









-
Simple Sequence Repeats (SSRs) are repetitions of 1–6 bp nucleotide motifs which are also named as microsatellites. SSRs distribute in both coding and non-coding regions in the genome and have been detected in various species[1−3]. SSRs are highly favored molecular markers in genetic research because of their wide genomic distribution, co-dominant inheritance, high levels of polymorphism, and can be easily analyzed by PCR. In addition, most linkage maps with important agronomic trait loci have been developed with primarily low-throughput markers, such as AFLP, RFLP, and RAPD or some SSR markers[4]. SNP is a high-throughput marker and widely used in genetic analysis. However, the development of SNP markers needs sequenced genomic information, errors in sequencing or assembling of the genome might lead to false SNPs. The development of SNP markers is time consuming, labor intensive, and expensive. SSR markers could be developed easily based on the known gene sequences. Currently, with the development of bioinformatics and next-generation sequencing technology, it is convenient and feasible to obtain a large number of SSR markers. Therefore, the SSR marker has been the preferential choice for various applications, such as the genetic analysis of population structure, association analyses, linkage map construction, fine mapping of QTL, and varietal identification, which is helpful to find gene linkage loci quickly and accurately[4−8]. SSR markers can provide clues for screening, identification, annotation and functional analysis of candidate genes.
Chinese cabbage (Brassica rapa L. ssp. pekinensis) is one of the most consumed vegetable crops in Asia. It is an economically important species, including many morphotypes cultivated as vegetable, oil, and fodder crops[9]. With the development of next-generation sequencing (NGS) technology and bioinformatics, in recent years genome-wide SSR markers have been developed in some plants[10, 11]. However, in Chinese cabbage, the development and characterization of genome-wide SSR loci have not yet been reported, which ultimately limits the molecular study of Chinese cabbage. Currently, with the complementation of whole-genome sequencing of Chinese cabbage 'Chiifu'[12], it is feasible to develop genome-wide SSR markers, and this will bring a revolution in the molecular marker-assisted breeding of Chinese cabbage[13−17].
In this study, SSRs were identified based on the whole-genome sequence information of Chinese cabbage and the characteristics of SSRs were also analyzed. Polymorphic analysis of SSR markers were performed and results were compared with previous research to check the variations. The SSR markers related to bolting and flowering time have also been identified, which provided a reference for the selection of candidate genes. This study provides a foundation for further utilization of SSR markers in Chinese cabbage.
-
The genome sequence of Chinese cabbage 'Chiifu' was used to analyse the distribution and characteristics of SSR loci. A total of 173,892 SSRs were detected throughout the Chinese cabbage genome. The abundance of repeat motifs from mono- to hexa-nucleotides were 131,636 (75.70%), 30,150 (17.34%), 10,885 (6.26%), 805 (0.46%), 225 (0.13%), and 191 (0.11%), respectively. The relative abundance of genome SSRs was 542.14 SSRs/Mb. The relative abundance of repeat motifs from mono- to hexa-nucleotides were 410.40, 94.00, 33.94, 2.51, 0.70, and 0.59 SSRs/Mb, respectively (Table 1). The average abundance/relative abundance from mono- to hexa-nucleotides repeat motifs at the chromosome level was 13,163.60/414.94, 3,015.00/95.23, 1,088.50/34.55, 80.50/2.54, 22.50/0.68, and 19.10/0.60, respectively. The greatest abundance from mono- to hexa-nucleotides repeat motifs occurred on chromosome 9, which were 20,423, 4,765, 1,581, 131, 47 and 31, respectively, while the lowest abundance were on chromosome 10, which were 7,725, 1,827, 742, 45, 12, and 11. The correlation analysis of SSR abundance and chromosome sizes indicated that the abundance of mono- and di-nucleotide repeat motifs positively correlated with chromosome size, with correlation coefficients of 0.913 and 0.955, respectively (Fig. 1). While abundance of repeat motifs from tri- to hexa-nucleotides were not correlated with chromosome size.
Table 1. Abundance and relative abundance of SSRs in the genome of Chinese cabbage.
