using Cytoscape (R > 0.8, P-value < 0.05). The result shown that 32 TFs were strongly correlated with CEP-biosynthetic genes in S. japonica. They may participate in regulating the biosynthesis of CEP or its precursors (Fig. 4).
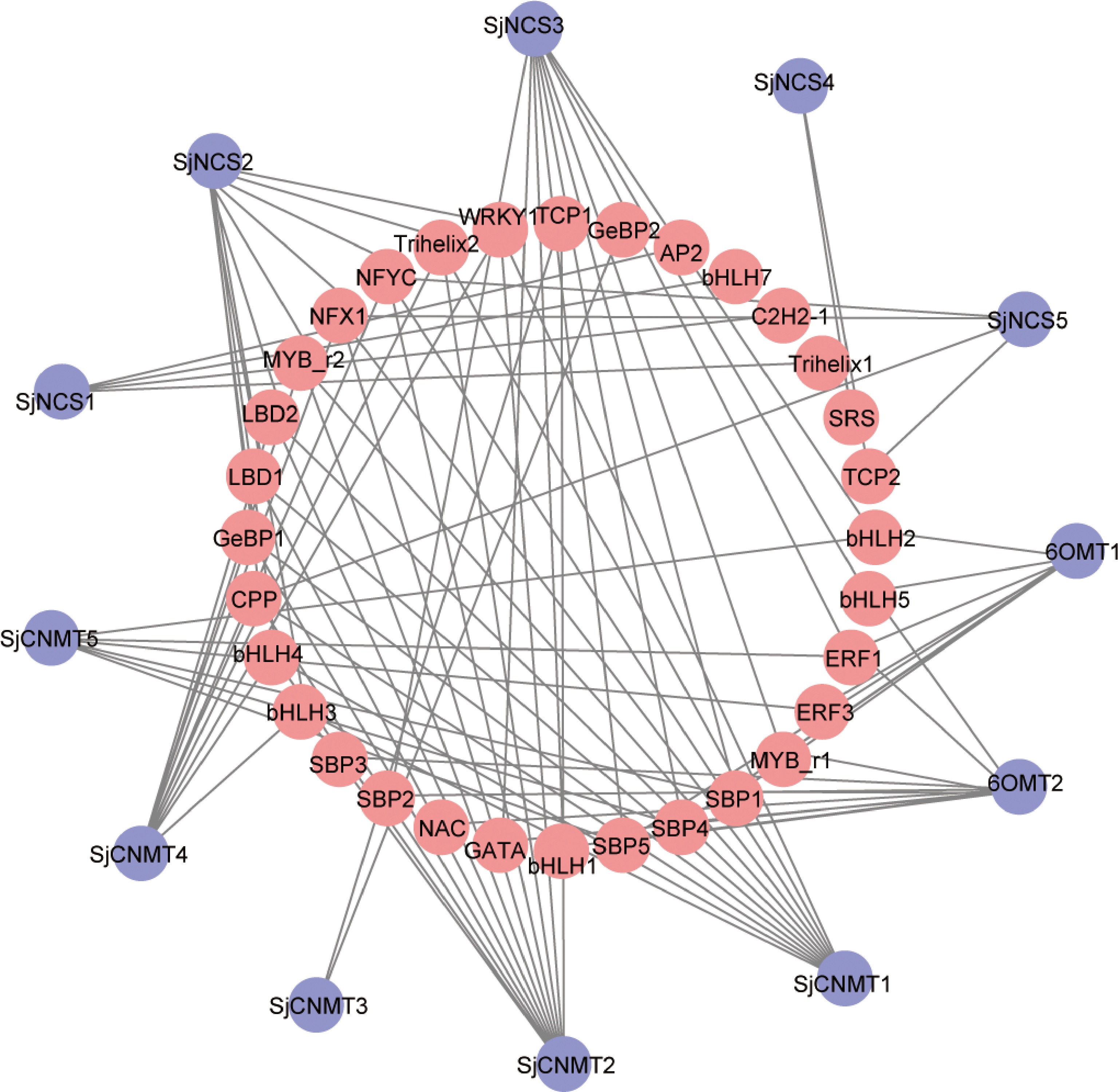
Figure 4.
