










-
Foxtail millet (Setaria italica L.), belonging to the poaceae family, was first cultivated in the green foxtail (S. viridis) about 16,000 years before present[1−3]. It has since become the most prominent miscellaneous cereal in northern China, especially in arid or semi-arid area[4,5]. Foxtail millet grains are rich in nutrients, including polyphenols[6], organic acids[7], vitamin E, and carotenoids[8], a variety of essential amino acids, and high-quality protein[9]. Additionally, it is a good source of trace elements such as zinc and iron[10]. Its bran is also rich in linoleic and linolenic acids[11,12], and is an excellent crude fiber source, which helps with intestinal digestion and promotes digestive health[13]. Recently, foxtail millet's small diploid genome, self-compatibility, strong stress tolerance, and low repetitive DNA content have made it a model crop for C4 monocot studies[2,3,14]. However, despite foxtail millet has many advantages, the planting area and total yield have been a declining trend, and its yield per unit area is significantly lower compared to other staple food crops such as maize, wheat, and rice. So attention should be paid to improving foxtail millet yield and quality.
Seeds are reproductive units of flowering plants, and the ultimate goal of seeds is to successfully establishing the progeny. The development of seed is not only the growing of the embryo, but also involves close coordination of different tissues, including seed coat, embryo, and endosperm which nourishes the embryo[15]. In cereals, the endosperm contains starch, protein, and lipids, accounting for approximately 42.5% of global food calories for humans[16]. In brief, seed development can be described in three stages: cellular division, morphogenesis, and maturation[17]. Morphogenesis encompasses all processes that form and develop the different parts of mature seed, and it is during this stage that resources are allocated[18]. Maturation is a physiological process that comes to an end when the seed begins the dormant state[19]. However, the developmental process of foxtail millet grains is not clearly defined morphologically. Therefore, the morphological description of foxtail millet grain development is particularly important.
Grain development is an important process in foxtail millet growth, related to seed setting, grain weight, yield, and quality[20]. This process is influenced by various factors including the delivery of photosynthetic products, transportation of stored substances, transport tissues and physiological activities of the grains themselves, regulation of various plant hormones, and restriction of various environmental factors[21]. The interaction of phytohormones, activity of starch biosynthesis enzymes, levels of polyamines, and synthesis and translocation of assimilates all play essential role in grain development[22−24]. In recent years, numerous genes and signaling pathways have been discovered to play a role in regulating grain development. The endosperm specific bZIP transcription factor (TF) O2 can bind the O2-box and transactivates the 22-kDa α-zein protein[25]. bHLH transcription factor OPAQUE11 (O11) can directly regulate the expression of various carbohydrate metabolizing enzymes, affecting carbohydrate accumulation, amino acid metabolism, and the transcription of stress response genes[26]. ZmNAC11 and ZmNAC29 are also involved in grain development as they directly activate the expression of ZmEXPB15, which promotes early grain development in maize[27]. Additionally, ZmNAC128 and ZmNAC130 are endosperm-specific TFs that affect starch biosynthesis and 16-kDa γ-zein content[28]. The specific expression of OsSUT1 in the aleurone layer indicates a role for this gene in sucrose uptake, and the spatial distribution of AGPL2 and AGPS2b is associated with the development of starch granules in the grain[29]. TA2 encoding DNA demethylase OsROS1 in rice, restricts the aleurone layer cell number by mediating DNA demethylation, providing means to improve rice nutrition[30]. Phytohormones can also affect seed development and regulate grain filling. DEP1/qPE9-1 can promote starch accumulation and prolong the grain filling process through auxin and cytokinin[31]. Recently, a new pathway, the sugar-auxin crosstalk signaling pathway, has been discovered that is involved in sugar metabolism and carbon partitioning[32]. Ethylene and abscisic acid have been shown to regulate the activity of enzymes involved in starch synthesis, ultimately affecting the grain filling process[33,34].
However, compared to major cereal crops, the molecular mechanisms and regulatory networks of grain development in foxtail millet are rarely reported and only a limited number of genes involved in seed development have been identified. Therefore, elucidating the regulatory networks and key regulators during foxtail millet grain development is important, which will help us to understand the molecular basis of foxtail millet grain yield and quality. LOOSE PANICLE1 (LP1) is known to encode a novel WRKY transcription factor, and the lp1 mutant can affect panicle development and seed size in foxtail millet[35]. F128 is the first seed-specific promoter gene reported in foxtail millet, encoding protein is likely a protease inhibitor / seed storage protein / lips-transfer protein. F128 was specifically expressed in immature and mature seeds and gradually decreased with seed maturity, and was no longer expressed in mature seeds of 25 DAP[36]. TRANSPARENT TESTA GLABRA1 (SiTTG1) is a WD40 repeat transcription factor, is able to induce the expression of genes associated with the accumulation of seed fatty acids and storage proteins, thereby plays an important role in seed metabolite production[37]. SiLEA14 is a late embryogenesis abundant (LEA) proteins, its transcription level was gradually increase with seed maturation, indicating that it had a potential role at the maturation and drying stages of seed development in foxtail millet[38]. SiDL is a member of the YABBY family, and the discovery that over-expression of SiDL leads to delayed flowering and reduced seed size confirms its function in foxtail millet seed development[39]. The transcriptional levels of genes SiPSY1/2/3, SiPDS, SiZDS, SiZ-ISO, SiCRTISO, SiLCYB, SiLCYE, and SiHYD, that play a role in the carotenoid pathway have been extensively studied. These studies have shown that up-regulated expression of SiPSY1 accompanied by down-regulated expression of SiCCD1 is the key to increased carotenoid accumulation in foxtail millet seeds[40]. The expression of SiGRAS41 increased gradually with the development of millet seeds, while the expression of SiGRAS01 decreased with fruit maturation[41]. SiMADS34 has been found to regulate inflorescence structure and panicle development in a variety of regulatory pathways[42]. Ring-type E3 ligase SGD1 and its E2 partner SiUBC32 control grain development by regulating grain weight and grain size in foxtail millet[43].
Although several genes involved seed development have been identified, few key regulatory genes have been cloned, and the molecular mechanisms and regulatory networks in foxtail millet seed development largely remain unclear. In recent years, several studies have used RNA-seq transcriptome analysis or metabolomics analysis to identify gene expression profiles in foxtail millet seed development[44−49]. Wang et al.[44] first identified the gene expression profiles of foxtail millet at five different developmental stages, including at 7, 14, 21, 28, and 35 days after anthesis (DAA), using RNA-seq analysis. These studies have identified a number of genes that exhibited dynamic or enriched expression patterns in foxtail millet seed development and highlighted many potential regulators that may be involved in seed filling and the accumulation of seed metabolites. However, detailed morphological analysis and description regarding the foxtail millet seed development process, is an important step in the study of the seed development process. In previous studies, transcriptome analysis of the seed development process was carried out according to the date of flowering, but detailed morphological analysis and description of the specific seed development process have not been provided. To improve our understanding of dynamic gene expression profiles of seed development, we conducted detailed morphological analysis and description of the development process of foxtail millet seeds, including the ovule stage of before pollination that have not been identified in previous studies. In our study, morphological analyses and definition of foxtail millet grain development were performed, and the dynamic regulatory network of grain development was depicted by RNA-seq analysis of grains at four developmental stages. Our studies have not only provided morphological and cytohistological analyses of grain development, but also developed a temporal transcriptome based on high-throughput RNA-seq atlas of grain development, and this study will provide a precious genetic resource for understanding grain developmental process for the future.
-
The cultivars 'Jingu 21' of foxtail millet were used in this study. Materials were planted and grown at Shenfeng experimental fields in Jinzhong, Shanxi, China (37°25′ N, 112°35′ E) from May to October in 2022. The surfaces of florets at anthesis were tagged using a marker pen to denote flowering dates. For morphological analysis, samples were collected daily over a period of 30 d of foxtail millet grain development. For staining assay, paraffin section analysis, and RNA-seq, samples from four stages (ovule stage, milk stage, dough stage, maturity stage) were collected. Three replicates were generated for each stage, and each replicate was made up of a mixture grain of five spikes. Samples were snap frozen in liquid nitrogen before processing and stored at −80 °C.
Confocal microscopy
-
For confocal microscopy, samples were fixed with 2.0% paraformaldehyde and 2.0% glutaraldehyde fixative (Coolaber, Beijing, China) for 48 h, rinsed three times in PBS buffer, then dehydrated in alcohol concentrations (20%, 50%, 70%, 80%, 90%, 95%, 100%, 100%, 100%) for 1 h, respectively. After completing the above operations, the gradient dehydration samples were immediately mounted in a mixture of benzyl benzoate (MACKLIN, Shanghai, China) and benzyl alcohol (MACKLIN, Shanghai, China) (2v:1v) and photographed using a confocal microscopy (DMI 8, Leica, Germany), or placed into the mixture above and stored away from light in refrigerator at 4 °C. The confocal microscopy system excitation wavelength is 568 nm and emission wavelength is 550−630 nm.
Stereo microscopic analysis
-
A stereo microscope (SZX16, Olympus, Japan) was used to record the daily growth changes of Jingu 21 developing grains throughout 1−30 DAP. For staining starch or lipid, median transverse sections of foxtail millet grains were manually cut with a sharp razor and immersed into solutions of Lugol's iodine, 0.1% (w/v) Evans blue, 0.1% (w/v) toluidine blue, and 0.5% (w/v) Sudan IV (dissolved in Chloroform), respectively. After staining, the hand-sectioned grains were viewed under microscope (BX51, Olympus, Japan), and immediately photographed.
Paraffin section observation
-
Paraffin section analyses were performed according to a method as described with a few modification[50]. Samples collected at four stages (ovule stage, milk stage, dough stage, maturity stage) were first fixed in 2.0% paraformaldehyde and 2.0% glutaraldehyde fixative. Fixed samples were washed, dehydrated, embedded into wax and cut into 10 μm-thick sections. Samples were redyed with Safranin O-Fast Green (Phygene, Fuzhou, China) or toluidine blue O (Coolaber, Beijing, China) and observed with microscope (BX51, Olympus, Japan).
Staining procedures
-
Hand sections from the central region of foxtail millet grains from ovule stage to maturity stage were made using sharp razor. Evans blue, toluidine blue, Sudan IV, and Lugol's iodine staining (Coolaber, Beijing, China) were performed according to the method as described with a few modification[51−53]. Sections were soaked in 0.1% Evans blue, 0.1% toluidine blue, and Lugol's iodine for 10 min, 4 h, and 5 min, respectively, and immediately washed with distilled water. For Sudan IV staining, sections were stained with 0.5% Sudan IV for 24 h, washed with 70% alcohol and immediately placed in distilled water. All samples were under the microscope for observation and photography (BX51, Olympus, Japan).
RNA-seq analysis
-
RNA-seq was performed by Shanghai OE Biotech Co., Ltd. (Shanghai, China). Raw reads were trimmed and mapped by Trimmomatic (version 0.36)[54] and HISAT2 (version 2.2.1.0)[55] to the reference genome of foxtail millet[56], respectively. DESeq package was used to analysis the differential expression of genes (using BaseMean value to estimate expression)[57]. Fold change > 2 and adjusted P-value < 0.05 as the criteria for differentially expressed genes (DEGs) screening. Hierarchical cluster, Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis were performed by the OE Biotech cloud analysis tools (
https://cloud.oebiotech.cn )[58].Quantitative RT-qPCR
-
Total RNA extracted by the FlaPure Plant Total RNA Extraction Kit (Genesand Biotech, Beijing, China). Four hundred ng total RNA was used to generate the cDNA by the Union Script First-strand cDNA Synthesis Kit (Genesand Biotech, Beijing, China). qPCR was carried out using the SYBR Green Super Mix (Mei5bio, Beijing, China) and Bio-Rad CFX96 (Bio-Rad CFX96, BIORAD, USA). The primers were listed in Supplemental Table S6. The gene expression was normalized to housekeeping gene Actin 1 (Seita.8G043100).
