









-
Ranunculales is one of the early-diverging lineages among the basal angiosperms. As a sister group to other eudicots, it had already formed an independent evolutionary branch before the divergence of core eudicots[1]. Within this order, plants of the families Berberidaceae and Ranunculaceae are commonly used as medicinal resources, characterized by their high accumulation of benzylisoquinoline alkaloids (BIAs), such as berberine, tetrandrine, morphine, papaverine, and sanguinarine[2]. These compounds exhibit diverse physiological activities and ecological functions, including antimicrobial properties, defense against herbivores, as well as pharmacological effects like anticancer and analgesic actions[3,4]. To date, the biosynthetic pathways of BIAs have been extensively studied in several species, particularly in Coptis japonica, Papaver somniferum, Nelumbo nucifera, and Thalictrum flavum[5−10]. In these plants, the intermediate products and key enzymes involved in the BIAs biosynthetic pathway have been elucidated progressively.
In this biosynthetic pathway, the core intermediate (S)-Reticuline is formed through the catalysis of key rate-limiting enzymes, such as (S)-norcoclaurine synthase (NCS), norcoclaurine 6-O-methyltransferase (6OMT), coclaurine N-methyltransferase (CNMT), N-methylcoclaurine-3'-hydroxylase (NMCH), and 4'-O-methyltransferase (4'OMT)[11]. These rate-limiting enzyme genes originated in ancient terrestrial plants and underwent multiple duplication events prior to the divergence of core angiosperms. However, throughout the course of evolution, the vast majority of monocots and core eudicots have systematically lost these duplicated gene copies, leading to the loss of their biosynthetic capabilities of BIAs[12].
Previous genome-based studies of representative Ranunculales species, such as Coptis japonica (Ranunculaceae), Papaver somniferum (Papaveraceae), and Epimedium pubescens[5,8,13], have shown that whole-genome duplication (WGD) and local gene duplication events are closely associated with the expansion of key enzyme gene families involved in alkaloid biosynthesis. Meanwhile, pronounced differences in alkaloid composition and types among Ranunculales species suggest that their secondary metabolic systems have undergone complex and lineage-specific diversification during evolution. However, most of these conclusions are derived from a limited number of model or representative species, and their general applicability at the order-wide scale of Ranunculales remains largely untested. Therefore, a comprehensive order-level analysis of the evolutionary dynamics of key enzymes in the benzylisoquinoline alkaloid biosynthetic pathway is essential for elucidating the mechanisms and evolutionary history underlying chemical diversity in Ranunculales plants.
Transcriptomic information for a total of 319 species was acquired by integrating leaf transcriptome sequencing data from 227 Ranunculales species with open-access data from National Center for Biotechnology Information (NCBI). A high-quality transcript dataset was constructed through a unified process. On this basis, functional annotation, structure analysis of motifs, selection pressure assessment, and expression level comparison were performed for several key enzymes involved in alkaloid biosynthesis, aiming to explore the sequence conservation and functional evolutionary trends of key enzymes in the benzylisoquinoline alkaloid biosynthetic pathway across different lineages within Ranunculales. This study not only provides rich genetic resources for research on secondary metabolism in Ranunculales plants, but also offers insights into understanding the intra-lineage evolutionary characteristics of key enzymes in specific metabolic pathways.
-
Fresh and healthy leaf tissues were collected from 227 species of Ranunculales. The samples were immediately flash-frozen in liquid nitrogen and stored at −80 °C for subsequent RNA extraction and analysis of key enzymes involved in alkaloid biosynthesis. Total RNA was extracted using the cetyltrimethylammonium bromide (CTAB) method, followed by DNase I treatment. After confirming RNA integrity with an Agilent 2100 Bioanalyzer, high-quality RNA was used to construct sequencing libraries following the standard library preparation protocol. Paired-end sequencing was performed on a BGISEQ-500 platform. To enhance data coverage and lineage representation, transcriptome data of an additional 92 Ranunculales species were downloaded from the NCBI database. In total, transcriptomic data from 319 species were processed and analyzed (the species list is provided in Supplementary Table S1).
Data preprocessing, transcriptome assembly, and annotation
-
Raw sequencing data were quality-controlled using fastp (v0.23.4)[14], which included adapter trimming, removal of low-quality reads, and filtering of sequences containing ambiguous bases (N). Quality control results showed high rates of valid data retention across all samples. The cleaned data were then used for de novo transcriptome assembly with Trinity (v2.15.1) under default parameters. After obtaining the initial transcript sets, CD-HIT (v4.8.1)[15] was used to cluster and remove redundancy at a 95% sequence similarity threshold, retaining representative transcripts. Subsequently, the Trinity 'script get_longest_isoform_seq_per_trinity_gene.pl' was employed to extract the longest isoform per gene as the representative sequence for annotation and comparative analysis.
To assess the completeness of the assemblies, BUSCO (v5.4.4) was run against the embryophyta_odb10 database, and the proportions of 'complete', 'fragmented', and 'missing' genes were calculated. For functional annotation, BLASTALL (E-value ≤ 1e-5)[16] was used to align transcripts against multiple authoritative protein databases, including NR (
www.ncbi.nlm.nih.gov/refseq/about/nonredundantproteins ), Swiss-Prot (www.uniprot.org ), Pfam (https://pfam.xfam.org ), KEGG (www.genome.jp/kegg ), COG (www.ncbi.nlm.nih.gov/research/cog-project ), and GO (https://geneontology.org ). Transcription factors (TFs) were identified via the iTAK tool[17] by matching to known TF family definitions. Meanwhile, the MISA[18] software was applied to detect simple sequence repeats (SSRs) in the transcripts, and their types and distribution patterns were summarized.Transcript quantification and prediction of open reading frames and proteins
-
Expression levels of each gene across all species were obtained using the quantification module provided by Trinity in combination with the Salmon expression estimation software. First, the TransDecoder.LongOrfs module of TransDecoder[19] (v5.5.0) was applied to predict open reading frames (ORFs) from the longest transcript sequences. To perform protein homology alignment, protein sequence data for Viridiplantae were downloaded from the UniProt database, and a local protein database was constructed using BLAST to generate alignment files. Finally, based on the preceding results, TransDecoder was used to generate the predicted protein files.