Chromosome no. Chromosome size (Mb) Repeat type Total Mono-
nucleotideDi-
nucleotideTri-
nucleotideTetra-
nucleotidePenta-
nucleotideHexa-
nucleotide1 32.85 13,291/404.60 3,092/94.12 1,113/33.88 85/2.59 22/0.67 20/0.61 17,623/536.47 2 29.51 13,111/444.29 2,974/100.78 1,035/35.07 77/2.61 18/0.61 22/0.75 17,237/584.11 3 35.35 16,379/463.34 3,424/96.86 1,484/41.98 84/2.38 28/0.79 20/0.57 21,419/605.91 4 22.75 9,743/428.26 2,330/102.42 771/33.89 69/3.03 13/0.57 11/0.48 12,937/568.66 5 35.02 12,994/371.05 3,083/88.04 1,106/31.58 72/2.06 19/0.54 24/0.69 17,298/493.95 6 38.65 13,949/360.91 3,192/82.59 1,154/29.86 91/2.35 30/0.78 19/0.49 18,435/476.97 7 28.86 12,633/437.73 2,914/100.97 1,003/34.75 86/2.98 14/0.49 16/0.55 16,666/577.48 8 26.88 11,388/423.66 2,549/94.83 896/ 33.33 65/2.42 22/0.82 17/0.63 14,937/555.69 9 52.89 20,423/386.14 4,765/90.09 1,581/29.89 131/2.48 47/0.89 31/0.59 26,978/510.08 10 17.99 7,725/429.41 1,827/101.56 742/41.25 45/2.50 12/0.67 11/0.61 10,362/575.99 Total 320.75 131,636/410.40 30,150/94.00 10,885/33.94 805/2.51 225/0.70 191/0.59 173,892/542.14 The number that before and after “/” represent SSRs abundance and relative abundance 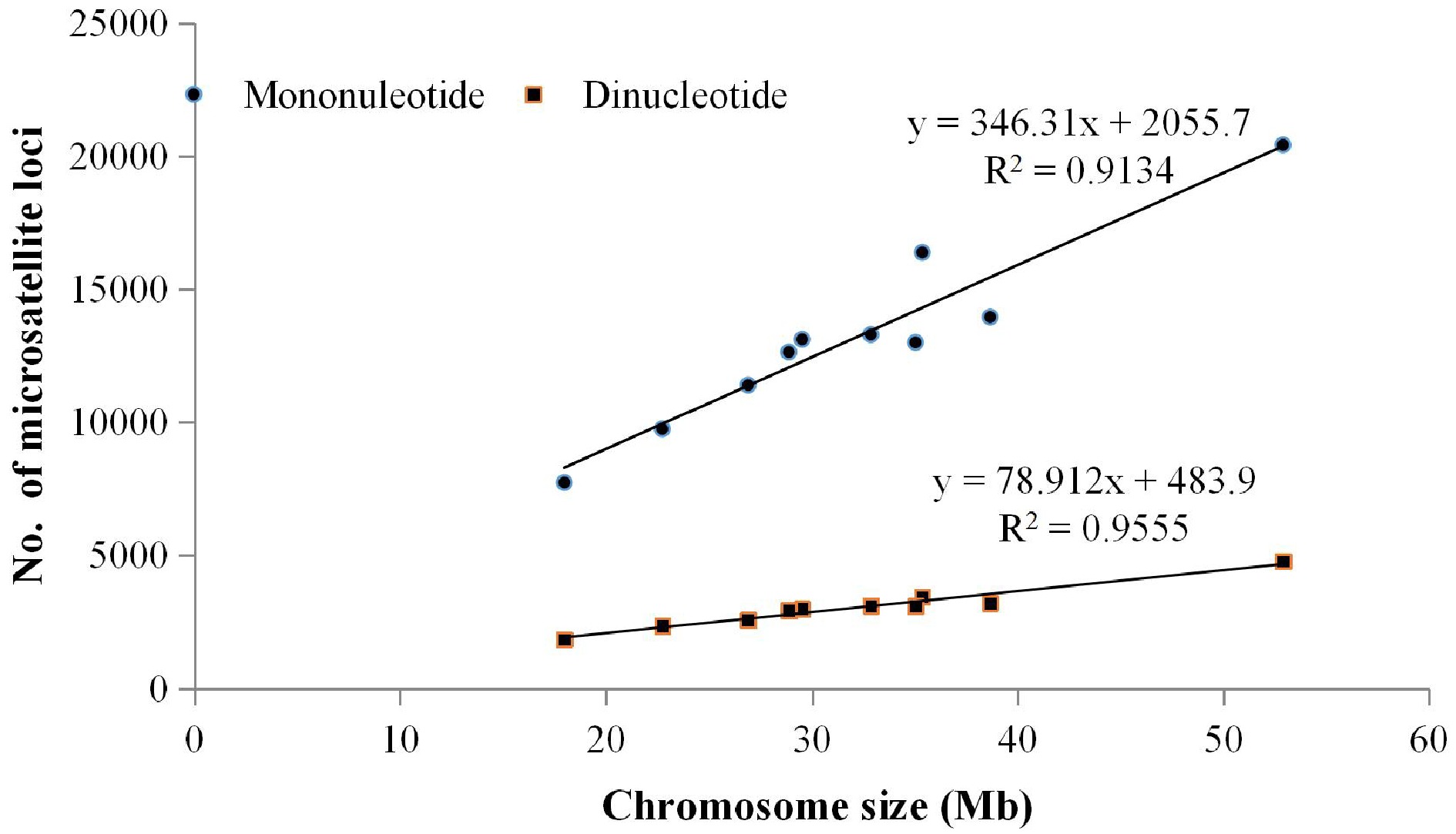
Figure 1.
Correlation analysis between chromosome size and abundance of the mono-nucleotide and di-nucleotide repeat motifs.
Types of SSR markers
-
MISA (MIcroSAtellite) software with Perl was used to identify the SSR sequence types. It was found that P type (perfect repeat sequences) were the main form of SSR sequence type with a total number of 166,827 sequences found in the 'Chiifu' genome (version 3.0), representing 95.94% of all SSRs, followed by C type (compound sequences) and I type (imperfect repeat sequences) sequences. The total number of the latter two types was 7,065, which represented 4.06% of the total SSRs. The number of P type SSR markers were significantly higher than C or I type markers; therefore, the distribution of P type markers in the Chinese cabbage genome was analyzed in this study. The number of P type markers differed across chromosomes (Fig. 2). Most P type SSR markers (25,866, 95.88%) occurred on chromosome 9, while the least occurred on chromosome 10 (9,947, 95.88%). The total number of P type SSR markers on chromosome 9 was more than twice as high as that on chromosome 10. Although the numbers of P type markers on each chromosome of the Chinese cabbage genome were quite different, the proportion of P type markers on each chromosome was slightly varied and ranged from 95.45% (chromosome 5) to 96.20% (chromosome 3) with a fluctuation range of only 0.75%.