Regulatory network of transcription factors genes with CEP biosynthetic genes in S. japonica. Red: transcription factors genes. Purple: CEP biosynthetic genes.
qRT-PCR analysis
-
To further investigate the correlation between microRNA and transcription factors based on expression levels, we examined the expression levels of four microRNA and six transcription factors (which were confirmed to play an important role in alkaloid biosynthesis in the previous studies) using quantitative reverse-transcription (qRT-PCR). qRT-PCR results indicated that four microRNA had higher expression levels in S. japonica root. Six transcription factors had higher expression levels in leaf and had lower expression levels in root and bud. The qRT-PCR results of six transcription factors were consistent with those obtained with RNA-seq, with high expressions in leaf (Fig. 5).
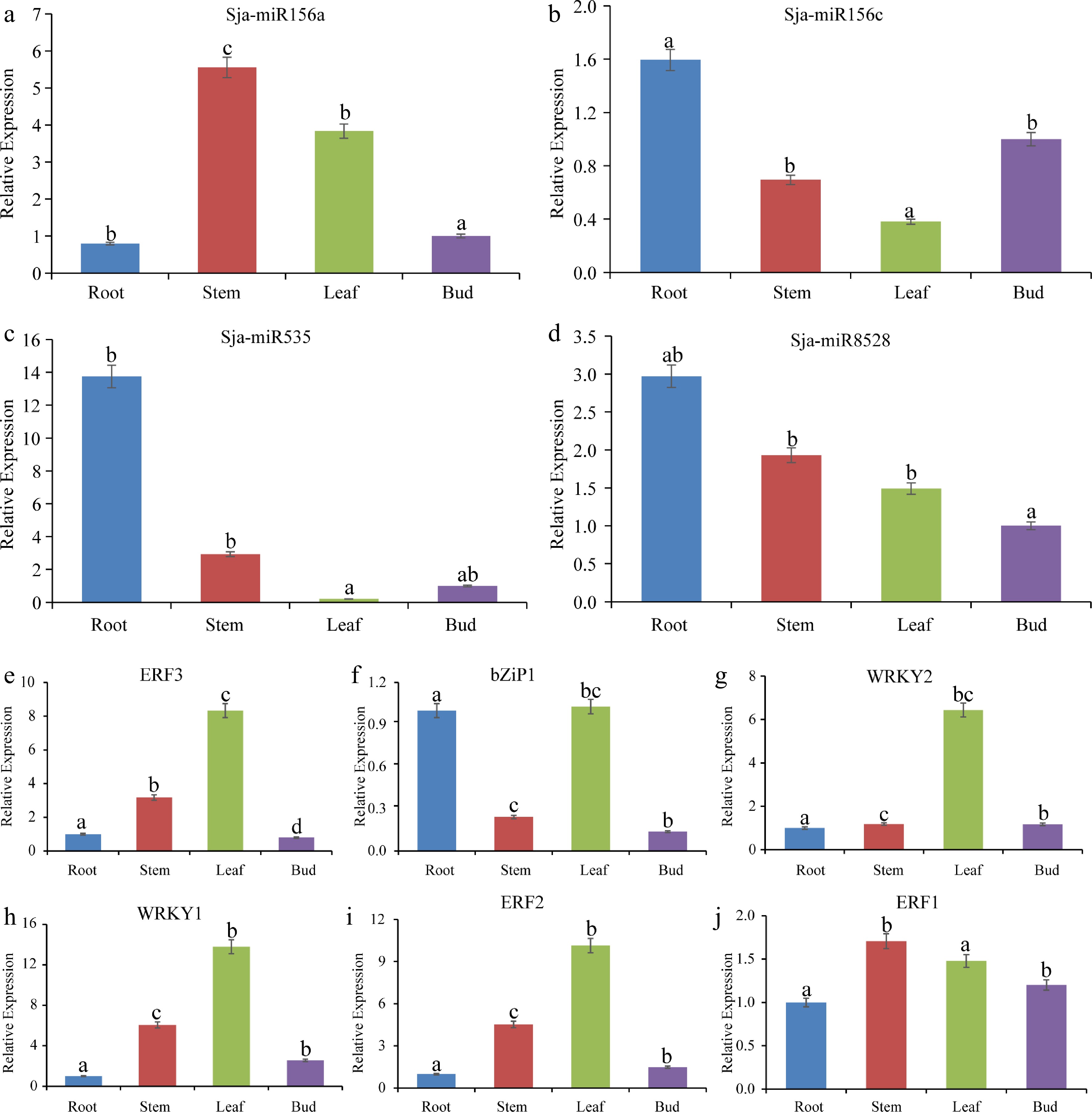
Figure 5.