Phylogenetic reconstruction and divergence time estimation
-
Based on the BUSCO assessment results, putative low-copy nuclear genes that were present in more than 80% of the sampled species were selected for phylogenetic analysis (corresponding to BUSCO-defined single-copy orthologs). For the coalescent-based approach, each single-copy gene was first aligned using MAFFT (v7.505)[20], and then TrimAl (v1.4.rev22)[21] was applied under stringent parameters to remove low-quality and highly gapped regions from the alignments, ensuring reliability. The processed alignments were separately input into IQ-TREE (2.1.4-beta)[22], where the model-selection option (-m MFP) was used to automatically determine the best substitution model. Phylogenetic inference was performed with 1,000 ultrafast bootstrap replicates to generate single-gene trees. All single-gene trees were further integrated with ASTRAL (v5.7.8)[23] to obtain a multi-gene consensus tree. For the concatenation-based approach, the aligned sequences of all single-copy genes were concatenated into a super-gene matrix after quality control. Model prediction and phylogenetic analysis were then conducted in IQ-TREE, with 1,000 ultrafast bootstrap replicates used to assess topological confidence. To obtain divergence time estimates, previously reported species divergence times from the TimeTree database (
https://timetree.org )[24] were incorporated as fossil calibration points. Specifically, the divergence time between Amborella trichopoda and Ranunculales was set to 196 Ma (95% confidence interval: 179.9–205 Ma). The inferred phylogenetic tree was subsequently time-calibrated using the r8s software[25], resulting in a time-scaled species phylogeny.Estimation of whole-genome duplication (WGD) events
-
To investigate ancient whole-genome duplication (WGD) events in Ranunculales, representative species were selected and analyzed using the WGD (v2.0) pipeline[26]. First, all-against-all BLAST searches were performed on the predicted protein sequences of each species, with an E-value set at '1e-5'[16], and homologous gene families were identified via the MCL clustering algorithm implemented in the WGD pipeline[27]. Phylogenetic trees were then constructed for each gene family by FastTree[28], and the distribution of synonymous substitution rates (Ks) between homologous gene pairs was estimated with the kd command in the wgd toolkit. In addition, using OrthoFinder(v2.5.5)[29] with default parameters, phylogenetic orthogroup inference was performed based on the protein sequences of all species to identify orthologous and paralogous relationships (Supplementary Table S2) and to construct the species tree. OrthoFinder assigns genes into orthogroups based on sequence similarity searches and graph-based clustering algorithms, and simultaneously infers gene trees for each gene family, thereby providing a robust homologous framework for subsequent WGD identification and evolutionary analyses. To further assess the reliability of potential WGD events, we performed Whale analyses based on the constructed gene family trees and the time-calibrated species phylogeny. WHALE was run with an MCMC chain length of 500,000 generations, sampling every 100 generations, with a burn-in of 100,000. Branches with posterior probability > 0.7 for gene duplication events were considered candidates for WGD.
Key enzyme profiling for alkaloid biosynthesis
-
To elucidate the biosynthetic mechanism of benzylisoquinoline alkaloids (BIAs), this study focused on two key enzymes in this pathway, norcoclaurine synthase (NCS) and N-methylcoclaurine-3'-hydroxylase (NMCH), as well as two key methyltransferase gene families, O-methyltransferases (OMT) and coclaurine N-methyltransferase (CNMT). Using previously collected known protein sequences as references, a combined BLASTP and HMMER strategy was employed to conduct homology searches and systematically identify homologous gene members for each target. Non-redundant sequences were further validated using the Conserved Domain Database (CDD;
www.ncbi.nlm.nih.gov/Structure/bwrpsb/bwrpsb.cgi )[30] to ensure accuracy.Based on the filtered sequences, phylogenetic trees were constructed separately with IQ-TREE to assess their lineage-specific evolutionary relationships. Conserved motifs in candidate proteins were identified using the MEME SUITE[31], and functional domains were annotated jointly with the Pfam database.
To investigate adaptive evolution of the genes, the aBSREL method in the HyPhy software package[32] was applied to detect signals of positive selection across branches, based on the phylogenetic trees and corresponding coding sequences (CDS). For lineages showing positive selection, the Hyphy MEME method was further used for site-specific analysis, and the detected codon sites were mapped to their corresponding positions in the protein sequences. To associate expression patterns with evolutionary features, gene expression levels were obtained from transcriptomic data and visualized in heatmaps. For proteins under positive selection, homology modeling was performed via the SWISS-MODEL server (
https://swissmodel.expasy.org/interactive )[33] to predict their three-dimensional structures. Finally, positive selection sites, key residues, catalytic sites, and predicted substrate-binding sites were spatially mapped and visualized on their three-dimensional structures via ChimeraX (v1.11)[34]. -
A total of 319 transcriptome datasets from plants of the order Ranales were obtained. (Supplementary Table S1). In total, 22.17 billion clean reads were obtained, corresponding to 3.23 Tb of clean bases, with an average data volume of 10.12 Gb per species (Supplementary Table S1). All transcriptome datasets were de novo assembled using Trinity[19], yielding a total of 61,487,655 transcripts with an average length of 858 bp and an N50 size of 1,489 bp (Fig. 1a, b; Supplementary Table S3). The completeness of the assembled transcriptomes was assessed using BUSCO (Benchmarking Universal Single-Copy Orthologs)[35]. The results showed that, on average, 89.73% of the conserved plant orthologs were present in the assembled transcriptomes (Fig. 1c), with detailed results provided in Supplementary Table S4. According to database annotations, 54% of the total transcripts could be annotated with known proteins or functional domains (Supplementary Table S5). A total of 530,459 transcription factors (TFs) were predicted from all transcripts, averaging 1,662 TFs per species. Among these, MYB/MYB-related constituted the most abundant TF family in the Ranunculales transcriptomes, followed by C2H2 and bHLH (Fig. 1d; Supplementary Table S6). Furthermore, 6,378,110 simple sequence repeats (SSRs) were identified within the Ranunculales transcriptomes (Fig. 1e; Supplementary Table S7).