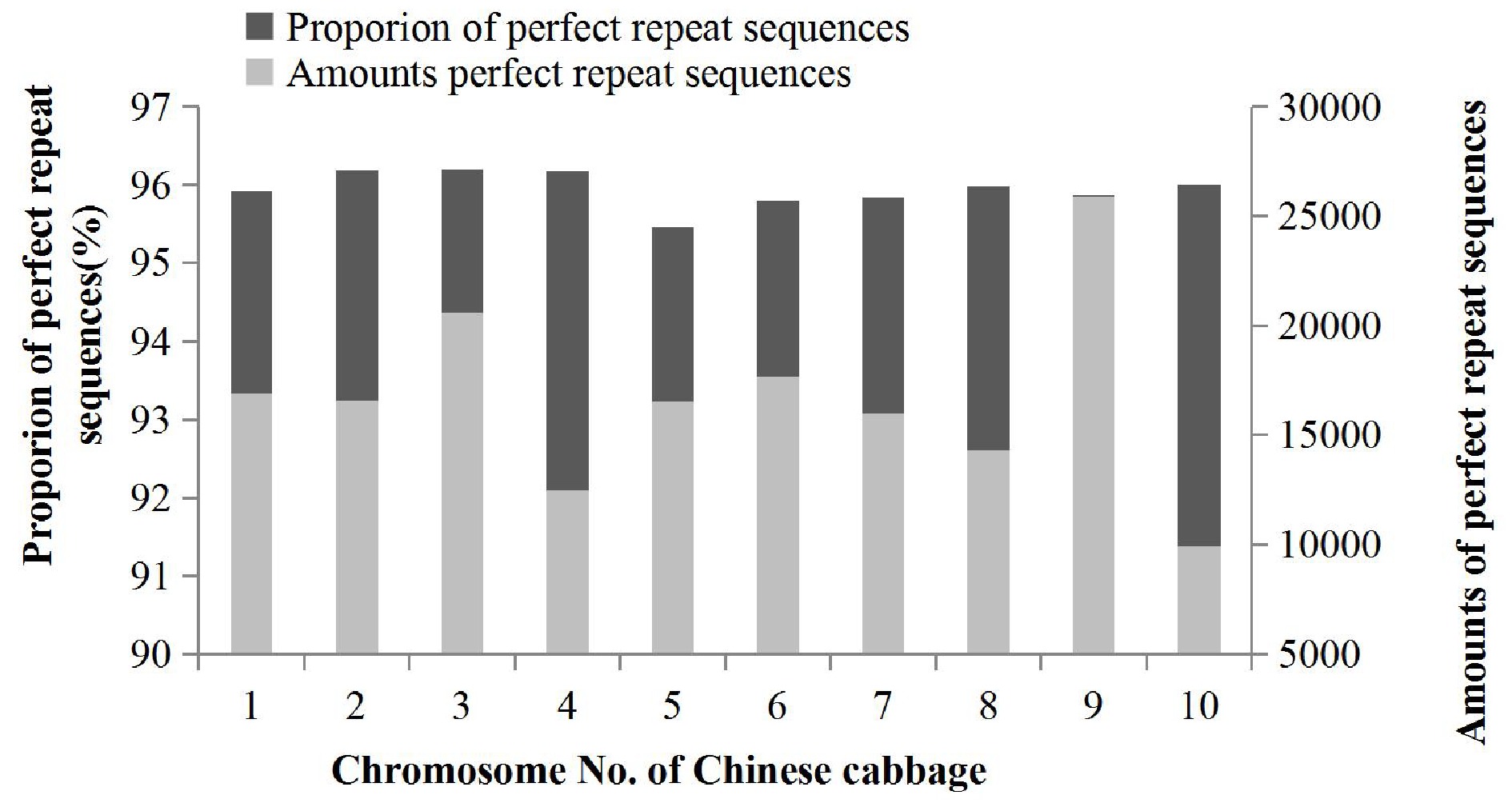
Figure 2.
Distribution of perfect repeat sequences in the Chinese cabbage genome.
Density of SSRs
-
Complete genome analysis of Chinese cabbage revealed that the SSR loci density was 1.89 kb/SSR. The densities of mono- to tri-nucleotide repeat motifs were 2.50, 10.89 and 30.18 kb/SSR respectively, and tetra- to hexa-nucleotides were 408.02, 1,459.81 and 1,719.67 kb/SSR respectively. The densities of mono- to tri-nucleotide repeat motifs were much higher than the others. The densities of SSR loci on 10 chromosomes were 1.91, 1.75, 1.69, 1.80, 2.07, 2.15, 1.77, 1.84, 2.01 and 1.78 kb/SSR. The maximum and minimum densities were found on chromosome 3 (1.69 kb/SSR) and chromosome 6 (2.15 kb/SSR) respectively. Further analysis revealed that the greatest densities of mono- to hexa-nucleotides repeat motifs were on chromosome 3 (2.21 kb/SSR), 4 (10.00 kb/SSR), 3 (24.39 kb/SSR), 4 (337.68 kb/SSR), 9 (1,152.28 kb/SSR) and 2 (1,373.59 kb/SSR) respectively. The smallest densities of mono- to tri-nucleotide repeat motifs were found on chromosome 6, which were 2.84, 12.40, and 34.29 kb/SSR respectively. While the smallest densities of tetra- to hexa-nucleotides were found on chromosome 5 (498.01 kb/SSR), 7 (2,110.86 kb/SSR) and 4 (2,118.18 kb/SSR). Additionally, the mono- to tri-nucleotide repeat motifs were more equally distributed across chromosomes compared with the others (Fig. 3).