Expression analysis of microRNA and transcription factors in different tissues by qRT-PCR. (a) - (d) Four microRNA expression levels. (e) - (j) Six transcription factors expression levels. Different lowercase letters indicate significant differences between different treatments (P < 0.05).
Correlation analysis of selected microRNAs and key transcription factors
-
In order to explore the correlation between microRNAs and transcription factors, we performed a correlation analysis on the expression levels of four microRNAs and six transcription factors. The correlation analysis showed that the expression pattern of Sja-miR8528 and six TFs present positive correlation. Additionally, the positive correlation trend has also been verified between the microRNA Sja-miR156a, Sja-miR156c. On the contrary, the Sja-miR535 expression was in negative correlation trend associated with the five TFs expect the SjERF3. Hence, the microRNA Sja-miR8528, Sja-miR156a and Sja-miR156c might played a certain role in participated regulate the program of the cepharanthine by regulating the expression of the transcription factors such as basic (region) leucine zippers (bZIP), WRKY, and ERF (Fig. 6).
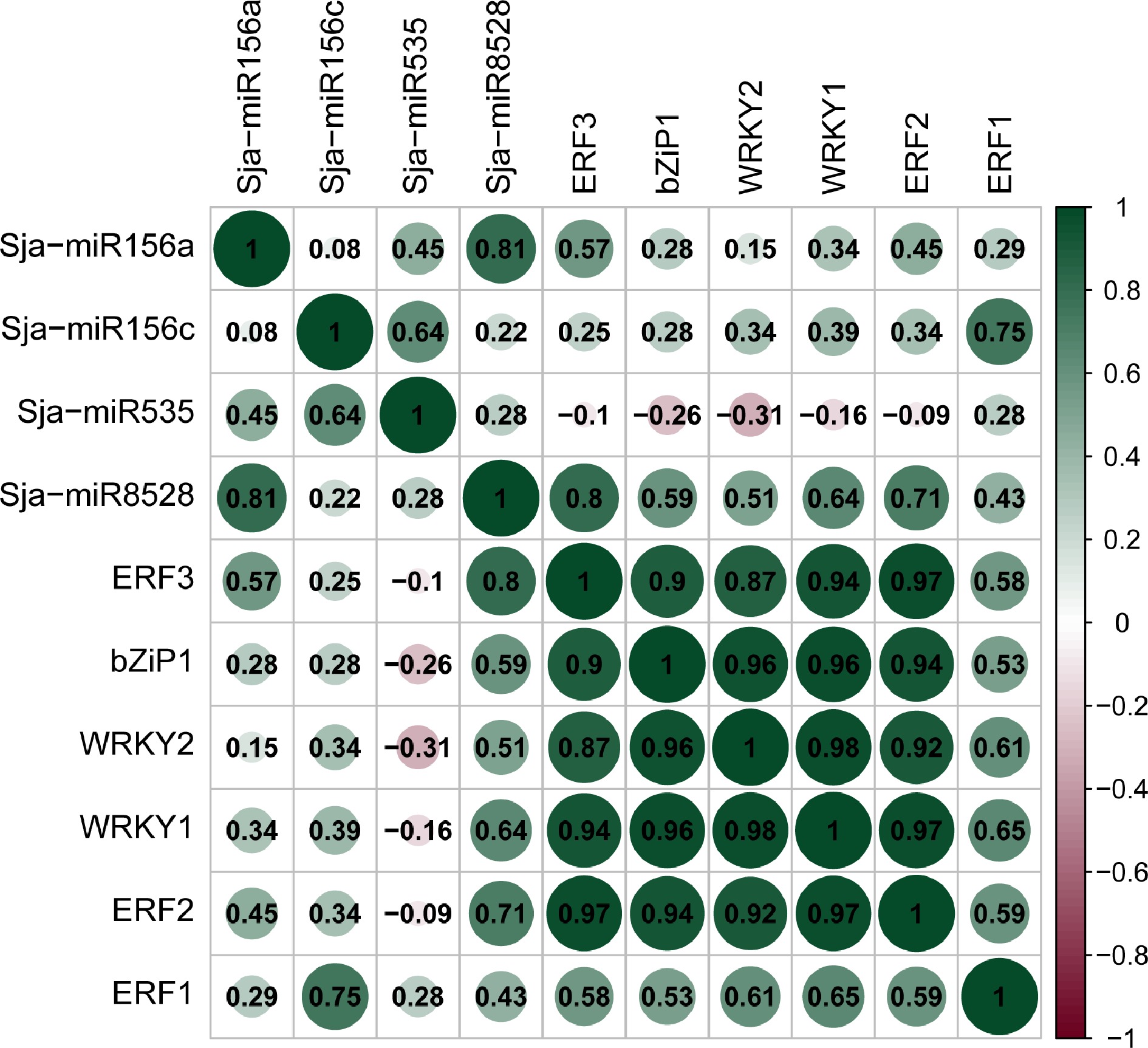
Figure 6.