Statistical analysis
-
Prism 7.0 (GraphPad Software, INC., USA) was used for statistical analysis. The statistical significance were evaluated by one-way analysis of variance (ANOVA) followed by Duncan's multiple comparison range test and were considered significant at p-values < 0.05 level.
-
To investigate the dynamic morphology of foxtail millet grain development after pollination. We utilized a stereo microscope to record the daily growth changes of foxtail millet grains developing during 30 d after pollination. We observed that the growth period of foxtail millet grain mainly occurred at 1−15 DAP, while grain development was initially characterized by longitudinal development before 4 DAP, and then transferred to lateral growth at 5−15 DAP. However, it was noted that the transverse and longitudinal growth was slower compared with the growth before 15 DAP (Fig. 1, Supplemental Fig. S1). We found that foxtail millet grains began to turn yellow at 8 DAP (Fig. 1b), which may be responsible for the accumulation of carotenoids and flavonoids during this process. Additionally, we also observed that the grains gradually became translucent, which could be caused by dehydration and crystallization of starch and storage proteins after 24 DAP (Fig. 1b). To investigate the development patterns of grain growth, area, perimeter, length, width, and length/width ratio of foxtail millet grains were measured everyday throughout the 30 d development period (Supplemental Fig. S1). It was observed that elongation of foxtail millet grains occurred sharply from 1 DAP, and the grain length and width reached their maximum at 15 DAP (Supplemental Fig. S1c). The grain perimeter and area were found to be consistent with the grain length and width, and also reached their maximum value at 15 DAP (Supplemental Fig. S1a, b). Grain change showed that the grain grew rapidly longitudinally in 1−4 DAP, consequently, the maximum length/width ratio was seen at 4 DAP, and lateral growth began after 5 DAP and aspect ratio change was not obvious after 15 DAP (Supplemental Fig. S1d). These apparent morphological results were consistent with the statistical data obtained from 1-30 d of seed development.

Figure 1.
Dynamics of foxtail millet grain development. (a) Time series of kernel development from 1 to 30 DAP; (b) Time series of grain development from 1 to 30 DAP without kernel hull. Numbers below denote the DAP. (a) Scale bars = 1 mm, (b) Scale bars = 500 μm.
To describe the developmental morphology of the ovule before pollination. The young spikes containing all developmental stages of ovules and florets were selected to examine the morphology of the developing florets and characterize the ovule structure using confocal microscopy (Fig. 2). The developmental process can be divided into eight periods according to appearance and morphological of foxtail millet spikelets (Fig. 2b). Morphological analysis of ovule development before pollination was also defined as eight stages, starting with the initial stage of ovule primordium (Fig. 2c, d). Similarly, the development of ovules was also defined as eight stages in cell morphology by confocal microscopy (Fig. 2e−l). The ovule developmental processes were divided into eight stages: Ovule primordium, megassporocyte, Functional megaspore (FM), Mono-nucleate embryo sac (Mnes), Two-nucleate embryo sac (Tnes), Four-nucleate embryo sac (Fnes), Eight-nucleate embryo sac (Enes), and Mature embryo sac (Mes). The cell morphological characteristics of foxtail millet ovule before pollination are similar to those of the embryo sac in rice[59].
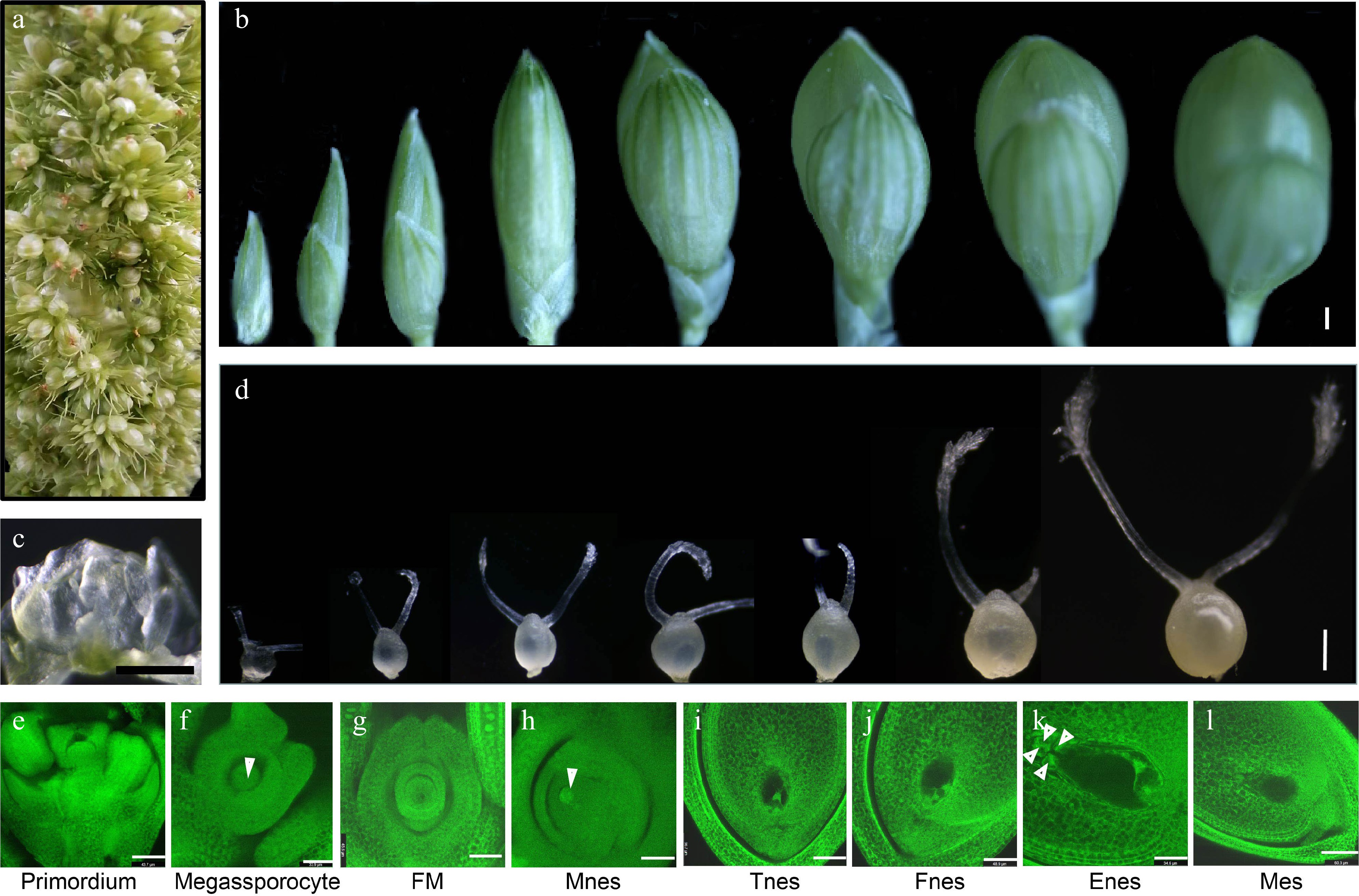
Figure 2.
Morphological analysis of the developing ovules and spikelets in foxtail millet before pollination. (a) The young foxtail millet spike contains all developmental stages of ovules and florets. (b) Appearance morphological analysis of florets development in foxtail millet. (c) Morphology of the ovule primordia in foxtail millet. (d) Morphology of ovule development in foxtail millet before pollination. (e)−(l) The structure of ovules observed by confocal microscopy in foxtail millet before pollination. FM: Functional megaspore; Mnes: Mono nucleate embryo sac; Tnes: Two-nucleate embryo sac; Fnes: Four-nucleate embryo sac; Enes: Eight-nucleate embryo sac; Mes: Mature embryo sac. (b)−(d) Scale bars = 10 μm, (e) Scale bars = 43.7 μm; (f) Scale bars = 33.9 μm; (g) Scale bars = 45.6 μm; (h) Scale bars = 45.3 μm; (i) Scale bars = 56.7 μm; (j) Scale bars = 48.9 μm; (k) Scale bars = 34.6 μm; (l) Scale bars = 60.3 μm.
We defined the development process of foxtail millet grains, referring to relevant research on the seed development process of rice, maize, wheat and other graminaceous cereal crop[60−66]. 1−5 DAP is defined as the watery stage, characterized by grains that can be easily squeezed to release transparent liquid. 6−11 DAP is defined as the milk stage, marked by grains gradually turning yellow and releasing milky white pulp substance when squeezed. 12−15 DAP is defined as the soft dough stage, and the green color of the seed coat begins to fade, and the grains squeeze out nutrients that have the consistency of dough. 16−22 DAP is defined as the hard dough stage, and the grains become hardened and could not be easily crushed, and the embryo grows to its maximum at this stage. 23−30 DAP is defined as the mature stage, and the grains become hard and transparent due to the loss of water, and the embryo becomes clearly visible and smaller. The mature ovule before pollination is defined as the ovule stage. To ensure the accurate collection of four different development stages, we systematically summarized the morphological characteristics of foxtail millet seed development (Supplemental Table S1). Our results provide detailed cytological and morphological support for the dynamic developmental process of foxtail millet grain, and offer reference for future research on specific periods of foxtail millet grain development.
Accumulation of storage products during foxtail millet grain development
-
Grain development is a critical process of nutrient accumulation, and nutrients were mainly accumulated in starchy endosperm and the aleurone layer[67]. Starchy endosperm mainly accumulates starch, while the aleurone layer primarily accumulates storage proteins, lipids, vitamins, and minerals[30,68]. Various staining methods were used to analysis the accumulation of storage products during grain development. Evans blue staining showed that programmed cell death (PCD) was initially present in the periphery zone of the mature ovule stage (Fig. 3a1), and extended to the entire starchy endosperm at the milk stage, dough stage, and maturity stage (Fig. 3a2−a4). In contrast, at the maturity stage, cells of the embryo-surrounding region (ESR) remain viable, as indicated by the absence of Evans blue staining (shown by light colour; Fig. 3a4). This suggests that active starch is still present in the starchy endosperm, and that apparently disrupting cytoplasmic membrane integrity does not affect starch biosynthesis. The most plausible explanation is that the nuclear, plastid, mitochondria, and ER membranes remain fully functional. The staining results of toluidine blue were similar to those of Evans blue (Fig. 3b1−b4). We observed a gradual increase in starch accumulation in the starchy endosperm, where starch is the main component (shown by brown/black; Fig. 3d1−d4, e1−e4). As shown in transversal sections of husked grain (Fig. 3e1−e4), lipids were predominantly stored in the aleurone layer and embryo (shown by orange-red; Fig. 3c1−c4, e1−e4). Through staining analysis of the developing grains, we also observed that starch grains changed from irregular state to regular crystalline starch (Fig. 3). Crystallization of starch and storage protein resulted in the formation of translucent endosperm, as has been reported[51,52].