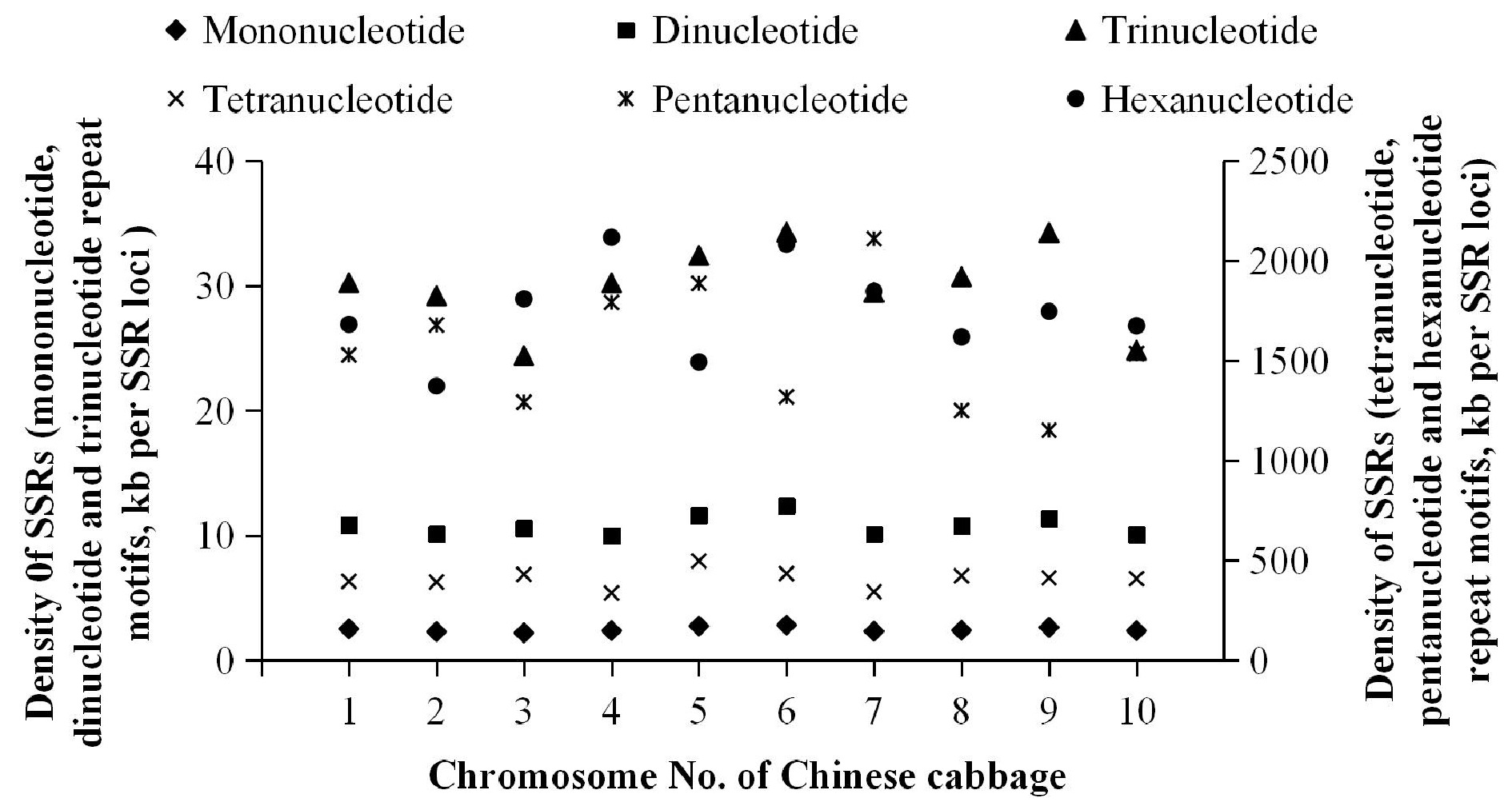
Figure 3.
Density of SSRs in Chinese cabbage.
SSR repeat motifs
-
A detailed analysis of the individual repeat motifs for each type of SSR in the genomic sequences of Chinese cabbage was conducted (Table 2). The number of dominant repeat motifs was 154,422 representing 95.94% of all motifs. A/T (128,489, 97.61%), AT/TA (19,311, 64.05%) and AAC/GTT/AAG/CTT/AAT/ATT (6,622, 60.84%) were the most frequent motifs from mono- to tri-nucleotides found in the Chinese cabbage genome respectively. Overall, the dominant repeat motifs from mono- to tri-nucleotides were rich in A/T. There were 10 repeat types of SSR loci with tetra- to hexa-nucleotides and the total number of repeat motifs was 154,422, accounting for only 0.70% of the whole genome. Many other kinds of dominant repeat motifs were identified but in minor quantities therefore they were not analyzed.
Table 2. Amounts of different SSR repeat motifs in the genome of Chinese cabbage.