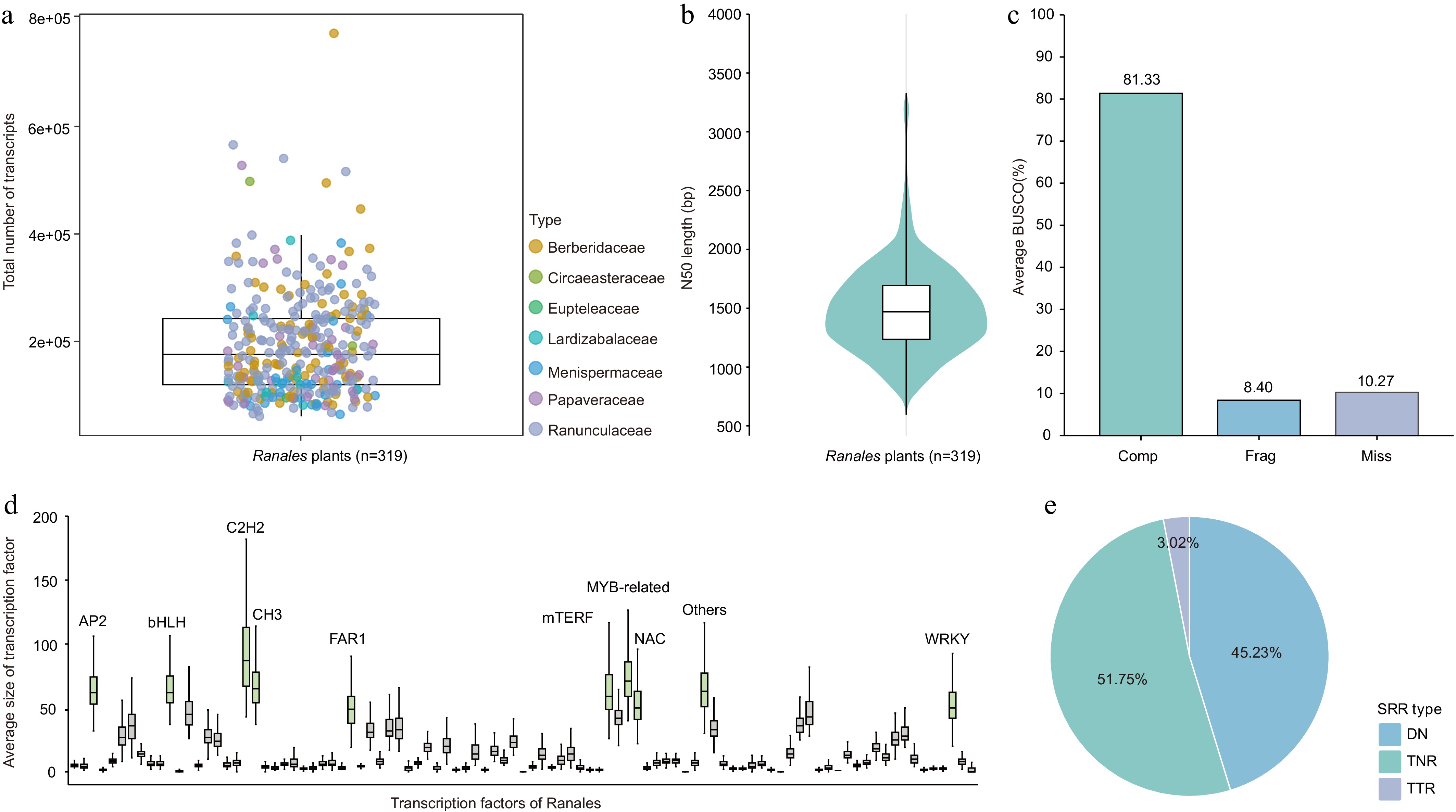
Figure 1.
Transcriptomic characterization of 319 Ranunculales species.
(a) Total assembled transcripts per plant. (b) Transcript N50 size across all plants. (c) BUSCO assessment of transcriptome assembly completeness: complete (Comp), fragmented (Frag), and missing (Miss). (d) Distribution of transcription factors in Ranunculales species, with TF families larger than 50 on average highlighted in green boxplots. (e) Proportional distribution of polymorphic SSR types across all plants, including dinucleotide (DNRs), trinucleotide (TNRs), and tetranucleotide (TTRs) repeats.Inferring phylogenetic relationships within Ranunculales from transcriptomes
-
The high-quality transcriptomic data of Ranunculales enabled the resolution of phylogenetic relationships within this order. First, Amborella trichopoda was selected as the outgroup for phylogenetic analysis. We then identified 733 putative low-copy nuclear genes that were present in more than 80% of the sampled species. These gene sequences were subjected to multiple sequence alignment and trimming to construct a sequence matrix for phylogenetic reconstruction. Finally, an appropriate evolutionary model was selected to infer the phylogenetic tree. The resulting (Fig. 2; Supplementary Fig. S1) showed that 79% of the branches received 100% bootstrap support. The analysis delineated three major clades within Ranunculales. Eupteleaceae occupied the earliest diverging basal position (bootstrap 98.2%), while Papaveraceae formed a distinct clade (bootstrap 100%); the remaining five families were divided into two groups, with Lardizabalaceae and Circaeasteraceae clustered closely together, while Menispermaceae served as the basal branch in the other group, with Berberidaceae and Ranunculaceae forming a sister group. Within this sister group, Berberidaceae clearly split into two subclades, whereas the relationships within Ranunculaceae were more complex. Divergence time estimation was performed to infer the timing of lineage differentiation within Ranunculales. The earliest divergence event, separating Eupteleaceae (Euptelea pleiosperma) from the remaining lineages, was estimated to have occurred approximately 119.97 (103.89, 128.9) million years ago (Mya). Overall, the major lineages of Ranunculales had completed their initial divergence by the Early Cretaceous, indicating that core Ranunculales underwent rapid radiation following the initial split of Eupteleaceae. Among them, the most important families, Berberidaceae and Ranunculaceae, diverged rapidly from 84.67 Mya to the present (Fig. 3).