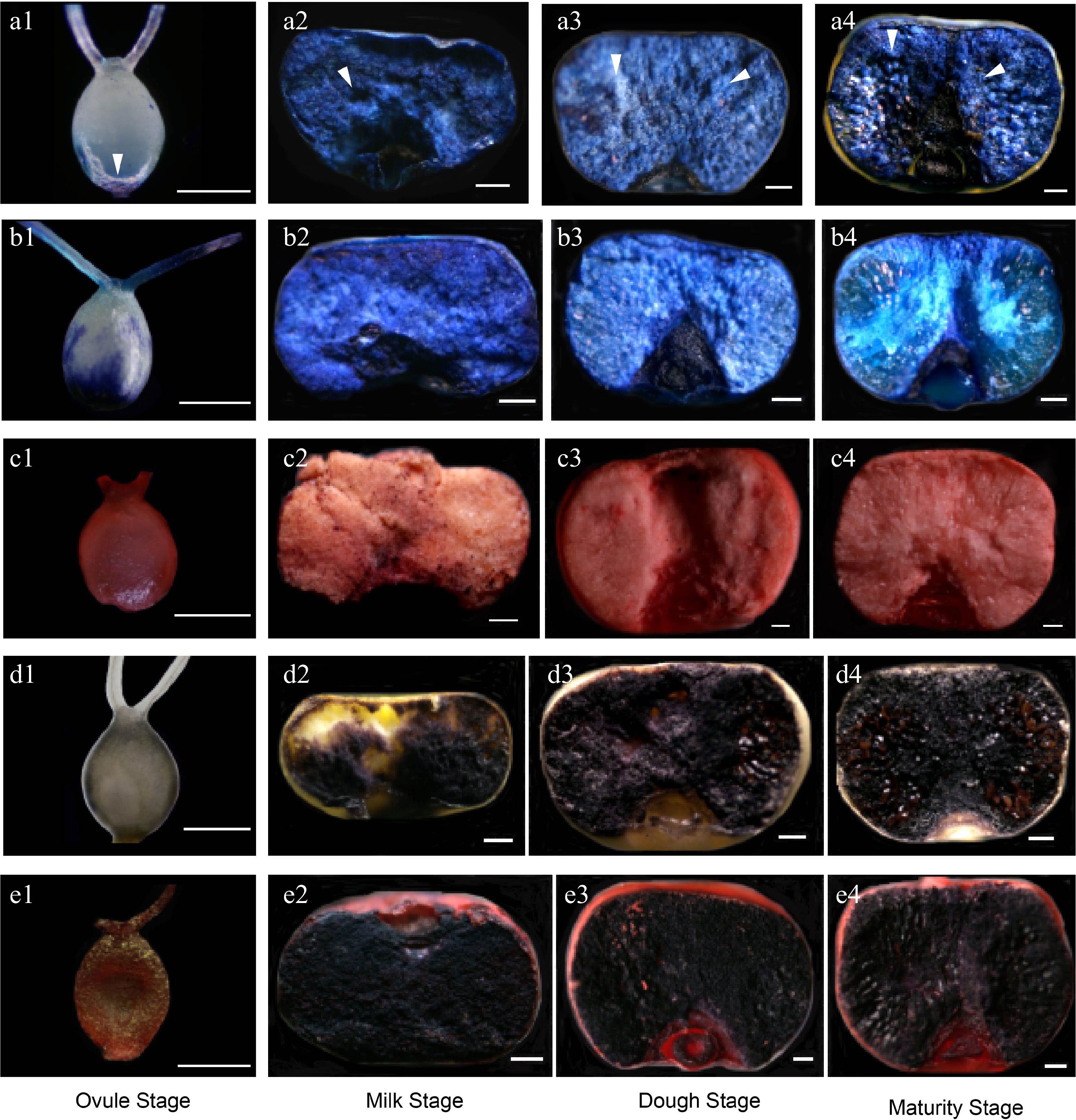
Figure 3.
Storage product accumulation in the foxtail millet hulled grain by staining analysis. (a1)−(a4) Transversely sectioned hulled grain stained with Evans blue to analyze the PCD of developing grain. (b1)−(b4) Transversely sectioned hulled grain stained with toluidine blue to observe endosperm storage product accumulation. (c1)−(c4) Transversely sectioned hulled grain dyed with Sudan Red IV to analyze the lipids accumulation. (d1)−(d4) Transversely sectioned hulled grain stained with Lugol's iodine to analyze the accumulation of starch. (e1)−(e4) Transversely sectioned hulled grain dyed with Sudan Red IV and Lugol's iodine to analyze the accumulation of lipids and starch. Scale bars = 10 μm.
To further observe the cytomorphology characteristics of foxtail millet grain development, two staining methods were used to make paraffin sections to observe the complete morphological changes of seeds from the ovule stage before pollination to the maturity stage. Cytological morphology of mature ovule before pollination is shown (Supplemental Fig. S2a, e), and morphology of mature ovule and mature pollen grains can be observed. Milk stage is the filling stage of foxtail millet grain, it is a process of endosperm starch accumulation and differentiation (Supplemental Fig. S2b, 2f; Fig. 3a2−e2). Dough stage is the process when the accumulation of foxtail millet grains storage products has been completed, at this stage foxtail millet grain has formed the complete embryo (Supplemental Fig. S2c, g; Fig. 3a3−e3). At the maturity stage the endosperm starch of the foxtail millet grains entered the maturation process, and the seed begins to lose water and become translucent (Supplemental Fig. S2d, 2h).
Global gene expression profiling of grain development in foxtail millet
-
To explore the molecular mechanisms of grain development, transcriptome profiling was used to detect genes potentially involved in foxtail millet grain development. According to the stage of grain development, samples used for RNA-seq were collected at four developmental phases, namely, the ovule stage before pollination (ovule stage), the milk stage after pollination (milk stage), nutrient storage stage after pollination (dough stage), and the grain maturity stage (maturity stage) (Fig. 4a). In this study, we collected 12 samples for RNA-seq analysis, with three biological replicates for each stage (Supplemental Table S1). Clean reads of the 12 samples ranged from 44.88 to 50.60 million. 94.78% to 99.02% of the clean reads were mapped to the reference genome (
http://foxtail-millet.biocloud.net/home ), and 83.14% to 95.09% of the reads were uniquely mapped (Supplemental Table S2). These data indicate that the quantity and quality of reads were sufficient to perform quantitative analysis of gene expression.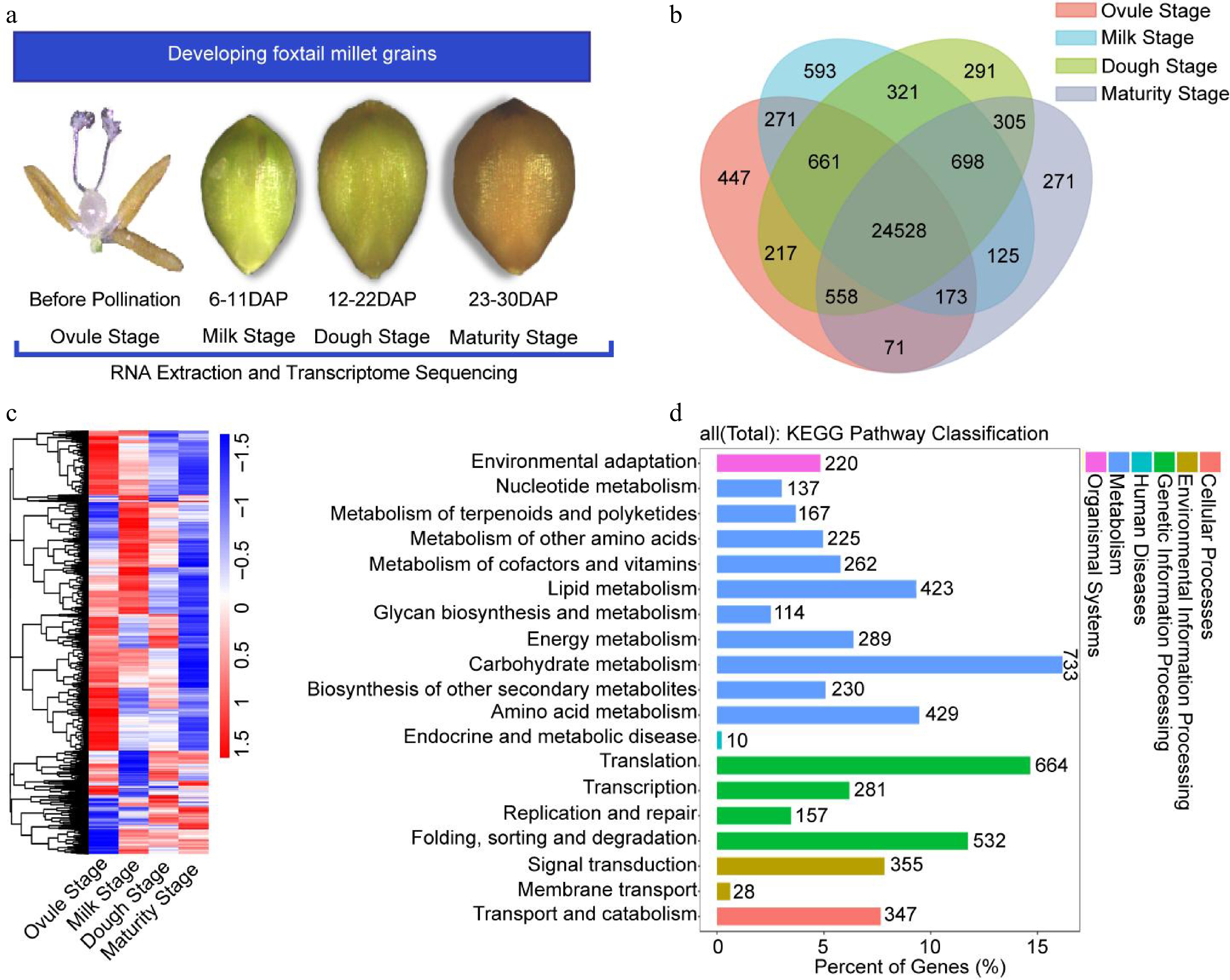
Figure 4.
Global gene expression profiling and KEGG pathway analysis. (a) Schematic overview of the experimental approach. RNA-seq was performed for four developmental stages. (b) Distribution of genes in four samples. (c) Hierarchical cluster analysis of genes expressed during four developmental stages. (d) KEGG pathway classification of the all expression gene in four developmental stages.
To understand the relationships between different groups, we used principal component analysis (PCA) on the full data set, which can visually demonstrate transcriptional features and developmental similarities (Supplemental Fig. S3a). To evaluate the global gene expression profiles of different samples, gene expression levels were assessed using fragments per kilobase of transcript per million mapped reads (FPKM), based on normalized read counts. Across these four stages, a total of 29,530 genes were measured (Supplemental Data Set S1). Compared to the dough and milk stages, fewer genes were detected in the remaining stages, especially in the maturity stage (Supplemental Fig. S3b). The relationship between the gene number of the four samples was plotted as a Venn diagram, with 24,528 genes identified as co-expressed genes (Fig. 4b). Based on their expression levels, we classified these genes into six groups. No expression FPKM < 1, extremely low expression FPKM < 10, low expression FPKM ranging between 10 and 30, medium expression FPKM ranging between 30 and 100, high expression FPKM ranging between 100 and 300, and very high expression FPKM > 300 (Supplemental Fig. S3c; Supplemental Table S3). Thus, the results revealed that 21,866 expressed genes with FPKM values > 1 (Supplemental Data Set S2). Hierarchical cluster analysis and KEGG pathway classification were performed on all expressed genes (Fig. 4c, 4d).