Chromosome
no.Mono-
nucleotideDi-nucleotide Tri-nucleotide 1 A/T(12977) AC/GT(168) AG/CT(952) AAC/GTT(150) AAG/CTT(313) AAT/ATT(206) ACC/GGT(86) ACG/CGT(22) C/G(314) AT/TA(1971) CG/GC(1) ACT/AGT(12) AGC/CTG(36) AGG/CCT(129) ATC/ATG(136) CCG/CGG(23) 2 A/T(12822) AC/GT(155) AG/CT(753) AAC/GTT(98) AAG/CTT(331) AAT/ATT(201) ACC/GGT(56) ACG/CGT(29) C/G(289) AT/TA(2064) CG/GC(2) ACT/AGT(18) AGC/CTG(18) AGG/CCT(117) ATC/ATG(147) CCG/CGG(20) 3 A/T(16090) AC/GT(208) AG/CT(1080) AAC/GTT(182) AAG/CTT(469) AAT/ATT(223) ACC/GGT(100) ACG/CGT(24) C/G(289) AT/TA(2136) CG/GC(0) ACT/AGT(34) AGC/CTG(46) AGG/CCT(162) ATC/ATG(217) CCG/CGG(27) 4 A/T(9505) AC/GT(127) AG/CT(653) AAC/GTT(89) AAG/CTT(242) AAT/ATT(140) ACC/GGT(68) ACG/CGT(11) C/G(238) AT/TA(1550) CG/GC(0) ACT/AGT(7) AGC/CTG(30) AGG/CCT(89) ATC/ATG(80) CCG/CGG(15) 5 A/T(12623) AC/GT(183) AG/CT(1001) AAC/GTT(125) AAG/CTT(314) AAT/ATT(221) ACC/GGT(74) ACG/CGT(11) C/G(371) AT/TA(1898) CG/GC(1) ACT/AGT(24) AGC/CTG(37) AGG/CCT(138) ATC/ATG(140) CCG/CGG(22) 6 A/T(13546) AC/GT(167) AG/CT(1033) AAC/GTT(138) AAG/CTT(348) AAT/ATT(235) ACC/GGT(63) ACG/CGT(21) C/G(403) AT/TA(1992) CG/GC(0) ACT/AGT(30) AGC/CTG(34) AGG/CCT(113) ATC/ATG(151) CCG/CGG(21) 7 A/T(12388) AC/GT(146) AG/CT(882) AAC/GTT(126) AAG/CTT(345) AAT/ATT(175) ACC/GGT(57) ACG/CGT(5) C/G(245) AT/TA(1885) CG/GC(1) ACT/AGT(18) AGC/CTG(36) AGG/CCT(87) ATC/ATG(142) CCG/CGG(12) 8 A/T(11168) AC/GT(119) AG/CT(799) AAC/GTT(90) AAG/CTT(295) AAT/ATT(162) ACC/GGT(74) ACG/CGT(11) C/G(220) AT/TA(1630) CG/GC(1) ACT/AGT(16) AGC/CTG(33) AGG/CCT(99) ATC/ATG(107) CCG/CGG(9) 9 A/T(19818) AC/GT(262) AG/CT(1383) AAC/GTT(161) AAG/CTT(505) AAT/ATT(269) ACC/GGT(116) ACG/CGT(26) C/G(605) AT/TA(3119) CG/GC(1) ACT/AGT(20) AGC/CTG(59) AGG/CCT(164) ATC/ATG(238) CCG/CGG(23) 10 A/T(7552) AC/GT(106) AG/CT(655) AAC/GTT(95) AAG/CTT(245) AAT/ATT(129) ACC/GGT(45) ACG/CGT(14) C/G(173) AT/TA(1066) CG/GC(0) ACT/AGT(19) AGC/CTG(23) AGG/CCT(74) ATC/ATG(87) CCG/CGG(11) Polymorphic analysis of SSR markers
-
To detect SSR polymorphisms, a total of 200 SSR primer pairs are selected randomly and screened using 12 Chinese cabbage accessions. The results showed that 30 (15.0%) are highly polymorphic (Supplemental Fig. S1a), 74 (37.0%) had low polymorphisms (Supplemental Fig. S1b), 81 (40.5%) were without polymorphisms (Supplemental Fig. S1c) and 15 (7.5%) resulted in no amplification products (Supplemental Fig. S1d). A total of 30 polymorphic primer pairs were used to amplify 190 Chinese cabbage accessions. Ninety-two polymorphic bands were gained and the number of polymorphic fragments ranged from 2–6 with an average of 3.1 for each primer. The primer pairs M51 and M127, located on chromosomes 2 and 6 respectively, resulted in the greatest number of alleles (6) . Most materials (average 74.2%) showed one allele (Supplemental Table S1).
Bolting and flowering times of the Chinese cabbage accessions
-
The 190 Chinese cabbage accessions are divided into two subpopulations by population structure analysis, which are shown as S1 and S2 (Supplemental Fig. S2). To analyze the association of the identified SSR markers with bolting and flowering time, a phenotyping analysis was performed and the results revealed that the bolting times of all accessions are in the range of 72–79 d (31), 80–89 d (80), 90–99 d (62), and 100–107 d (17). The bolting time of most accessions in S1 (50.0%) ranged from 80–89 d, while most in S2 (45.7%) ranged from 90–99 d (Supplemental Fig. S3). The flowering times of all accessions ranged from 83–89 d (43), 90–99 d (75), 100–109 d (68), and 110–112 d (4). The flowering time of most accessions in S1 (42.9%) ranged from 90–99 d, while most in S2 (54.3%) ranged from 100–109 d (Supplemental Fig. S4).