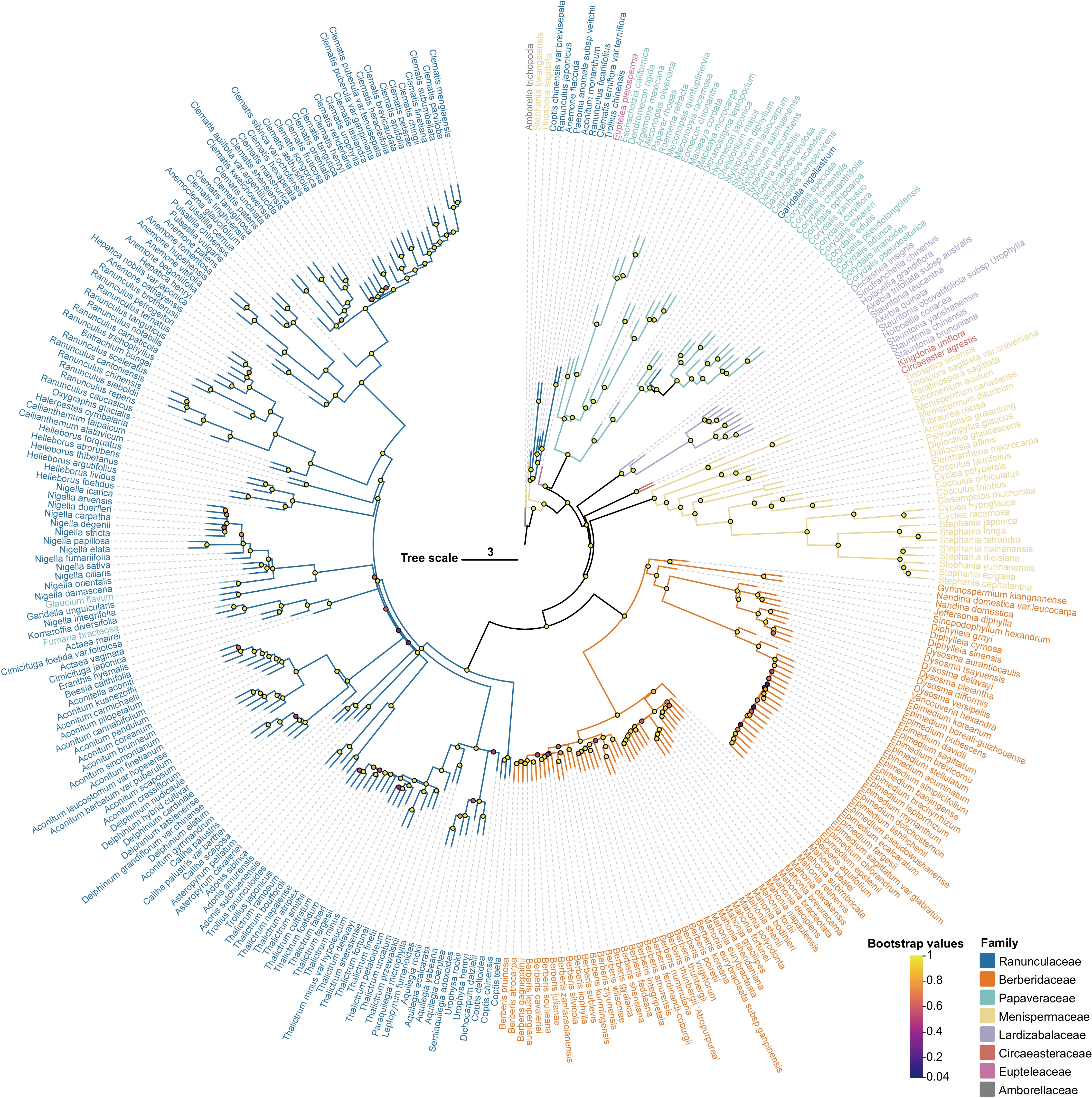
Figure 2.
Phylogenetic reconstruction of 319 Ranunculales species using maximum likelihood.
Bootstrap values are color-coded on the branches of the phylogeny. Excluding the outgroup, seven distinct colors represent the major families within Ranunculales: Ranunculaceae, Berberidaceae, Papaveraceae, Menispermaceae, Lardizabalaceae, Circaeasteraceae, and Eupteleaceae.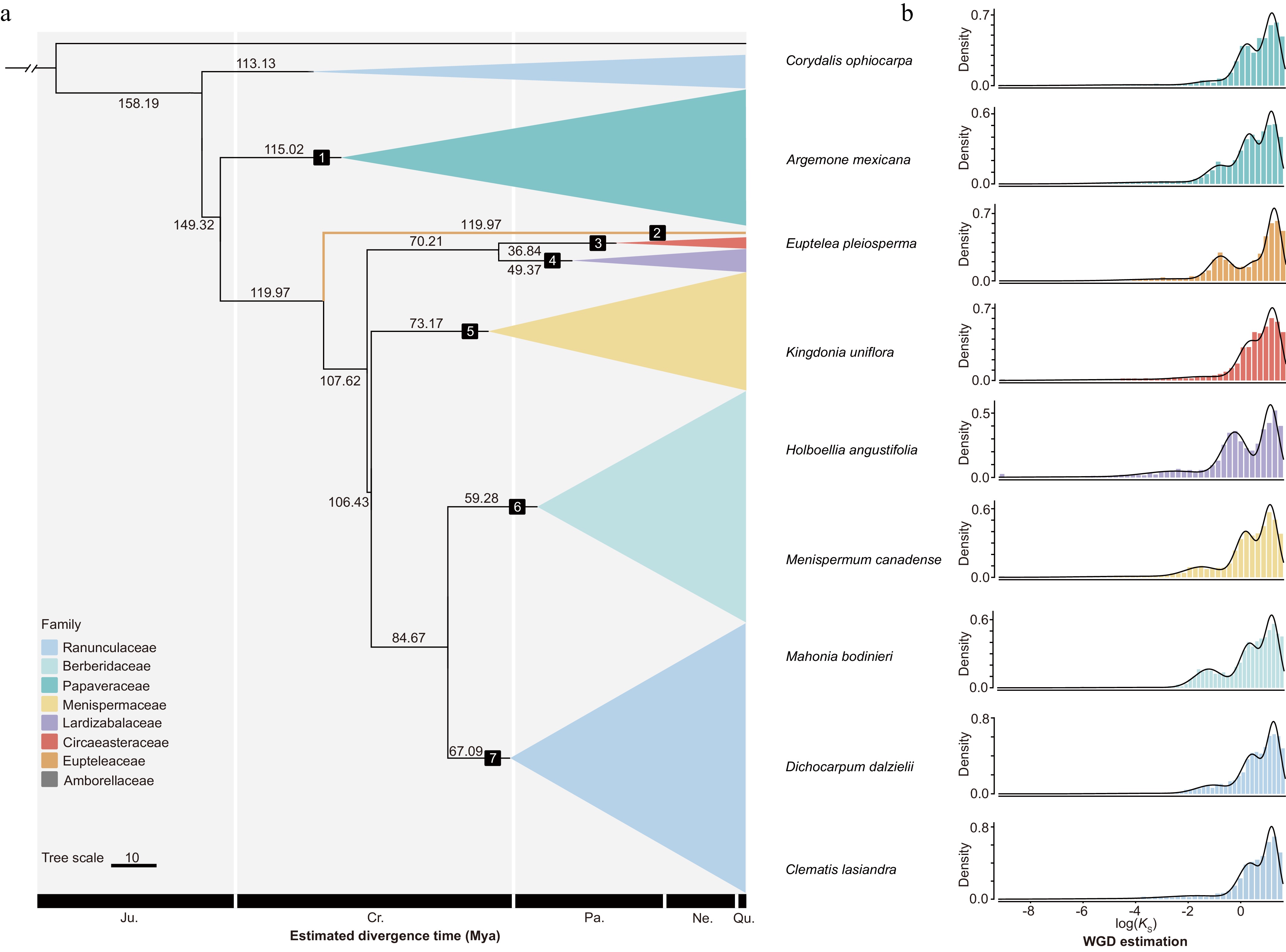
Figure 3.