Multiple TFs involved in grain development in foxtail millet
-
The expression dynamics of TFs during grain development were investigated. At least one developmental stage, 509 TFs were detected, and they belonged to 42 families and other categories of TFs (Supplemental Data Set S3; Supplemental Table S4). The genes encoding putative TFs was obtained from the Plant Transcription Factor Database (
http://planttfdb.gao-lab.org/index.php )[69]. One hundred and six differentially expressed TFs were identified, including MADS-box family, WRKY family, bHLH family, MYB family, AP2/ERF family, and NAC family. Based on their expression patterns at four developmental stages, the 106 TFs were classified into four groups (Fig. 5). Among these TFs, 38 exhibited significantly higher expression levels specifically during the ovule stage, compared to the remaining three stages. These TFs were assigned as ovule stage preferential genes (Fig. 5a). In the milk stage, 60 TFs exhibited differential expression, with significantly higher expression compared to the other three stages (Fig. 5b). Only two TFs displayed considerably higher expression at the dough stage compared to other developmental stages, and these were designated as dough stage predominantly expressed genes (Fig. 5c). There were six genes whose expression were significantly higher in the maturity stage, and these genes were designated as the main TFs expressed in this stage (Fig. 5d). WRKYs that regulate seed development have been reported[35,70−72]. Our results showed that eight WRKYs TFs were detected during grain development. Among these, Si3g13930-WRKY53 was found to be enriched at the ovule stage, while six WRKYs TFs (Si3g27390-WRKY24, Si2g18500-WRKY30, Si5g26980-WRKY48, Si2g18510-WRKY70, Si1g06940-WRKY71 and Si3g17470-VQ4) were enriched at the milk stage. Additionally, Si5g14790-VQ4 was enriched at the dough stage. The MADS-box is a crucial regulator of various processes of plant reproductive development and play important roles in the control of seed development[73]. SiMADS34, an E-class MADS-box protein, played a crucial role in regulating foxtail millet grain yield[42]. Among the MADS-box genes, SiMADS17 and SiMADS2 showed high expression in foxtail millet seeds[74]. Our findings demonstrated the presence of 13 MADS-box genes, with seven genes (Si6g22290-MADS7, Si2g27130-MADS8, Si3g10210-MADS13, Si7g22110-MADS17, Si5g40600-MADS21, Si3g07900-MADS58 and Si1g07790-ZMM17) being enriched at the ovule stage, and six genes (Si9g47800-MADS1, Si5g40400-MADS2, Si4g06820-MADS5, Si4g26480-MADS16, Si5g30750-MADS32, Si9g09150-MADS34) being enriched at the milk stage. An important regulatory role in plant development is played by the bHLH[75]. Opaque11 (O11) and ZmZHOUPI (ZmZOU), encoding bHLH TFs, are activated after pollination and regulate several biological processes during endosperm development in maize[26,53]. ZmbHLH121 can bind to the G-box cis-element ABA response element (ABRE) to positively regulate maize grain size and weight[76]. An-1 and OsbHLH107, also regulate grain size in rice[77,78]. Our analysis results showed that 11 bHLH genes were detected during foxtail millet grain development, including five at the ovule stage (Si9g05580-bHLH89, Si2g26780/Si9g10860-bHLH49, Si7g29990-LHW, Si1g35350-bHLH82), five at the milk stage (Si2g03880-PIF4, Si9g09960-bHLH148, Si6g19250-BIM1, Si5g08020-bHLH168, Si5g41500-bHLH128), and only one at the maturity stage (Si3g08580-bHLH68). Opaque2 (O2) and Opaque3 (O3), an extensively studied bZIP TF, its functional studies has roles in regulating grain protein accumulation, amino acid and sugar metabolism, through the transcriptional regulation of certain zein genes[79,80]. Our results also revealed the presence of a small amount of the bZIP TFs Si1g10450-HBP-1a and Si1g33540-bZIP23.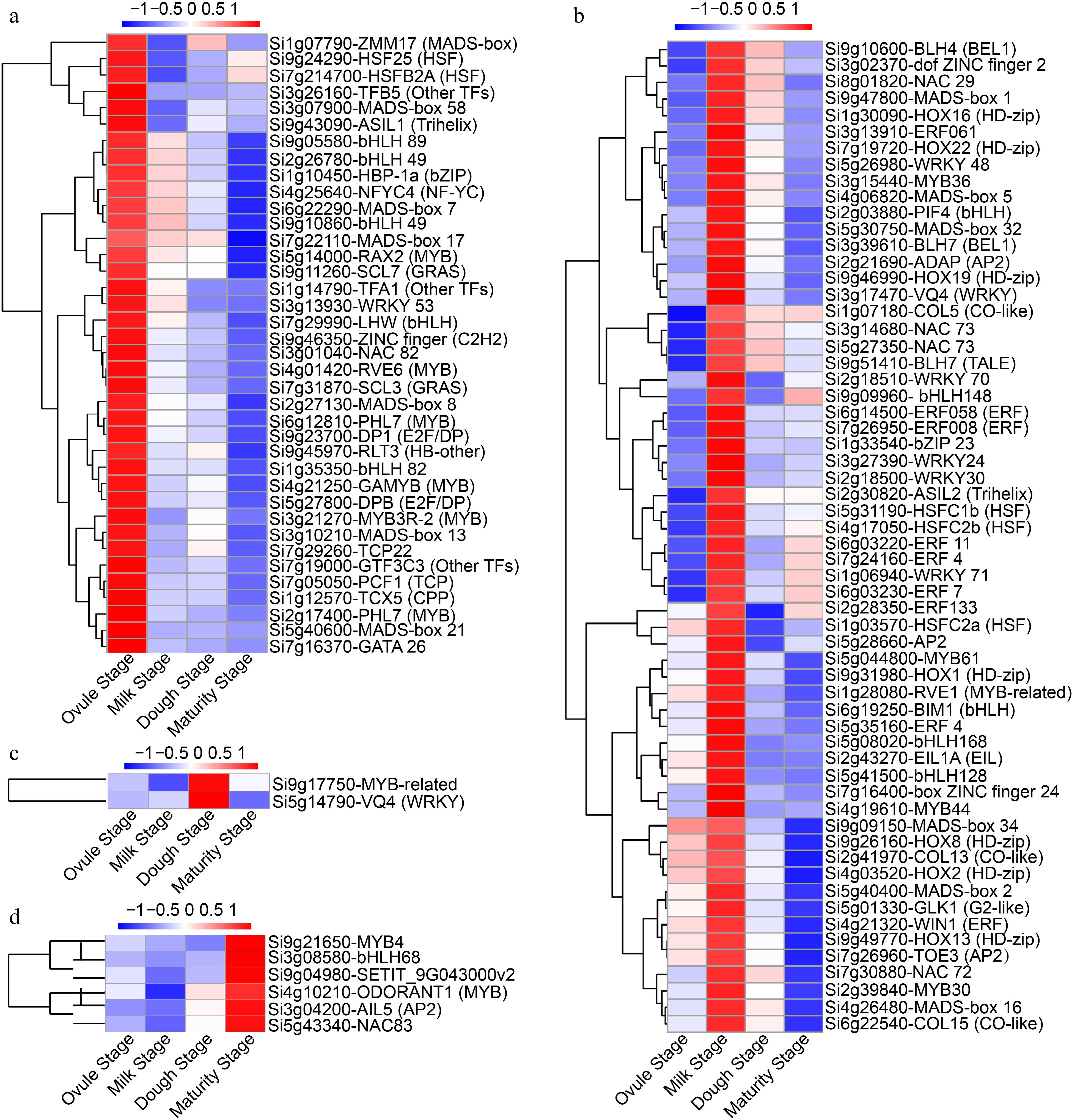
Figure 5.
Hierarchical clustering of TFs expression levels during four developmental stages. TFs predominantly expressed at (a) the ovule stage, (b) the milk stage, (c) the dough stage, and (d) the maturity stage, respectively.
Genes enriched in the hormone signaling pathway during foxtail millet grain development
-
KEGG pathway classification analysis was performed on TFs, revealing a significant enrichment of TFs in hormone signaling (Supplemental Fig. S4). Our results identified 221 genes involved in phytohormones signaling, including auxin, cytokinin (CTK), gibberellin (GA), abscisic acid (ABA), and brassinosteroid (BR) signaling pathways. To investigate the temporal expression patterns of phytohormone-related genes, we investigated gene expression profile clustering. Among the phytohormones, auxin plays crucial roles in ovule and seed development[81]. Therefore, our focused primarily on genes related to auxin signal transduction, including those participating in auxin biosynthesis, polar transport, and response (Fig. 6a). Plasma membrane influx and efflux transporters mediate polar auxin transport, including three transporters[82]. These include the influx carrier AUXIN1/LIKE-AUX1 family, the efflux carrier PIN-FORMED family, and the P-glycoprotein (PGP) subfamily of the ATP-binding cassette transporter family. The auxin signaling pathway during grain development in foxtail millet included eight PIN genes, four AUX genes, and nine PGP genes (Fig. 6b). Among these, six efflux carrier genes (SiPILS2, SiPIN1a, two SiPIN1b, SiPIN1c, SiPIN3a) were preeminently expressed at the ovule and milk stages, but their expression significantly down-regulated in the remaining samples. Whereas the expression levels of the following two efflux carrier genes (SiPIN5a, SiPIN5b) increased at the dough and maturity stages. The influx carrier genes SiLAXs (SiLAX1, SiLAX2x1, SiLAX2x2, SiLAX3) was found to be up-regulated at the milk stage. SiPGP4 (Si3g06120), SiPGP11, SiPGP19, SiPGP21 (Si3g15930) showed preferential expression at the ovule stage and significantly down-regulated at other stages of grain development. However, SiPGP1, SiPGP4 (Si5g28820), SiPGP11, SiPGP14, and SiPGP21 (Si5g04220) exhibited significantly higher expression at the milk stage than other stages. It is worth noting that the tryptophan-dependent pathway is the primary pathway responsible for the production of indole-3-acetic acid (IAA)[83]. Our transcriptome data showed that 22 IAAs genes, five IAAs genes were preferentially expressed at the ovule stage, while 16 IAAs genes showed the highest expression at the milk stage (Fig. 6c). Auxin response factor (ARF) and Small Auxin-Up RNA (SAUR) families are the most important family of auxin-responsive proteins, potentially playing an important role in the regulation of foxtail millet grain development and its response to abiotic stress[84,85]. Several ARFs and SAURs genes were found to be enriched at the ovule and/or milk stages during foxtail millet grain development (Fig. 6d, f). The Gretch Hagen 3 (GH3s) family not only plays an essential role in auxin signaling, but is also involved in the plant defense response systems[86,87]. Three GH3s genes were markedly down-regulated at the milk stage, while the expression of two GH3s genes gradually increased during foxtail millet grain development (Fig. 6e). These data suggest that these auxin signaling related genes are involved in the grain development. Cytokinin, an important phytohormone, plays a key role in regulating various physiological processes such as seed production, cell proliferation, and differentiation[88]. It also plays a significant role in the expression of several important genes during grain development. There genes include three histidine kinases (Si5g431500-HK3, Si9g11580-HK4, Si9g26610-HK5), four histidine-containing phosphor transfer proteins (Si6g24170-AHP1, Si2g14080-AHP2, Si5g31910-PHP5, Si3g17280-PHP5X1), and the corresponding 10 response regulators type-A/B (ARR) family members (Si7912360-ARR1, Si1g20230-ARR2, Si1g37750-ARR3, Si5g45190-ARR4, Si3g02790-ARR6, Si8g03520-ARR9, Si9g47340-ARR21, Si1g35420-ARR23, Si1g06860-ARR24, Si5g41810-ARR26) (Supplemental Fig. S5). GA is an important endogenous hormone that regulates multiple developmental processes in plants[89,90]. Research has shown that GAs are particularly important for ovule formation in early seed development and can also regulate embryo development in late seed development[91]. The GA receptor GID1 specifically recognizes DELLA protein and prevents it from being trans activated, forming the GA-GID1-DELLA complex, which can facilitate interaction with GID2[92]. GA signaling is a complex process and involves 12 GA oxidase genes responsible for GA biosynthesis, two DELLA proteins (Si9g50510-GA1 and Si4g27600-RGL2) and one GA receptor (Si3g23940-GID1) involved in GA signal transduction. Additionally, our transcriptome data revealed the presence of numerous TFs involved in GA signaling (Supplemental Fig. S6). ABA serves as the key regulator of seed dormancy and germination[93]. ABA signaling promotes PP2Cs binding to the PYR/PYL families of ABA receptors, dephosphorylates SnRK2 kinase activity, and activates SnRK2 to phosphorylate and autophosphorates downstream effector genes[94]. Our transcriptome data revealed the genes involved in these ABA signaling pathways were highly enriched (Supplemental Fig. S7). Specifically, we detected six ABA receptor PYR/PYL family genes, including one PYR gene (Si9g39880-PYR1) and five PYL genes (Si4g22990-PYL2, Si1g02490-PYL3, Si9g42890-PYL4, Si5g37100-PYL5, Si3g08200-PYL8). Furthermore, we identified eight protein phosphatase genes (Si9g33810-PP2C27, Si9g45010-PP2C30, Si3g16340-PP2C50, Si3g12370-PP2C1, Si5g21960-PP2C6, Si2g18270-PP2C4, Si7g02990-PP2C3.1, Si5g03330-PHS1), a protein kinase SnRK2 gene (Si3g36320-SnRK2.3), and Si3g24450-ABF2 serves as a binding factor of the ABA responsive element. BR are novel and potent plant hormones that have a wide range of biological activities, which can regulate flowering time, promote anthocyanin accumulation, cell division and elongation, stimulate vegetative growth, promote flowering and fruit ripening, and improve fruit quality and yield[95,96]. In foxtail millet, studies have shown that a LRR receptor-like kinase DROOPY LEAF1 (DPY1) can regulate leaf droopiness by interacting with SiBRI1-SiBAK1, thereby achieving the purpose of regulating plant architecture[97]. RING-type E3 ligase SiSGD1 gene can interact with E2 ubiquitin-conjugating enzyme SiUBC32 and ubiquitinates the BR signaling receptor SiBRI1, affecting foxtail millet grain size[43]. These data suggested potential involvement of these BR signaling genes in foxtail millet grain development. Altogether 72 genes related to BR signaling were detected, including a series of enzymes such as receptor kinase, protein phosphatase, protein kinase, casein kinase, hydrolase, and some TFs (Supplemental Fig. S8). Among these, four receptor kinases (Si9g12450-SERK2, Si5g30180-BRI1, Si8g01300-BRI1, Si6g05160-BAK1), eight protein kinases (Si9g43630-PBL7, Si4g16710-GSK4, Si1g03360-GSK4, Si5g15090-GSK1, Si9g03700BSK5, Si3g01910-BSK3, Si9g31580-BSK2, Si9g34630-ASK8), and three hydrolases (Si9g54960-XTH24, Si4g23660-XTH22, Si4g23650-TCH4) were identified.