Association analysis of bolting and flowering time with SSR markers
-
To reveal loci associated with bolting and flowering time, we performed an association analysis and detected 12 alleles from eight SSR markers located on chromosomes A01, A02, A03, A06, and A07 (p < 0.01). Among these alleles, nine were associated with bolting time and the M127(a) allele had the highest association degree (p < 1 × 10−5; Supplemental Table S2), while 10 were associated with flowering time and the M127(a) allele had the highest association degree (p = 3 × 10−5). Overall, seven alleles were associated with both traits.
-
In this study, mono- to tri-nucleotide repeat motifs of the entire genome were found to account for 73.89%, 11.11%, and 3.81%, respectively, while tetra- to hexa-nucleotide motifs accounted for only 0.70%. The frequencies of mono- to tri-nucleotide repeat motif SSRs were significantly higher than those of tetra- to hexa-nucleotide repeats, indicating that as the number of nucleotides in the repeat motifs increased, the frequency of SSRs decreased. This phenomenon is consistent with the findings in many other plant genomes, including Arabidopsis thaliana, tomato, apple, and Chinese jujube[10, 11, 18−20]. By contrast, in the genomes of alfalfa rhizobium, soybean rhizobium, bacillus rhizobium, and Caenorhabditis elegans, tetra- to hexa-nucleotide repeat motifs are significantly more than other types[21, 22]. Previous studies suggest that species with a large number of short repeat-type SSR loci generally exhibit a higher genomic mutation rate or evolution level[21] while the opposite result has been obtained from species with long repeat-type as the dominant SSR loci[22, 23]. Therefore, the highest proportion of short repeat-type SSR loci in the Chinese cabbage genome indicated that its genome has a long evolutionary history or a high mutation rate.
Distribution characteristics of SSRs
-
The P type SSRs were found to be the main SSR sequence type identified in the 'Chiifu' genome, accounting for 95.94% of all SSRs. The number of P type SSRs on each chromosome in the Chinese cabbage genome differed considerably (9,947–25,866), while the proportion of P type sequences on each chromosome had little variation (95.45%–96.20%). These results were consistent with the findings in the apple genome[10], however the underlying reasons require further investigation. The abundance and density analysis of repeat motifs indicated that the abundances of mono- and di-nucleotide repeat motifs positively correlated with chromosome size while the abundances of tri- to hexa-nucleotide repeats did not. The mono- to tri-nucleotide repeat motifs were equally distributed across chromosomes; however tetra- to hexa-nucleotide repeats did not show obvious abundance or distribution patterns due to their diverse types and small quantities.
The base composition analysis of repeat motifs indicated that the mono-nucleotide repeats exhibited a strong bias toward A/T motifs (97.61%) and the base frequency of A/T was significantly higher than that of C/G in di-nucleotide and tri-nucleotide motifs. These findings were consistent with the results from a previous study on eukaryotic genomes[21] and it was speculated that high A/T frequency resulted from the conversion of cytosine to thymine by DNA methylation[24]. In some monocotyledons such as wheat, corn and rice CCG/CGG was the primary tri-nucleotide motif while the proportion of CCG/CGG motif was very small in some di-cotyledons such as Arabidopsis thaliana, soybean, apple and Chinese jujube[10, 11, 25]. The high GC content of tri-nucleotide repeats in monocotyledons is possibly caused by codon bias[26, 27].
Usefulness of the newly developed genome-wide SSRs
-
Brassica rapa contains several subspecies such as Chinese cabbage (B. rapa ssp. pekinensis), non-heading Chinese cabbage (NHCC) and turnip (B. rapa ssp. rapa). Chinese cabbage and NHCC differ in morphological characteristics, planting areas and growth habits. NHCC is without heading and is widely used as a leaf vegetable and cultivated in Southeast Asia, Japan, USA, and Europe[28]. Chinese cabbage is with leafy heads and curling, crinkling leaves. It is grown worldwide, with main production and consumption area's in Asia and Europe. Compared to the non-heading accessions, the heading types can be transported easily, stored for a long time and have better cold tolerance and higher yield[9]. In a previous study, the systematic analysis and identification of SSRs was carried out in NHCC[28].With the update of the genome information of Chinese cabbage, we report, for the first time, the development of whole-genome SSR markers with the version 3.0 genome of Chinese cabbage 'Chiifu'.
Our study identified SSR loci with a higher total amount (173,892 SSRs vs. 20,836 in NHCC) and density (542.14 SSRs/Mb vs. 62.93 SSRs/Mb). This is likely due to the new version of genome information of Chinese cabbage. The repeat types is abundant in NHCC, which varied from mono-nucleotide to nona-nucleotide. The repeat types varied from mono-nucleotide to hexa-nucleotides in Chinese cabbage. In NHCC, the most type was AG/CT, followed by A/T, and di-nucleotides were the most common SSR type, followed by mono- and tri-nucleotides. In Chinese cabbage, the most type was A/T , and followed by AT/TA. Mono-nucleotide was the most common SSR type, followed by di- and tri-nucleotides. The results showed that A/T was the main type both in NHCC and Chinese cabbage. It was speculated that high A/T frequency resulted from the conversion of cytosine to thymine by DNA methylation[24]. These findings were consistent with the results from a previous study on eukaryotic genomes[21]. The polymorphisms are also different between the two studies. In NHCC, 74 primer pairs were randomly selected for validation. Among these, 60 (81.08%) were polymorphic in 18 Cruciferae. The number of polymorphic bands ranged from two to five, with an average of 2.70 for each primer. In Chinese cabbage, for the amplification of 12 Chinese cabbage germplasm, 200 SSR markers were used in which 30 (15.0%) were highly polymorphic. The number of polymorphic bands ranged from two to six, with an average of 3.1 for each primer. The polymorphisms of SSR markers in NHCC are much higher than that in Chinese cabbage. However, the number of polymorphic bands is much more in Chinese cabbage. The genome-wide SSR markers developed could provide a useful tool to marker-assisted selection in Chinese cabbage breeding.