Estimation of divergence times and whole-genome duplication events in Ranunculales.
(a) Divergence time inference: geological time scale abbreviations are shown at the bottom, with numbers on branches indicating divergence times in million years ago (Mya). (b) Inferred whole-genome duplication events in representative species of each Ranunculales family, based on KS distributions.Inference of multiple major duplication events
-
Previous studies have identified whole-genome duplication (WGD) as a major driver behind the origin and diversification of angiosperms[36−38], and genomic duplication events in Ranunculales were likely as complex as those in core eudicots[39]. In this study, we constructed Ks distributions and performed Gaussian Mixture Model (GMM) fitting based on homologous genes from 319 Ranunculales species. The analysis revealed multiple ancient and lineage-specific duplication signals, consistent with putative WGD events across the entire order.
The results of GMM decomposition revealed six major peaks across all Ks data, each corresponding to distinct duplication signals potentially associated with WGD events (Fig. 3; Supplementary Table S8).
Within individual species, the number of inferred Ks peaks ranged from one to three.
The GMM decomposition identified six major peaks across all Ks datasets; however, only five of these peaks were supported by Whale, with posterior probabilities greater than 0.7. Apart from the first peak, which was shared by all species, the second peak was widely distributed across multiple families: it was detected in 58.3% of Papaveraceae species, 51.7% of Menispermaceae, 83.75% of Berberidaceae, and 38.4% of Ranunculaceae, making it the most broadly distributed Ks signal aside from the common ancient peak. The third peak was exclusive to Papaveraceae. The fourth peak occurred only in Lardizabalaceae and was detected in all of its species. The fifth peak was found specifically in the genera Berberis and Mahonia within Berberidaceae.
Conservation and lineage-specific divergence in key enzymes for alkaloid biosynthesis
-
To identify genes potentially involved in the BIAs pathway (Fig. 4a), we investigated two key enzymes (Supplementary Table S9), norcoclaurine synthase (NCS) and N-methylcoclaurine 3'-hydroxylase (NMCH), based on the classical alkaloid biosynthetic pathway. The study encompassed phylogenetic analysis, alignment of key residues and catalytic determinants, as well as conserved motif analysis. From all transcripts, we identified 647 NCS members and 297 NMCH members (Supplementary Table S10). Among these, species from Ranunculaceae and Berberidaceae accounted for 72% and 73% of the total NCS (Supplementary Fig. S2) and NMCH members (Supplementary Fig. S3), respectively. Notably, NCS members were more abundant in Ranunculaceae, while NMCH members were more prevalent in Berberidaceae. Multiple sequence alignment revealed that only a minority of NCS sequences conserved key residues (20.7%) and catalytic determinants (43.8%) (Fig. 4c; Supplementary Table S11). However, certain sites showed species-specific variations. According to previous reports, NCS members can be classified into two main subtypes: NCS-I and NCS-II[10]. These two subtypes were distributed relatively evenly in Berberidaceae and Ranunculaceae, with strong branch support in phylogenetic reconstruction. Compared to the NCS-II clade, NCS-I contained 72 distinct sites, some of which were highly variable (Fig. 4d). Conservative motif analysis demonstrated that Motif1 through Motif6 exhibited extremely high statistical significance and were present in over 80% of sequences. Notably, Motif8 was exclusively identified within the Berberidaceae-specific NCS-I clade. In contrast, members of the NMCH family belong to the cytochrome P450 (CYP) superfamily[40]. Multiple sequence alignment similarly showed that 90% of sequences retained only one key residue, while 86.8% of catalytic determinants were highly conserved; however, characteristic variations were still observed at individual sites across different species (Supplementary Table S11, Supplementary Fig. S4).
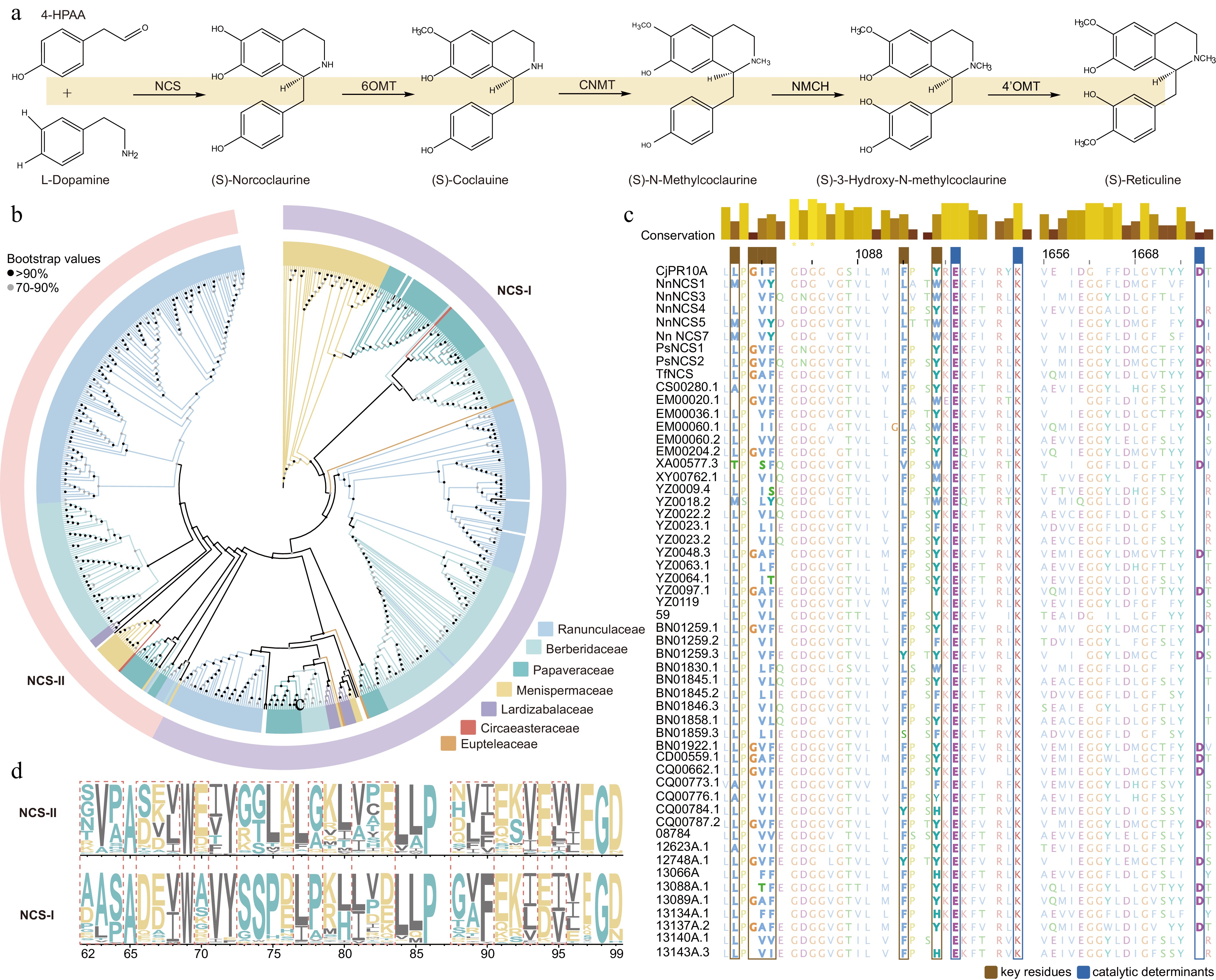
Figure 4.