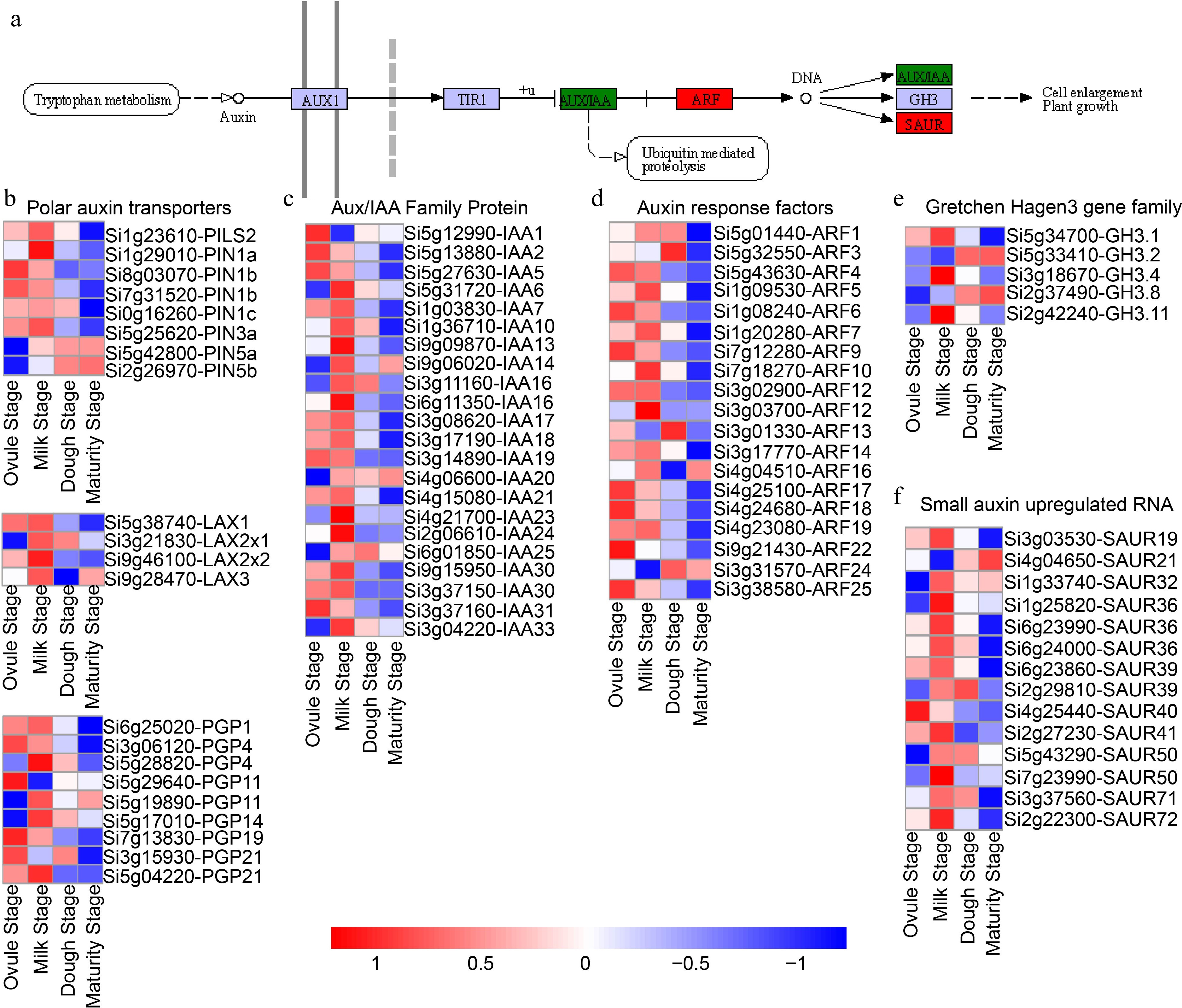
Figure 6.
Expression patterns of genes enriched in the auxin signaling during the four developmental stages. (a) Schematic of the auxin pathway in plants. (b) Polar auxin transporter gene expression patterns, (c) IAAs, (d) ARFs, (e) GH3s, and (f) SAURs respectively. AUX1: auxin influx carrier (AUX1 LAX family); TIR1: Transport inhibitor response 1; AUX/IAA: auxin-responsive protein IAA; ARFs: Auxin response factors; GH3: Indole-3-acetic acid-amido synthetase (auxin responsive GH3 gene family); SAUR: Small auxin up-regulated RNA (SAUR family protein). PINs: Auxin efflux carrier component; LAXs: Auxin transporter-like protein; PGPs: ABC transporter B family member; IAAs: Indole-3-acetic acid.
Sucrose and starch metabolism in foxtail millet during grain development
-
A critical process in grain filling is the conversion of sucrose to starch[98]. Starch is an important component of foxtail millet grains[99]. This study identified 164 genes related to starch and sucrose metabolism were identified (Fig. 7 ; Supplemental Fig. S9). The expression of SiSUS2 and SiSUS7 gradually increased and peaked at the milk stage, whereas the expression of SiSUS1 peaked at the dough stage and then gradually dropped. SiSUS4 expression increased progressively with a peak at the maturity stage. Four SiSPSs, one SiTPP, one SiAGPase, one SiGBSS, four SiSSs, two SiSBEs, one SiAMY, and one SiBMY gene were highly expressed at the ovule stage, and their expression gradually decreased. One SiSPS, three SiTPSs, six SiTPPs, one SiAGPase, one SiAMY, and one SiBMY gene were higher expressed at the milk stage. The highest at the dough stage was observed for three SiTPSs, one SiTPP, one SiAGPase, four SiSSs, one SiSBE, three SiAMYs and one SiBMY. At the maturity stage, they were highest expressed by one SiTPP, one SiGBSS, one SiSS, one SiSBE and one SiAMY. Increasing AGPase activity may improve the sink strength of developing grains and ultimately promote grain weight at the maturity stage.
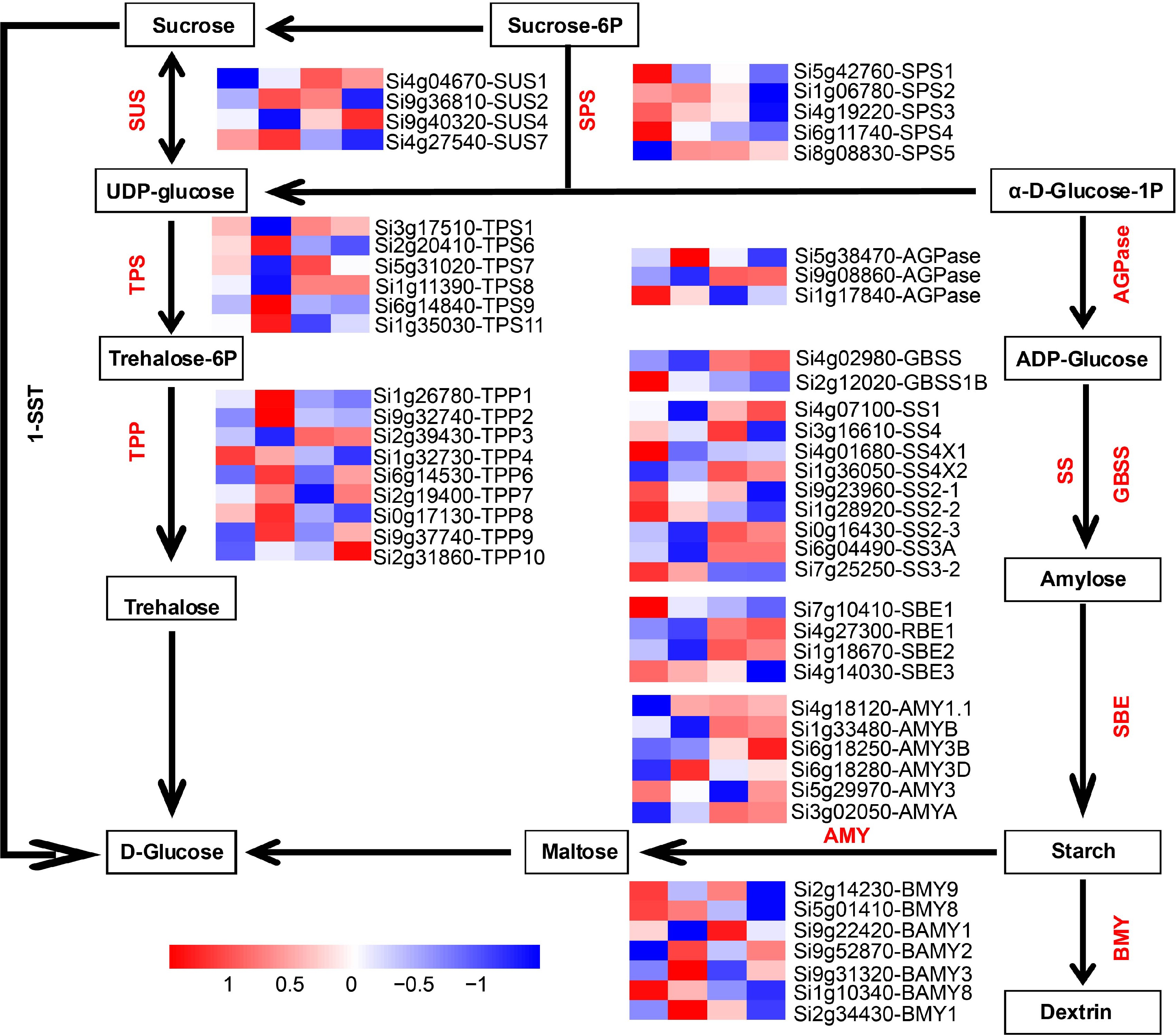
Figure 7.