Most genetic research in Chinese cabbage has been performed using low-throughput markers such as RAPD, RFLP and AFLP, which is laborious and time-consuming, and only a few important economic genes or loci have been detected in Chinese cabbage[29]. SSR markers have been proven useful for population genetics of numerous species[6, 30, 31]. With the development of bioinformatics and NGS technology, identification of a large number of SSR markers by genome sequencing has become convenient and feasible[32]. In soybean, 323 cultivar accessions and 101 SSR markers were used to detect the marker-phenotype associations revealing 79 marker-trait associations including 44 SSR markers related to the traits of 100-pod weight, 100-seed weight, sucrose content and free amino acid content[33]. In another study, eight unique loci were discovered by analyzing phenotypes and whole-genome InDel/SSR loci between the elite cabbage inbred-line '01-20' and its sister line '01-07-258' with five loci associated with important agronomic traits based on the QTL association results[34]. The developed SSRs revealed that significant loci associated with excellent agronomic traits.
The same Chinese cabbage accessions were used to find out the bolting and flowering time related loci in 2012[35]. Fifty-seven SSR and InDel markers were selected for the amplification of 190 accessions of Chinese cabbage. By using population genetic structure analysis, accessions were divided into three subpopulations (S1, S2 and S3). S1 and S3, S2 were consisted on 48 F1 and 70 inbred, 62 F1 and 10 inbred lines respectively. Previously, using 13 primers 17 bolting and flowering time related alleles were identified which were present on chromosome numbers 1, 2, 3, 6, 7, 8 and 10. In this study, the same accessions were divided into only two subpopulations (S1 and S2). S1 and S2 were consisted on (62, 48) F1 and (30, 50) inbred lines respectively. The result was consistent with the research carried out in previously[35].
To compare association analysis 30 polymorphic SSR markers were used, 12 alleles were detected by 8 SSR markers which were located on Chromosome numbers 1, 2, 3, 6 and 7. The SSR makers M2, M10, M50, M69, M127, M141 and M144 were related to flowering. Among them, M50 and M69 located on Chromosome 2 and 3, which are near to the gene FLC that has been known to control flowering. M2 and M141 located on Chromosome 1 and 6, which near to the gene AGL24 and FT that are also regulators of flowering. We also found that M10, M127 and M144 located on Chromosome 1, 6 and 7 were related to flowering, which provided a reference for the selection of candidate genes. These studies support the convenient applications of SSR markers.
SSR loci are highly conserved among species which have a closer genetic relationship[36] , and SSR markers are universal among closely related species. Analysis of SSR loci in the genomes of sequenced species could be helpful for the development and genetic analysis of SSR markers in unsequenced closely related species. SSR markers developed based on the sequenced genome information which have a clear position, the corresponding SSR markers can be found quickly and accurately when a certain sequence on the chromosome is known. It is helps to share the SSR information and avoid repetitition. These SSR markers enhance the density of the existing genetic Chinese cabbage maps, which could also be a useful source for high-throughput QTL mapping and marker-assisted Chinese cabbage improvement.
-
A collection of 190 Chinese cabbage accessions was used in this study, including 80 inbred lines, 83 F1 which was obtained by crossing the aforementioned inbred lines, and 27 commercial varieties[35].
DNA extraction and analysis
-
DNA was extracted from the young leaves of 190 Chinese cabbage accessions using an improved cetyltrimethyl ammonium bromide (CTAB) method[37]. After extraction, 5–10 μL DNA solution was loaded on a 1.0% agarose gel to assess sample quality. Then, DNA quality and concentrations were further assessed using a NanoDrop2000 spectrophotometer.
Identification and characteristic analysis of SSRs
-
The genome sequence of Chinese cabbage 'Chiifu' was downloaded from the Brassica database (BRAD,
http://brassicadb.cn/#/ ). Identification of the SSRs in the Chinese cabbage genome was performed using the MISA (MIcroSAtellite) software with Perl. The following search criteria was implemented: ≥ 10 repeat units for mononucleotides, ≥ 7 repeat units for di-nucleotides and ≥ 5 repeat units for tri-, tetra-, penta-, and hexa-nucleotides. Interrupted compound SSRs were also selected when the bases interrupting two SSRs were ≤ 10 repeat units. SSRs are divided into three types[38]: perfect repeat sequences (P type), imperfect repeat sequences (I type) and compound sequences (C type). P type sequences were defined as alternating, tandem repeat motifs without interruption and without adjacent repeats of another sequence, such as (AG)10. I type sequences were defined as 2+ runs of uninterrupted repeats motifs separated by more than three consecutive non-repeat bases, such as (AG)6gc(AG)3gg(AG)7cg(AG)3. C type sequences were defined as runs of repeat motifs separated by more than three consecutive non-repeat bases from a run of ≥ 5 uninterrupted dinucleotide or longer sequence repeats, other than (XX)n, or from ≥ 10 uninterrupted mononucleotides, such as (AG)9gttagtta(AT)12.The proportion of three SSR repeat types of the total SSRs, the proportion of P type SSRs on 10 chromosomes, the abundance and relative abundance of SSRs, the density of SSRs, and in the end, types and quantities of SSR repeat motifs were analyzed.