Identification and comparative analysis of key alkaloid biosynthetic enzymes in Ranunculales.
(a) Initial steps of the classical alkaloid biosynthetic pathway leading to the core intermediate (S)-Reticuline. (b) Phylogenetic analysis of the Bet v1 gene family showing its division into two subfamilies: NCS-I and NCS-II. (c) Sequence alignment of key residues and catalytic determinants in selected important NCS sequences. (d) Sequence logo of multiple sequence alignments for the NCS-I and NCS-II clades, with dashed boxes highlighting residues that show significant differences between the two clades.Using the same methodology, we investigated the CNMT and OMT gene families, identifying 779 members in CNMT family and 676 members in OMT family (Supplementary Table S10). The CNMT gene family can be divided into two main evolutionary clades, CNMT-I and CNMT-II, with all known functional CNMTs belonging to CNMT-I[41]. Phylogenetic analysis revealed that genes from Berberidaceae dominate in CNMT-I, while genes from Ranunculaceae are more abundant in CNMT-II (Supplementary Fig. S5). Statistical analysis indicated 117 divergent sites between the functional CNMT members in the CNMT-II clade, 42 of which are located in conserved domains, with multiple regions exhibiting high-density variation (Supplementary Table S11, Supplementary Fig. S6). The OMT gene family involved in the BIA pathway was classified into 6OMT (257 members), 4'OMT (192 members), and 7OMT (268 members) based on the position of methylated groups in BIAs and specific amino acids in conserved catalytic residues[6,9,11,42]. Statistical results showed that Berberidaceae plants account for the highest proportion across these three subclasses, approximately 45%, indicating strong representation in BIA metabolism (Supplementary Table S11, Supplementary Figs S7, S8). The diversity and species-specific distribution of these genes suggest that some members may have undergone adaptive evolution. Therefore, we further conducted positive selection analysis to explore potential functional innovation and mechanisms of metabolic optimization.
Positive selection drives the functional diversification of keyenzymes
-
To further investigate the selective pressures acting on key genes in the BIAs pathway, we first conducted positive selection tests on the filtered key genes within their respective phylogenetic branches. The results revealed that among the homologous sequences, nine NCS sequences showed signals of positive selection, seven of which originated from Ranunculaceae. Similarly, nine NMCH homologous sequences were under positive selection, with four from Berberidaceae and three from Papaveraceae. Furthermore, 36 CNMT and 32 OMT homologous sequences exhibited positive selection, with the highest proportions of these sequences coming from Ranunculaceae, Berberidaceae, and Papaveraceae (Supplementary Table S12). Subsequently, we quantitatively compared these positively selected genes with their sister-group genes by analyzing the FPKM values of the homologous sequences for NCS, NMCH, CNMT, and OMT. The results indicated that the expression levels of genes in the positively selected branches did not show consistent or systematic changes compared to other genes across the major tissues examined (Fig. 5a; Supplementary Table S13).
Further, we conducted positive selection tests at the site-level on the positively selected sequences and identified multiple codon sites under episodic positive selection (Supplementary Table S11). Among these, positively selected sites were relatively fewer in NCS sequences, with a maximum of only 20 sites, whereas NMCH, CNMT, and OMT sequences contained more positively selected sites, exhibiting uneven distribution. By integrating homology modeling, we predicted the three-dimensional protein structures and mapped the positively selected sites, key residue positions, and catalytic determinant sites onto the structural models (Fig. 5b; Supplementary Fig. S9).