Analysis of expression patterns of genes in the sucrose-starch metabolism pathway during four developmental stages. AGPase: ADP glucose pyrophosphorylase; GBSS: Granule-bound starch synthase; SSs: Starch synthase; SBE: Starch-branching enzyme; BMY: Beta-amylase; AMY: Alpha-amylase isozyme; SUS: Sucrose synthase; TPS: Alpha trehalose phosphate synthase; TPP: Trehalose phosphate phosphatase; SPS: Sucrose phosphate synthase; 1-SST: Sucrose 1-fructosyltransferase.
Expression of zein genes in foxtail millet during grain filling
-
Encoded by several different genes and families, zein can be divided into α-, β-, γ-, and δ- types zein, which are the most important storage proteins[100,101]. In maize, α-zein is further divided into 19-kD (Z1A, Z1B, Z1D) and 22-kD α-zeins (Z1C), β-zein contains only a class of 14-kD β-zeins, whereas the γ-zein subfamily contains 16-kD, 27-kD, 50-kD zeins, and δ-zein includes 10-kD, 18-kD zeins[102]. The expression profiles of the 16 zein genes clearly showed that zeins were strongly induced during the dough and maturity stages of endosperm development, which is characteristic of storage product accumulation (Fig. 8 & Supplemental Table S5). Our study showed a total of seven 19-kd-α-zein genes, notably Si8G18820-PMS1-19-kDα exhibited the highest expression level of 100,000 during the maturity stage. There were also seven 22-kD α -zein genes, with three of them showing expression levels exceeding 10,000 in the dough and maturity stages. Furthermore, we observed the presence of one β-zein gene (Si3g11730-β-zein) and one γ-zein gene (Si2g22180-γ-zein). Surprisingly, all the detected 19-KD-α-zein genes in the transcriptome data were found to be located on chromosome 8 of foxtail millet, forming a tandem array in a single gene cluster (Fig. 8b). Therefore, we speculate that α-zein genes in foxtail millet may be arranged in tandem in one or more clusters within the genome, which play a role in regulating the accumulation of storage proteins. Amino acids are crucial components of proteins and serve as essential nutrients for humans. Foxtail millet grains are abundant in various amino acids, including essential amino acids needed by human, such as leucine, phenylalanine, isoleucine, tryptophan, methionine, and lysine[103]. Therefore, we further analyzed the expression of genes involved in amino acid synthesis and metabolism during foxtail millet grain development. These genes can be divided into those associated with the synthesis and metabolism of essential amino acids, non-essential amino acids synthesis and metabolism, and those involved in both pathways. We found that a majority of these genes were highly expressed during the initial three stages of grain development (Supplemental Fig. S10).
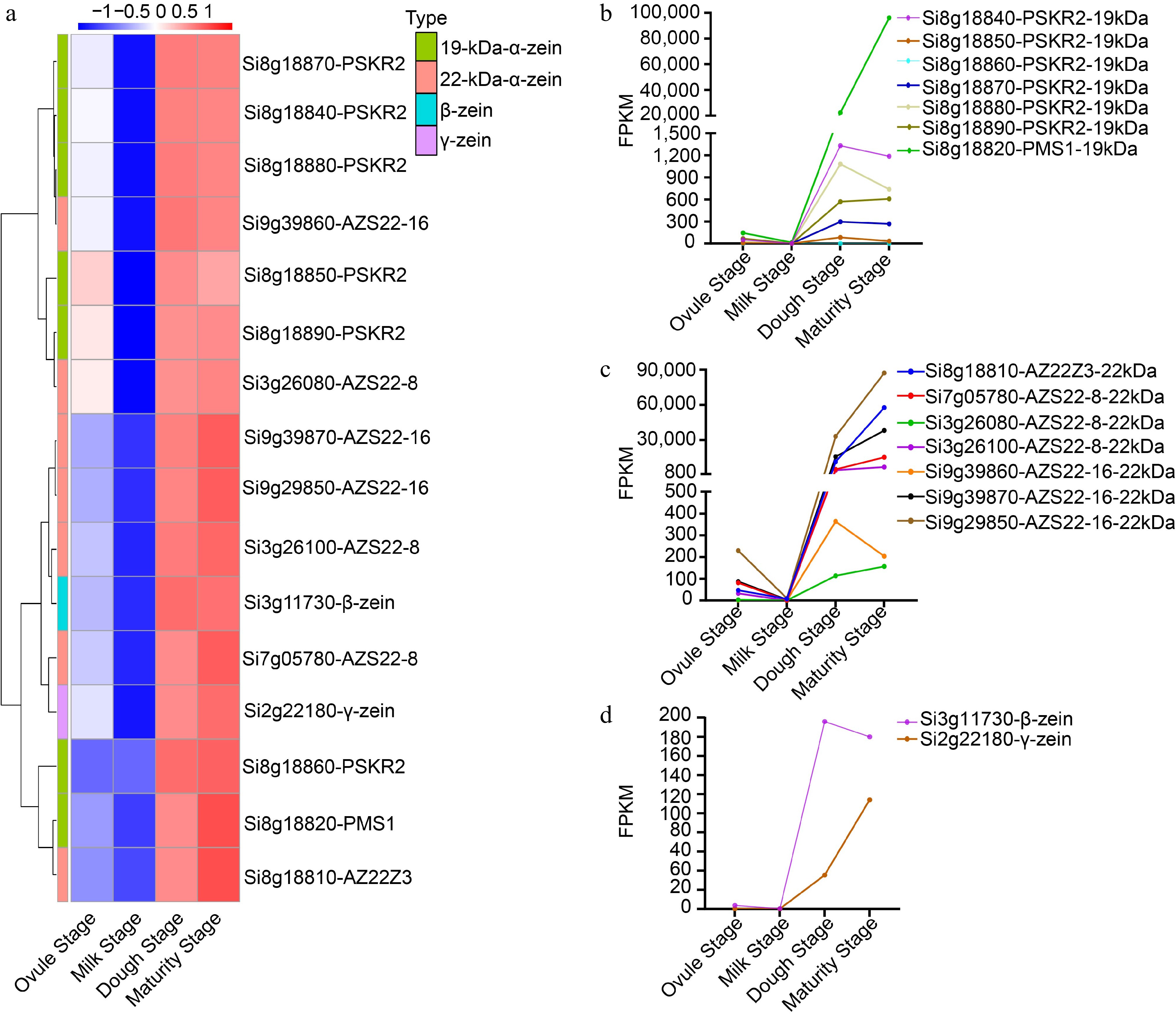
Figure 8.
Analysis of zein family genes expression during four developmental stages. (a) Cluster Heat map of the FPKM values of 16 zein genes at the four developmental stages. (b) Dynamic transcript levels of 19-kDa-α-zein genes at four developmental stages. (c) Dynamic transcript levels of 22-kDa-α-zein at four developmental stages. (d) Dynamic transcript levels of β- and γ-zeins at four developmental stages.
Dynamic changes in carotenoid, flavonoid, folate metabolism pathways in foxtail millet grain filling
-
The selection of yellow grain in the domestication of foxtail millet presents a striking aspect, especially when compared to major cereal crops such as wheat (Triticum aestivum) and rice (Oryza sativa). Grain colour and quality were important breeding traits for foxtail millet domestication. The yellow color of most foxtail millet landraces and varieties, is typically produced by the accumulation of carotenoids and flavonoids in the pericarp, aleurone, and endosperm[104]. This study identified 25 genes, including a bunch of enzymes, that are related to the carotenoid pathway (Fig. 9a). Schematic representation of the carotenoid pathway during foxtail millet grain development (Fig. 9b). Carotenoids play a critical role in maintaining good health, its biosynthesis requires many enzymes, phytoene synthase (PSY) is the most essential rate-limiting enzyme in the carotenoid pathway[105]. Most genes involved in carotenoid biosynthesis accumulated significantly during foxtail millet grain development. This indicating that the carotenoids began to accumulate at the milk stage, which aligned with previous findings, and the foxtail millet grains started to turn yellow after 8 DAP (Fig. 1b). One of the candidate genes, SiPSY1, which affects foxtail millet grain color and quality, was identified as a potential gene for foxtail millet color variation that can be utilized in breeding[104]. In this study, we detected three PSY genes (Si4g27520-PSY1, Si3g38930-PSY2, and Si2g30580-PSY3) in the carotenoid metabolic pathway of foxtail millet. Furthermore, we further analyzed the expression levels of genes involved in flavonoid pathways from ovule stage to maturity stage. In the flavonoid biosynthesis pathway, a total of 36 genes related this pathway were identified (Supplemental Fig. S11a). Among these genes, Si2g44100-C4H, Si8g14530-CHS, two CHI genes (Si7g30460-CHIL, Si9g04210-CHI-1), Si1g33590-FLS, Si9g38590-F3'5'H, and four ANR genes (Si7g25720, Si7g25730, Si7g25740, Si7g25760) showed the highest levels at the milk stage, and then gradually decreased. Si3g03560-F3H, Si9g23880-F3'H, Si5g25220-DFR, and Si1g01130-LAR showed higher levels at the ovule and dough stages, but lower levels at the milk and maturity stage (Supplemental Fig. S11). Folate metabolism process is conserved and requires the catalysis of a variety of specific enzymes[47]. This study detected most genes related to the folate metabolism pathway in foxtail millet (Supplemental Fig. S12a). SiADCS, SiDHPS1/2, SiDHFR1/2, two SiFPGS, SiDHNA2, SiDHN-P, and SiGGH expression levels gradually decreased, SiGCHI expression levels gradually increased, SiDHFS, SiDHNA1, SiDHN-P3 expression levels decreases and then increases during foxtail millet grain development (Supplemental Fig. S12b).