Primer design for SSR markers
-
Based on the 200-bp flanking sequences of the developed SSRs, primer pairs were designed using Primer 5.0 with Perl. The parameters were set as follows: (a) GC content was ~50%; (b) the melting temperature (Tm) ranged from 45 to 65°C and the difference between upstream and downstream primers was < 5 °C; (c) primer lengths ranged from 22–25 bp and 19–21 bp; (d) the predicted target Polymerase Chain Reaction (PCR) products ranged from 100 to 400 bp; and (e) all of the other parameters were set to default values. A total of 200 SSR primers were selected from the newly designed primers and synthesized by Sangon Biotech Co., Ltd. (Shanghai, China).
PCR and fragment analysis
-
A total of 200 SSR primer pairs were used to detect SSR polymorphisms among the 12 accessions selected from the 190 Chinese cabbage accessions. PCR was performed with a total volume of 10.0 μL containing 1.0 μL 50 ng/μL genomic DNA, 0.1 μL 2.5 U/µL Taq DNA polymerase (TaKaRa, Dalian, China), 0.5 μL 50 ng/µL each of forward and reverse primers, 1 µL 10 × PCR Buffer (Mg2+), 0.8 µL 2.5 mmol/L dNTPs, and 6.1 µL H2O. PCR optimization was as follows: initial denaturation at 94 °C for 3 min, 35 cycles at 94 °C for 45 s , 30 s at the appropriate annealing temperature, extension at 72 °C for 45 s, and a final elongation at 72 °C for 5 min. Finally, the PCR products were assessed for SSR polymorphisms on 7% denaturing polyacrylamide gels and visualized by silver nitrate staining. The high polymorphisms SSR primer pairs which could separate the parents, amplify multiple alleles, the amplification fragments obtained are clear and with great difference in different accessions. The opposite are the primers with low polymorphism.
This work was supported by the fundings from the National Natural Science Foundation of China (31930098, 32172594), Hebei Provincial Department of Human Resources and Social Security (C20210363), Hebei Provincial Department of Science and Technology (216Z2904G), and the Natural Science Foundation of Heibei (C2020204111).
-
The authors declare that they have no conflict of interest.
-
# These authors contributed equally: Ying Gao, Na Li
- Supplementary Figure S1 Polymorphism screening result of some SSR primer pairs from Chinese cabbage accessions. A, results from primer M143 with high polymorphism, B, results from M173 with low polymorphism, C, results from M112 without polymorphism, D, results from M20 without amplification products, 1~12 represent 12 Chinese cabbage inbred lines.
- Supplementary Figure S2 Result from population structure analysis of Chinese cabbage Red and green show the estimated membership fractions of each individual in S1 and S2.The classification of accessions is based on the ratio of different colors.
- Supplementary Figure S3 Bolting time of Chinese cabbage accessions.
- Supplementary Figure S4 Flowering time of Chinese cabbage accessions
- Supplementary Table S1 Alleles of 30 SSR primers amplified in 190 Chinese cabbage accessions
- Supplementary Table S2 Marker loci associated with bolting and flowering time and their P values.
- Copyright: © 2022 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Gao Y, Li N, Li X, Lu Y, Feng D, et al. 2022. Genome-wide development and utilization of Simple Sequence Repeats in Chinese cabbage (Brassica rapa L. ssp. pekinensis). Vegetable Research 2:9 doi: 10.48130/VR-2022-0009 

Genome-wide development and utilization of Simple Sequence Repeats in Chinese cabbage (Brassica rapa L. ssp. pekinensis)
- Received: 26 March 2021
- Accepted: 18 July 2022
- Published online: 29 July 2022
Abstract: With the complementation of whole-genome sequencing of Chinese cabbage, it is necessary to develop genome-wide Simple Sequence Repeat (SSR) markers and analyse their characteristics, which will bring a revolution in the molecular marker-assisted breeding of Chinese cabbage. In this study distribution and characteristics of SSR loci in the genome of Chinese cabbage 'Chiifu' was analyzed. Finally, a total of 173,892 SSR markers that occurred in 10 chromosomes were identified, and Perfect Repeat Motifs (P type) which is a main form of SSR loci with a mean distance of 1.89 kb/SSR was identified. Among them, mono-, di-, and tri-nucleotide repeat motifs were equally distributed across chromosomes compared with tetra-, penta-, and hexa-nucleotide repeat motifs. The largest proportion of SSR loci consisted of mono- and dinucleotide repeat motifs, which accounted for 93.04% of the total SSR loci. A/T and AT/TA were the most abundant motifs, accounting for 97.61% and 64.05% of the mono- and dinucleotide repeats, respectively. Furthermore, 200 SSR markers were used for the amplification of 12 Chinese cabbage germplasm, in which 30 were highly polymorphic. These SSR markers were further used for the association analyses of flowering time of 190 Chinese cabbage accessions. The results revealed that 9 and 10 alleles were associated with bolting and flowering time, respectivley. The results show that development of SSR markers is feasible and useful in marker assisted selection of Chinese cabbage.
-
Key words:
- Chinese cabbage /
- Whole genome /
- SSR /
- Marker assisted selection