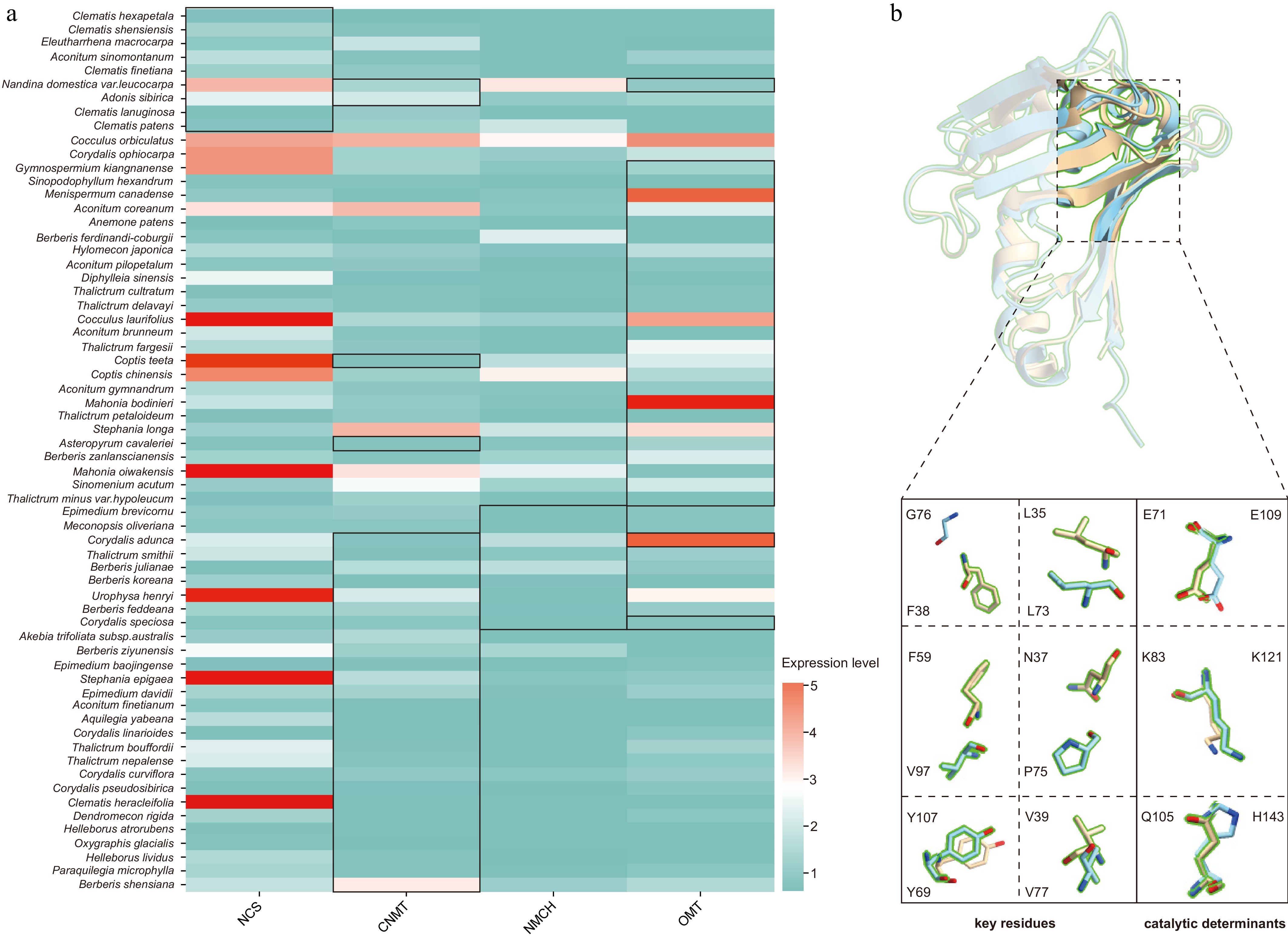
Figure 5.
Gene expression and structural variation of positively selected key enzymes.
(a) Expression levels of key sequences visualized using log2-transformed FPKM normalized counts, with sequences under positive selection outlined in black. (b) Structural comparison of TfNCS and NdNCS (XA00077_TR556_c0_g1_i1_p1) proteins. TfNCS and NdNCS are shown in blue and yellow, respectively. Enlarged views below highlight key residues and catalytic determinants. -
Across diverse ecosystems in the Northern Hemisphere, from forests to meadows, plants in Ranunculales have continuously expanded their habitats through vibrant floral displays and sophisticated chemical defense strategies. Their genomes and transcriptomes preserve long-undeciphered evolutionary and metabolic codes. However, transcriptomic data covering the entire Ranunculales order remain relatively limited to date[43,44]. In this study, we have, for the first time, compiled a large-scale transcriptome dataset comprising 319 Ranunculales species. From this resource, we identified 733 low-copy nuclear genes, establishing a standardized genetic resource system that provides a solid foundation for phylogenetic research within this group. Based on this dataset, phylogenetic reconstruction clarified the deep relationships within Ranunculales with high statistical support. It confirmed Eupteleaceae as the earliest-diverging basal lineage, Papaveraceae as an independent early branch, and Berberidaceae and Ranunculaceae as a stable sister group, consistent with previous studies[45−47].
The analysis of divergence time places the differentiation of major Ranunculales lineages in the Early Cretaceous (approximately 120–107 Mya). This period aligned closely with the global radiation of angiosperms, the ecological opportunities arising from the breakup of Pangaea[48], and an ancient whole-genome duplication (WGD) event shared by core angiosperms[36]. This temporal correspondence suggests that these three factors collectively drove the early rapid radiation of Ranunculales. Ks-based analyses further revealed a prominent duplication signal in the common ancestor of core Ranunculales, consistent with a putative whole-genome duplication (WGD) event corresponding to the second Ks peak, dated to the late Early Cretaceous (approximately 110–120 Mya)[49]. Notably, the inferred history of genome-wide duplication signals within Ranunculales appears highly complex, exhibiting patterns comparable to those reported in core angiosperms, with multiple putative duplication events showing distinct lineage-specific retention patterns.
The series of genomic and ecological events above has left a profound imprint on the evolutionary trajectories of key metabolic genes. Functional annotation of representative species revealed that duplicated genes associated with alkaloid biosynthesis are preferentially retained, suggesting that genome-wide duplication events may have provided important genetic substrates for the diversification of alkaloids in Ranunculales. Further in-depth analysis of the NCS, NMCH, CNMT, and OMT gene families demonstrated a clearly uneven distribution across different lineages within Ranunculales: NCS genes are more abundant in Ranunculaceae, while NMCH genes are enriched in Berberidaceae. Phylogenetic analysis of CNMT further showed that genes from Berberidaceae dominate the CNMT-I clade, whereas genes from Ranunculaceae display greater diversity within CNMT-II. The formation of this distribution pattern may be related to the divergence between Ranunculaceae and Berberidaceae around 84 Mya during the Late Cretaceous, a period marked by global cooling from extreme greenhouse conditions and a systematic restructuring of land and sea configurations[50], which may have driven functional specialization in the BIAs biosynthetic pathways of the two families. Alternatively, it could be associated with rapid intra-family diversification promoted since the Cenozoic era (0–23 Mya) by a combination of factors including continued global cooling, habitat fragmentation due to orogeny, and Quaternary glacial cycles[51−53].