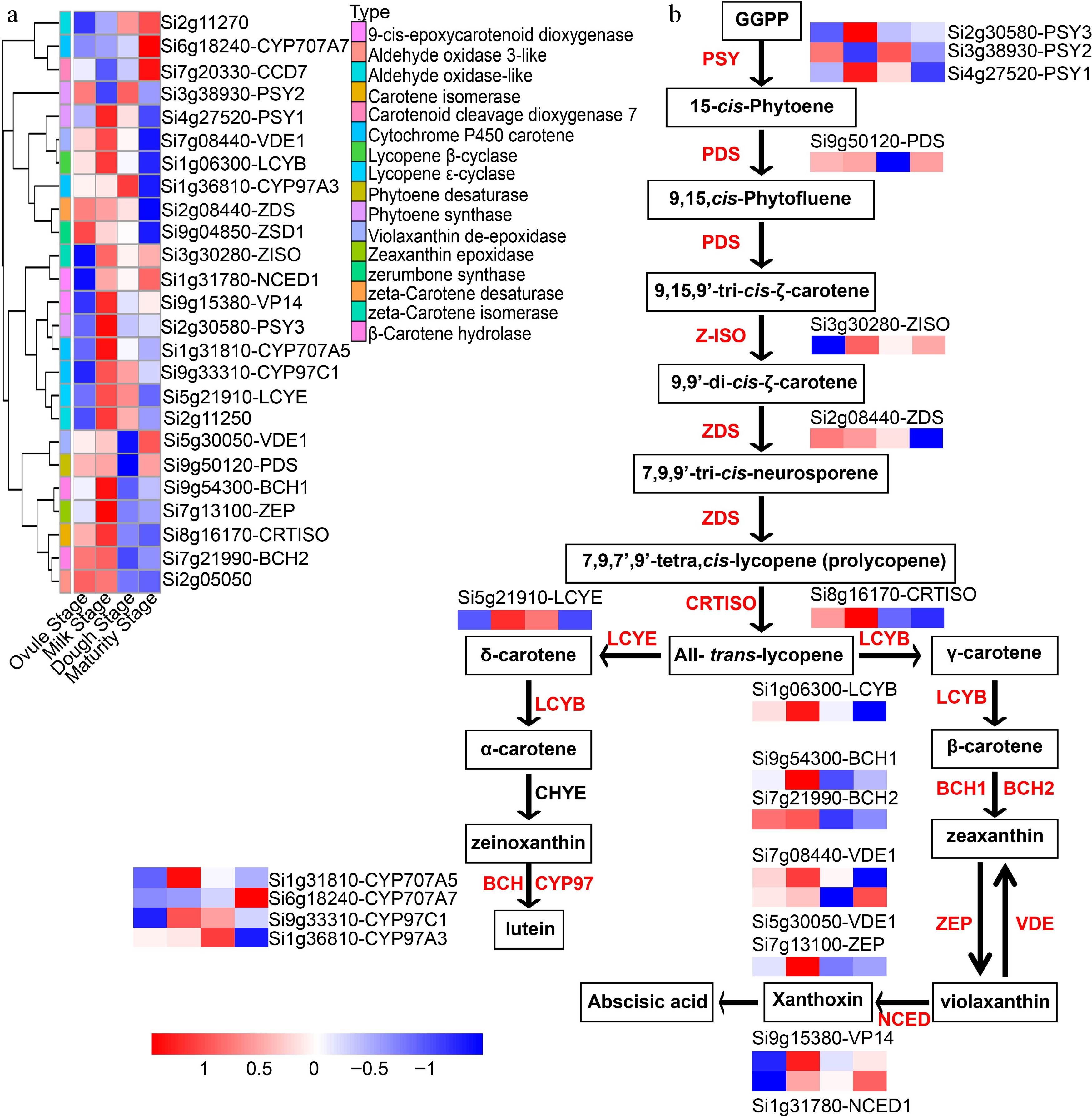
Figure 9.
Hierarchical clustering analysis of carotenoid metabolic pathway genes at the four developmental stages. (a) Heat map analysis of carotenoid metabolic pathway genes in the four developmental stages. (b) Schematic diagram of carotenoid metabolic pathway. PSY: phytoene synthase; PDS: phytoene desaturase; Z-ISO: ζ-carotene isomerase; ZDS: ζ-carotene desaturase; CRTISO: carotenoid isomerase; LCYE: lycopene ε-cyclase; LCYB: lycopene β-cyclase; LCYE: lycopene ε-cyclase; BCH: β-carotene hydroxylase; ZEP: zeaxanthin epoxidase; VDE: violaxanthin de-epoxidase; NCED: 9-cis-epoxycarotenoid dioxygenase; CYP97A: cytochrome P450 carotene hydroxylase.
RT-qPCR verification of RNA-seq data
-
To ensure the reliability of our transcriptome data, we selected 14 key metabolic genes by RT-qPCR validation. Specifically, we analyzed the expression patterns of three zein genes (Si9g29850-AZS22-16, Si3g11730-β-zein, Si2g22180-γ-zein), the rate-limiting enzyme gene (Si2g30580-PSY3) of carotenoid biosynthesis, five auxin signaling pathway-related genes (Si5g13880-IAA2, Si6g23990-SAUR36, Si1g25820-SAUR36, Si5g43630-ARF4, Si9g21430-ARF22), five transcription factors (Si9g47800-MADS1, Si6g22290-MADS7, Si3g10210-MADS13, Si5g406000-MADS21, Si6g14500-ERF058), and a house-keeping gene (Seita.8G043100). The results of our RT-qPCR analysis demonstrated that the observed variation in gene expression levels aligned with those obtained from the RNA-seq data (Supplemental Fig. S13). This concurrence strongly suggests the reliability of our analytical findings.
-
Foxtail millet grain development is crucial for grain quality and yield, although previous studies have identified several genes that regulate starch biosynthesis, grain color, carotenoid, flavonoid, and folate metabolism[40,106,107]. However, detailed morphological analyses of foxtail millet grain development process are scarce and comprehensive description of transcriptome dynamics based on morphological analyses is lacking. Therefore, conducting a, detailed morphological analysis of the ovule before pollination and the grain development stages after pollination can provide detailed morphological support for the dynamic developmental process of foxtail millet grains (Fig. 1, Fig. 2; Supplemental Fig. S1). This study provides a valuable reference for studying the specific periods of ovule and grain development in foxtail millet. In addition to the morphological analysis of grain development of Jingu 21, the accumulation characteristics of main storage substances (protein and fat) during foxtail millet grain development were also analyzed (Fig. 3; Supplemental Fig. S2). On the basis of morphological analysis and storage material analysis of foxtail millet grain development, we have established a time-dynamic transcriptome of grain development in Jingu 21. This transcriptome provides valuable genetic resources for understanding the genetic control of grain development in foxtail millet. Additionally, we have analyzed the integrated dynamic transcriptome of TFs, plant hormone signaling, starch and sugar metabolism, zein related genes, carotenoid metabolism, flavonoid metabolism, and folate metabolism in grain development. This comprehensive analysis deepens our understanding of the changes in important metabolic pathways during foxtail millet grain development, and help us provide new insights for developing better high-yield cultivars.
Seed development is regulated by numerous TFs, many of which have been extensively reported[108,109]. In our RNA-seq analysis, we aimed to identify regulatory networks that play important role in grain development. We have discovered 106 grain-specific TFs (Fig. 5). These results are similar with the previous study on the gene expression profile of foxtail millet grain filling process[44]. MADS-box were responsible for the regulation of the development of flower organs, embryos, endosperm, and seeds[73]. SiMADS17/33/34/37/46/52 were considered key regulatory factors for grain yield in foxtail millet, and they are highly expressed in seeds. Moreover, SiMADS2/12/26/28/60 were thought to be highly expressed in pericarp and mainly expressed at the filling stage[42,74]. Many SiMADS TFs were analyzed in our transcriptome data. Combined with previous reports and our research results, we have identified highly homologous genes from the transcriptome data, namely Si1g07790-ZMM17, Si4g06820-MADS5, Si4g26480-MADS16. SiMADS34 (Si9g09150-MADS34), which has been reported to regulate foxtail millet inflorescence structure, was also found to be highly up-regulated in the ovule and milk stages of grain development, but its role in the grain filling process is remains unknown. It is noteworthy that the four TFs Si9g47800-MADS1, Si6g22290-MADS7, Si3g10210-MADS13, Si5g406000-MADS21 exhibit different expression patterns during grain development. Further investigation is required to determine their functions and roles. In maize, OPAQUE11 is a central hub in endosperm development and nutrient metabolism[110,111]. Among the numerous TFs we have discovered, 11 belong to the bHLH family. It is noteworthy that only Si3g08580-bHLH68 shows differential expression at the maturity stage, however, no studies have been reported on its regulatory mechanism at the maturity stage of foxtail millet grain development. Our dynamic transcriptome analysis of TFs enables us to identify key regulators of seed development, and gain insights into the mechanisms underlying grain development.
Phytohormones play an extremely crucial role in seed development, so far, numerous plant hormone-related genes have been identified[112,113]. BR has been found to promote foxtail millet grain development, it has been discovered that SiBZR1 and SiUBC32-SGD1-SiBRI1 of the BR signaling pathway, can regulate grain yield in foxtail millet and other gramineous crops[43,114]. These findings provide a new strategy for BR in breeding and improving important traits in foxtail millet. In our transcriptome data, we observed the presence of several enzymes and TFs within the BR signaling pathway (Supplemental Fig. S8). Four BZR1 genes were detected in our study, among which Si2g36710-SiBZR1 gene was consistent with the genetic results reported in previous studies. The Si5g14880-SiBZR1, Si1g03480-SiBZR1, Si3g37600-SiBZR1 genes were found to be homologous genes of SiBZR1. Further analysis indicated that Si1g03480-SiBZR1 and Si3g37600-SiBZR1 were strongly induced at maturity stage, while Si5g14880-SiBZR1 was expressed throughout grain development. Based on these findings, it can reasonably speculate that these SiBZR1 genes may play distinct regulatory roles in different stages of foxtail millet grain development. What's more, our transcriptome data indicated the preferential expression of auxin influx carriers, efflux carriers, and auxin-response factors in early foxtail millet grain development, specifically at the ovule and/or milk stages (Fig. 6). Genes associated with the auxin signaling pathway may be valuable resources for foxtail millet molecular breeding.
Starch is the main component of seeds, and its synthesis is a complex process involving many enzymes[115]. The activity of key enzymes participating in converting sucrose to starch in seeds was shown to regulate grain filling[116,117]. In our study, we identified several genes associated with starch and sucrose metabolism (Fig. 7). These genes include SiSUS, SiAGPase, SiGBSS, SiSSs, SiSBE, SiAMY, and SiBMY, which are important for grain formation. It is important to note that the amylose content is the main factor affecting the nutritional and eating quality of foxtail millet[13]. Recessive mutations in the waxy (Wx) gene, encoding granule-binding starch synthetase (GBSS), an enzyme required for amylose synthesis, control the waxy characteristic of foxtail millet grain[118,119]. However, the waxy gene not only reduces amylose content but also affects the gelatinization, aging, adhesiveness, and physicochemical properties of starch[120]. In our study, we detected two genes in granule binding starch synthetase (Si4g02980-GBSS and Si2g12020-GBSS1B). Specifically, the expression levels of the Si4g02980-GBSS gene at the milk and maturity stages were found to be 300 times and 650 times higher, respectively, compared to earlier stages. This observation suggests that the GBSS gene is essential for seed filling and starch accumulation. This study also showed that the Wx gene is an important target for domesticating of foxtail millet to improve its nutritional quality. During foxtail millet grain development, the expression of zein genes was highest at the milk stage, which is consistent with characteristics of nutrient accumulation (Fig. 8). High levels of zein expression play a key role in endosperm hardness, but it contains almost no essential amino acids such as lysine and tryptophan[121]. In maize, zein gene transcription is regulated by O2, PBF, OHP1, OHP2, ZmMADS47, and others, among O2 having the greatest effect on α-zein gene expression[102,122]. Maize and sorghum storage protein genes exist in the form of gene clusters, which are tandem array by gene family members, and this arrangement leads to the accumulation of storage proteins at extremely high levels[123,124]. Our study identified a gene cluster consisting of seven 19-KD-α-zein genes tandemly arrayed. However, it remains unclear whether other genes are also involved in this gene cluster. These results indicate that the accumulation of foxtail millet storage proteins may also be regulated by complete tandem gene clusters. Therefore, to gain a better understanding the accumulation of foxtail millet storage proteins, it is crucial to consider the zein gene cluster as a whole. Zein, a seed storage protein that is specifically expressed at the endosperm filling stage, is a representative substance for studying storage proteins at the endosperm filling stage. Despite the importance of zeins, their regulatory mechanisms are still unclear, and further investigation is needed to elucidate the molecular mechanisms of zeins in the endosperm.
Carotenoid and flavonoid, together as nutrients of foxtail millet, greatly impact its beige color, and the more carotenoids, the more yellow the beige color and the better the quality of foxtail millet[104]. SiPSY gene plays an important role in carotenoid accumulation in foxtail millet, it was found to be up-regulated, while the SiCCD1 gene was down-regulated, resulting in continuous carotenoid accumulation in foxtail millet[40,125]. Our data analysis revealed three SiPSY genes (SiPSY1/2/3), with two genes showing the highest expression at the milk stage, and another gene being most expressed at the dough stage. This suggests that the SiPSY gene is expressed at different stages of foxtail millet grain development and may have different regulatory roles. Understanding the key genes that control grain colour and carotenoid accumulation in foxtail millet is crucial for breeding high-quality varieties.