Correlation analysis of microRNAs and transcription factors.
-
MicroRNAs have been widely found to play regulatory roles in plants, which play an important role in many aspects such as plant development, reproduction and genome reprogramming[47]. In addition to regulation of gene expression, microRNA have also been found to be involved in key steps such as post transcriptional modification, processing and export steps, other regulatory mechanisms may also be involved[48]. MicroRNA was once ignored and thought to function only in non-mammalian species[20]. With the discovery of that microRNA plays an important role in many steps, the research on microRNA will be possible to solve more unsolved mysteries of biology.
In the present study, we predicted seven microRNAs which belong to three microRNA families (miR156, miR535, miR8528). The miRNA156 was found in A. thaliana firstly, with highly conserved regions[49] which are involved in the growth and development process of plants by regulating the SQUAMOSA promoter binding proteins/ SQUAMOSA promoter-binding protein-like (SBP/SPL) gene[38]. It’s reported that there is highest expression of miR156 family in seeds and mature embryos, most of which are expressed during embryonic development, and the expression volume shows a steady upward trend with seed maturity[50]. In the study, 47 TFs have been verified which were regulated by the seven microRNAs, and the six target genes including one SBP gene and two bHLH gene has been predicted of the five members of miR156 family, as well as bHLH (7), SBP (5), TCP (4), TALE (3), and ERF (3) account for a large proportion. The SBP (Squamosa promoter binding protein) is the unique transcription factors in the plant which play a vital role in the regulating plant-type, flowering, yield, hormone signal transduction and stress response[51]. However, in the analysis of chromosome location distribution of seven microRNAs and 47 transcription factors, we found that microRNA and the regulated genes were distributed on all chromosomes, without centralized distribution, and there were intra-chromosome regulation and inter-chromosome regulation. Most of the microRNA sites are located at the edge of chromosome (Fig. 3). Therefore, in this work, the five microRNAs of miR156 family may participate in the growth and development of the S. japonica by regulating the SBP. There are few related research of the miR535 family and the miR8528 family, the research showed that the miR535 family may play a considerable role in fine tuning SPL gene expression in banana[52]. In the study, although 47 target genes of miR535 and the miR8528 were predicted, the function of the two transcription factors need to further verified.
The bZIP transcription factors are characterized by a basic DNA-binding region and an adjacent so-called leucine zipper, enabling bZIP to dimerize[53]. They play an important role in plant signal transduction, response to biological and abiotic stress[54], regulation of growth and development, and biosynthesis of secondary metabolites[55]. The WRKY family is a unique TF family of higher plants and algae, which play important roles in many life processes, particularly in response against biotic and abiotic stress, which was first found in Impoea batatas[56]. It is reported that FtWRKY42 is highly expressed in the root and may perform a similar function with its homologous gene AtWRKY75, which play a role in the formation of lateral and hairy roots. FtWRKY9, FtWRKY42 and FtWRKY60 are highly expressed in fruit and may play an important role in fruit development[57]. The recent research showed that the WRKY was involved in the biosynthesis of Artemisinin[58]. ERF transcription factors regulate fruit ripening by effecting the changes of pigment and softening, and control the longevity of flowers and leaves by regulating the process of senescence and abscission[59]. In the present study, we analyzed the relationship between four microRNA and six transcription factors (bZIP, WRKY, ERF), the correlation analysis showed the Sja-miR8528, Sja-miR156a and Sja-miR156c expression was in positive association with the six TFs, which may mean these microRNAs participate in the process of the growth and development of S. japonica by regulating the expression of transcription factors. On the contrary, the Sja-miR535 expression was in negative associated with the five TFs expect the ERF-2. Thus, the microRNA might play a certain role in regulation of plant secondary metabolism through TFs.