Regarding the OMT gene family, Papaveraceae shows a particularly prominent representation, likely originating from lineage-specific whole-genome duplication (WGD) events that shaped its independent evolutionary trajectory. For instance, the third Ks peak, unique to Papaveraceae, aligned with its reported additional WGD events[54,55] and may have directly facilitated biosynthetic innovations leading to the production of highly specialized BIAs, such as berberine and morphine. In contrast, the fourth Ks peak, exclusively observed in Lardizabalaceae, may be associated with the distinct evolution of fruit morphology in this family.
Crucially, positive selection analyses provide strong support for adaptive evolution of these key genes. Multiple sites under positive selection were identified in NCS, NMCH, CNMT, and OMT, and these sites were significantly enriched within or near the core functional regions or catalytic centers of the proteins. This suggests that natural selection may have contributed to functional optimization by altering the intrinsic chemical properties of the key enzymes, rather than relying solely on the regulation of expression levels. This aligns with the classical mechanism in secondary metabolism where 'a few key mutations drive new functions'[56,57]. Notably, the uneven distribution of these positively selected genes among Ranunculaceae, Berberidaceae, and Papaveraceae is likely related to the distinct genetic raw material provided for positive selection by the unique WGD history of each group.
In summary, this study systematically delineates the evolutionary landscape of key enzymes in the alkaloid biosynthetic pathway of Ranunculales. Ancient and lineage-specific paleopolyploidization events likely provided important genetic resources for the expansion and diversification of key enzyme gene families, such as NCS, NMCH, CNMT, and OMT. Subsequently, natural selection acted upon these genes, leaving strong signals of positive selection particularly in the core functional regions of the proteins, thereby achieving a 'fine-tuning' of enzyme function. This evolutionary trajectory, from gene duplication to natural selection, systematically explains the mechanism generating functional diversity in the key enzymes for alkaloid biosynthesis in Ranunculales. These findings not only deepen the understanding of the evolutionary patterns governing plant secondary metabolic pathways but also provide important genetic resources and a theoretical foundation for molecular breeding and synthetic biology research related to medicinal plants.
-
The authors confirm contributions to the paper as follows: study conception and design: Dong Y, Hu Y; sample collection: Qiao Z, Zhou Z, Zhang G; data collection: Wei X, Wang W, Qiao Z; draft manuscript preparation: Dai Z, Xu F; manuscript revision and editing: Guo D, Li X. All authors reviewed the results and approved the final version of the manuscript.
-
The sequencing data can be accessed through the BIG Data Center BioProject PRJCA054011.
-
This research was funded by the Guangxi Major Science and Technology Project of China (Grant No. GuikeAA22096021).
-
The authors declare that they have no conflict of interest.
-
#Authors contributed equally: Zijian Dai, Feng Xu
- Supplementary Table S1 Summary of the RNA sequencing data of Ranunculales plants.
- Supplementary Table S2 Orthogroup assignment statistics per species.
- Supplementary Table S3 Transcriptome assembly of Ranunculales plants and its close species.
- Supplementary Table S4 Completeness evaluation of transcriptome assembly of Ranunculales using BUSCO.
- Supplementary Table S5 Transcriptome annotation of Ranunculales plants using six public protein databases.
- Supplementary Table S6 Transcription factors of Ranunculales plants identified using iTAK.
- Supplementary Table S7 Simple sequence repeats of Ranunculales plants identified using MISA.
- Supplementary Table S8 Estimated Ks distribution for Ranunculales species.
- Supplementary Table S9 List of functionally validated genes from previous studies.
- Supplementary Table S10 Names of sequences from the four identified gene families in Ranunculales.
- Supplementary Table S11 Statistics of conserved amino acid sites in the four screened gene families.
- Supplementary Table S12 Critical amino acid sites in positively selected sequences.
- Supplementary Table S13 Expression values (FPKM) of sequences from the four gene families in Ranunculales.
- Supplementary Fig. S1 Phylogenetic reconstruction of 319 Ranunculales species using a concatenation approach.
- Supplementary Fig. S2 Phylogenetic analysis of the Bet v1 gene family showing its division into two subfamilies: NCS-I and NCS-II.
- Supplementary Fig. S3 Phylogenetic Analysis of CYP80B/NMCH.
- Supplementary Fig. S4 Alignment of key amino acid sites in major CYP80B/NMCH genes sequences.
- Supplementary Fig. S5 Phylogenetic analysis of the CNMT gene family showing its division into two subfamilies: CNMT-I and CNMT-II.
- Supplementary Fig. S6 Alignment of key amino acid sites in major CNMT gene family sequences.
- Supplementary Fig. S7 Phylogenetic analysis of the OMT gene family showing its division into three subfamilies: 6OMT, 7OMT and 4'OMT.
- Supplementary Fig. S8 Alignment of key amino acid sites in major OMT gene family sequences.
- Supplementary Fig. S9 Prediction of 3D structural models for positively selected sequences in key enzymes.
- Copyright: © 2026 by the author(s). Published by Maximum Academic Press on behalf of Yunnan Agricultural University. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Dai Z, Xu F, Wei X, Qiao Z, Wang W, et al. 2026. Evolution of key enzymes in the alkaloid biosynthetic pathway of Ranunculales. Agrobiodiversity 3(1): 31−40 doi: 10.48130/abd-0026-0003 

Evolution of key enzymes in the alkaloid biosynthetic pathway of Ranunculales
- Received: 31 December 2025
- Revised: 26 February 2026
- Accepted: 05 March 2026
- Published online: 31 March 2026
Abstract: The order Ranunculales is an early-diverging group of angiosperms with abundant medicinal plant resources, attracting considerable attention due to its diverse capabilities of alkaloid biosynthesis. In this study, we integrated transcriptomic data from 319 Ranunculales species to construct a well-supported phylogenetic framework. In addition to previously reported whole-genome duplication events, we identified a lineage-specific duplication event in Berberidaceae. These events significantly drove the expansion of key enzyme gene families involved in the benzylisoquinoline alkaloid (BIA) pathway, particularly prominent in Berberidaceae and Ranunculaceae. Furthermore, we found that NCS genes in Ranunculaceae have undergone strong positive selection, suggesting potential functional adaptive innovations during evolution. These results elucidate the evolutionary mechanisms underlying alkaloid diversity in Ranunculales from both phylogenetic and genomic perspectives, providing a theoretical foundation for the development of relevant medicinal components and synthetic biology.
-
Key words:
- Ranunculales /
- Transcriptomics /
- Gene family /
- Alkaloid biosynthesis