-
Overall, we conducted a detailed morphological analysis of foxtail millet grain development, defining the grain development process, and described the characteristics of the accumulation of storage products during grain development. Subsequently, we performed transcriptome analysis to elucidate the dynamic mechanisms of TFs, plant hormone signaling, starch and sugar metabolism, carotenoid metabolism, flavonoid biosynthesis, and folate metabolism pathways during grain development in foxtail millet. The expression of key genes involved in important metabolic pathways during foxtail millet grain development were investigated using RT-qPCR. It was found that some genes exhibited high expression levels at the early stages (ovule and milk stages), whereas others showed high expression levels at the later stages (dough and maturity stages). These findings suggest different genes play different roles in grain development. The results of this study provide valuable insights into the developmental process and molecular mechanism of foxtail millet grain, as well as establish a strong theoretical foundation for its improvement.
-
The authors confirm contribution to the paper as follows: study conception and design: Wang JG, Wang D; draft manuscript: Wang D, Wang JG, Du H, and Yang G; experiments performance and manuscript revision: Wang D, Su M, Hao JH, Li ZD, Dong S, Yuan X, Li X, Gao L, and Chu X. All authors have read and agreed to the published version of the manuscript.
-
All data generated or analyzed during this study are included in this published article and its supplementary information files.
This work was supported by National Natural Science Foundation (32200222 for Wang JG); High-level Talents Start-up Fund of Shanxi Agricultural University (J242198006 for Wang JG); Shanxi Province Outstanding Doctoral and Post-Doctoral Scholarship Award Foundation (SXBYKY2021055 for Wang JG) and Hou Ji Laboratory Foundation (202204010910001-32 for Wang JG); Shanxi Province Key R&D Program Project (2022ZDYF119 for Du H); Shanxi Province Outstanding Doctoral and Post-Doctoral Scholarship Award Foundation (SXBYKY2021059 for Yang G).
-
The authors declare that they have no conflict of interest.
- Supplemental Fig. S1 Dynamics of foxtail millet grain development: from 1 to 30 days after pollination (DAP). (a) Changes in the grains area of during foxtail millet grains development; (b) Changes in the perimeter of during grains development; (c) The growth of the foxtail millet grains in the length and width; (d) The change in the length/width ratio during grains development. Values for (a) , (b) , (c) and (d) are means ± s.d. (from 1 to 15 days after pollination n = 40, from 16 to 30 days after pollination n = 20).
- Supplemental Fig. S2 Cell morphological analysis of the developing grains in foxtail millet: Paraffin section analysis. (a) and (e) Morphology of mature ovule cells before pollination; (b) and (f) Milk stage of grain development at 6-11 DAP; (c) and (g) Dough stage of grain development at 12-22 DAP; (d) and (h) Maturity stage of grain development at 23-30 DAP. DAP: days after pollination. (a-d) stained with 0.1% Toluidine Blue-O to observe. (e-f) stained with Safranin O and Fast Green to observe. Scale bars =10 μm.
- Supplemental Fig. S3 Analysis of different samples global gene expression. (a) The principal component analysis (PCA) of the RNA-seq data of the 12 samples; (b) Total number of genes expressed at each developmental stage; (c) Percentage of gene numbers in different categories according to their expression levels in each tissue, based on FPKM values; (d) Gene ontology (GO) enrichment analysis of the all expression gene in four developmental stages.
- Supplemental Fig. S4 Hierarchical clustering analysis of the relative expression levels of transcription factors during grain development. (a) Hierarchical clustering analysis of the relative expression levels of transcription factors during grain development; (b) Gene ontology (GO) enrichment analysis of the TFs in foxtail millet grain development; (c) KEGG pathway classification top 20 of the TFs in foxtail millet grain development.
- Supplemental Fig. S5 Clustering expression patterns of cytokinin signaling pathway related genes in foxtail millet grains development. (a) Diagram of the cytokinin signaling pathway; (b) Expression patterns of histidine kinase in the cytokinin signaling pathway; (c) Expression patterns of histidine containing phosphotransfer protein in the cytokinin signaling pathway; (d) Expression patterns of the response regulator in the cytokinin signaling pathway; (e-f) The dynamic transcript levels of cytokinin signaling pathway in the different development stages of the foxtail millet grain. HK4/CRE1: AHK4/CYTOKININ RESPONSE1; AHP1: Histidine-containing phosphotransfer protein 1; AHP2: Histidine-containing phosphotransfer protein 2; A-ARR: two-component response regulator ORR belong to ARR family Type-A subfamily; B-ARR: two-component response regulator ORR belong to ARR-B family.
- Supplemental Fig. S6 Dynamic transcriptome analysis of genes associated with gibberellin signaling pathway. (a) Diagram of the gibberellin signaling pathway; (b) Hierarchical clustering analysis of the relative expression levels of gibberellin signaling pathway genes during grain development; (c-d) The dynamic transcript levels of gibberellin signaling pathway in different development stages. GID1/2: GA-insensitive dwarf 1/2; DELLA: DELLA protein; TFs: Transcription factors.
- Supplemental Fig. S7 Dynamic transcriptome analysis of genes involved in abscisic acid signaling pathway of foxtail millet grains development. (a) Diagram of the abscisic acid signaling pathway; (b) Hierarchical clustering analysis of the relative expression levels of abscisic acid signaling pathway genes during grain development; (c-f) The dynamic transcript levels of abscisic acid signaling pathway DEGs in the different development stages of the foxtail millet grain. PYRs: Abscisic acid receptor PYR family; PYLs: Abscisic acid receptor PYL family; PP2C: Protein phosphatase 2 C; SnRK2: serine/threonine-protein kinase SnRK2; ABF: ABA responsive element binding factor.
- Supplemental Fig. S8 Dynamic transcriptome analysis of genes involved in brassinosteroid signal transduction of foxtail millet grains development. (a) Diagram of the brassinosteroid signal transduction pathway; (b) Hierarchical clustering analysis of the relative expression levels of brassinosteroid signal transduction pathway genes during grain development; (c-e) The dynamic transcript levels of brassinosteroid signal transduction pathway in the different development stages of the foxtail millet garins. BAK1: Brassinosteroid insensitive 1-associated receptor kinase 1; BRI1: Brassinosteroid insensitive 1; BKI1: BRI1 kinase inhibitor 1; BSK: BR-signaling kinase; BSU1: serine/threonine-protein phosphatase BSU1; BIN2: Brassinosteroid insensitive 2; BZR1/2: Brassinosteroid resistant 1/2; TCH4: xyloglucosyl transferase TCH4; CYCD3: Cyclin D 3.
- Supplemental Fig. S9 Heat map analysis of genes associated with sucrose-starch conversion during grain filling from Ovule stage to maturity stage.
- Supplemental Fig. S10 Dynamic analysis of genes involved to amino acid metabolism pathways in foxtail millet grains development. (a) Dynamic analysis of genes involved in amino acid biosynthesis during foxtail millet grains development. These genes are divided into essential amino acid biosynthesis genes, non-essential amino acid biosynthesis genes and genes synthesized by both; (b) Dynamic analysis of genes involved in amino acid metabolism and degradation during the development of foxtail millet grains. These genes can be divide into essential amino acid metabolism genes, non-essential amino acid metabolism genes and genes involved in the common metabolism of both.
- Supplemental Fig. S11 Dynamic analysis of the flavonoid biosynthesis pathway during foxtail millet grains development. (a) Hierarchical clustering analysis of the relative expression levels of the flavonoid biosynthesis pathway genes during grain development; (b) Flavonoid biosynthesis pathway during foxtail millet grain development. The rectangles represent the expression level of genes. C4H: Trans-cinnamate 4-monooxygenase; CHS: Chalcone synthase; CHI: Chalcone isomerase; F3H: Flavanone 3-hydroxylase; F3’H: flavonoid 3'-hydroxylase; F3’5’H: flavonoid 3',5'-hydroxylase; FLS: flavonol synthase; DFR: Dihydroflavonol 4-reductase; LAR: Leucoanthocyanidin dioxygenase; ANR: Anthocyanidin reductase.
- Supplemental Fig. S12 The biosynthetic pathway and expression patterns of folate synthesis pathway genes during foxtail millet grain development. (a) Hierarchical clustering analysis of the relative expression levels of folate metabolism pathways genes during seed development; (b) Folate synthesis pathway during foxtail millet seed development. The rectangles represent the expression level of genes. ADC: Aminodeoxychorismate; pABA: para aminobenzoic acid; GTP: guanosine-triphosphate; DHN: dihydroneopterin; ADCS: aminodeoxychorismate synthase; ADCL: aminodeoxychorismate lyase; HPPK: hydroxymethyldihydropterin pyrophospho kinase; DHPS: dihydropteroate synthase; DHFS: dihydrofolate Synthetase; DHFR: dihydrofolate reductase; FPGS: folylpolyglutamate-synthase; GGH: gamma glutamyl hydrolase; GTPCHI: GTP cyclohydrolase I; DHNA: dihydroneopterine aldolase.
- Supplemental Fig. S13 RT-qPCR was used for quantitatively verification of the key genes during grain development. (a-e) Relative expression levels of five key TFs (SiMADS-box21, SiMADS-box13, SiMADS-box7, SiMADS-box1, SiERF058) during grain development. (f-j) Relative expression levels of fix auxin signaling pathway related genes (SiIAA2, Si6g23990-SAUR36, Si1g25820-SAUR36, SiARF4, SiARF22) during grain development. (l-n) The relative expression levels of rate-limiting enzyme gene SiPSY3 and three zein genes.
- Supplemental Table S1 Morphological analyses of foxtail millet grain.
- Supplemental Table S2 Summary of RNA-Seq read mapping results.
- Supplemental Table S3 The classification of gene expression levels from the four tissues in this study.
- Supplemental Table S4 Multiple TFs involved in grain development in foxtai millet.
- Supplemental Table S5 Zein gene in transcriptome data.
- Supplemental Table S6 Primers used for RT-qPCR.
- Supplemental Data Set S1 The total number of genes detected in the four RNA-seq samples.
- Supplemental Data Set S2 All expressed genes with FPKM value >1 in the four RNA-seq samples.
- Supplemental Data Set S3 Many transcription factors were detected, belonging to 42 transcription factor families and other types of transcription factors.
- Copyright: © 2023 by the author(s). Published by Maximum Academic Press on behalf of Hainan Yazhou Bay Seed Laboratory. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Wang D, Su M, Hao JH, Li ZD, Dong S, et al. 2023. Dynamic transcriptome landscape of foxtail millet grain development. Seed Biology 2:19 doi: 10.48130/SeedBio-2023-0019 

Dynamic transcriptome landscape of foxtail millet grain development
- Received: 15 July 2023
- Accepted: 24 October 2023
- Published online: 24 November 2023
Abstract: Grain development of foxtail millet (Setaria italica L.) is essential for yield and quality. However, its transcriptional dynamics molecular mechanisms and morphological analyses remain scarcely described. Thus, we conducted detailed daily morphological analyses of foxtail millet grain development throughout the 30 d post-fertilization development period. On the basis of the morphological analyses, we used RNA-sequencing (RNA-seq) to examine the transcript dynamics involved in foxtail millet grain development at four stages. These genes included those associated with transcriptional regulation, hormone signaling, sucrose and starch metabolism, zein family members, amino acid metabolism, carotenoid metabolism, flavonoid biosynthesis, and folate synthesis. We have validated the accuracy of the transcriptome data by means of reverse transcription quantitative polymerase chain reaction (RT-qPCR). This study provides precious genetic resources for understanding grain developmental process in the future. These results expand our understanding of the molecular mechanisms of grain development in foxtail millet and contribute to the functional studies of genes related to grain development in the future.
-
Key words:
- Foxtail millet /
- Grain development /
- Temporal transcriptome /
- Regulatory network