-
In the present study, seven conserved microRNAs were identified in the S. japonica genome, belonging to three microRNA families (miR156, miR535, miR8528), and 47 predictive target transcription factors were obtained. Co-expression network of transcription factors and CEP-biosynthetic genes indicated that microRNAs may involve in regulating the biosynthesis of CEP and its precursors by regulating potential target transcription factors. The tissue expression level of four selected microRNAs (Sja-miR156a, Sja-miR156c, Sja-miR535, Sja-miR8528) and six transcription factors (bZIP, WRKY and ERF) were determined using the qRT-PCR method. Through correlation analysis, it was found that the expression pattern of Sja-miR8528 was positively correlated with the six TFs. Our study is the first to identify microRNAs and their targets in S. japonica and will be helpful for strengthening the research on microRNA-mediated regulation in herbal plants.
This work was supported by introduces the talented person scientific research start funds subsidization project of Chengdu University of Traditional Chinese Medicine (030040017) and Hubei science and technology planning project (2020BCB038).
-
The authors declare that they have no conflict of interest. Chi Song is the Editorial Board member of Medicinal Plant Biology who was blinded from reviewing or making decisions on the manuscript. The article was subject to the journal's standard procedures, with peer-review handled independently of this Editorial Board member and his research groups.
-
# These authors contributed equally: Hanting Yang, Zhaoyu Liu
- Supplemental Fig. S1 Cepharanthine biosynthesis pathway. The red dotted line shows the unidentified enzyme.
- Supplemental Fig. S2 Secondary Structure of MicroRNA.
- Supplemental Fig. S3 Expression patterns of 47 transcription factors in different tissues and CEP biosynthetic pathway genes in S. japonica.
- Supplemental Table S1 List of specific primer sequences in this study.
- Supplemental Table S2 47 potential regulating relationship were predicted between microRNA and transcription factors.
- Supplemental Table S3 Protein sequence that has catalytic activity and participates in CEP biosynthesis in other species.
- Copyright: © 2023 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Yang H, Liu Z, Yu C, Song C, Wang C. 2023. Expression relationship between microRNA and transcription factors in Stephania japonica. Medicinal Plant Biology 2:7 doi: 10.48130/MPB-2023-0007 

Expression relationship between microRNA and transcription factors in Stephania japonica
- Received: 13 December 2022
- Accepted: 06 May 2023
- Published online: 02 August 2023
Abstract: MicroRNAs are a group of endogenous small non-coding RNAs, they can guide silencing complex to degrade mRNA or hinder its translation by pairing with the target gene mRNA base. In this study, applying high stringent genome-wide computational-based approaches, a total of 31 putative microRNAs were predicted in Stephania japonica. After strict secondary structure screening, seven microRNAs with high reliability were identified and classified into three microRNA families. Using Targetfinder tool, a total of 47 potential target transcription factors of microRNAs were identified, including bHLH, SBP, TCP, ERF transcription factors. The co-expression network of transcription factors and three important cepharanthine-biosynthetic genes shown that microRNAs might be involved in regulating the biosynthesis of cepharanthine and its precursors by regulating potential target transcription factors. Additionally, correlation analysis of microRNAs and transcription factors indicated that the tissue expression patterns of Sja-miR8528, Sja-miR156a and Sja-miR156c showed positive correlation with six transcription factors, while Sja-miR535 showed a negative correlation with five transcription factors. This is the first study explored putative microRNA and analyzed their predictive target transcription factors in S. japonica, providing useful information for future microRNA research in herbal plants.
-
Key words:
- MicroRNA /
- Transcription factor /
- Stephania japonica /
- Correlation analysis