









-
As the world's third pole and Asia's water tower, the Qinghai-Xizang Plateau (QXP) acts as a vital ecological security barrier for the world[1]. In addition, the QXP is also one of the important biodiversity hotspots and harbours many rare resource plants[2]. In recent decades, with global climate change and human activities, the alpine meadow ecosystem on the QXP is facing great risks of grassland degradation and land desertification, which would greatly affect the safety of ecosystems around the world[3]. Therefore, there is an urgent need to repair the desertified lands on the QXP. Among the numerous prevention and control technologies/methods for desertified lands, the construction of stable vegetation adapted to the local climate environment has become the consensus on the sustainable development of desert ecosystems[4]. Due to the long-term stresses of low oxygen, strong ultraviolet radiation, drought, and the harsh conditions of alpine environments, plants on the QXP typically grow very slowly. Therefore, once vegetation degradation occurs on the QXP, restoration efforts become considerably more challenging. Compared to other plant species, desert-adapted plant species exhibit greater resilient to the harsh environmental conditions of the QXP, making them ideal for restoring degraded vegetation. Therefore, plant species naturally adapted to deserts on the QXP are the optimal choice for vegetation reconstruction in alpine desertified lands.
Over the past decades, numerous researches have been carried out to dissect the genetic basis of plateau adaptation in species endemic to the extreme environments on the QXP[5,6]. Among these studies, most were focused on human and animal species[7−9], while studies on plant adaptation to the QXP have started to gain more attentions in recent years, driven by an increased recognition of the importance of plant diversity for ecosystem resilience[10−12]. Based on comparative transcriptome, metabolome, and/or genome analysis, an increasing number of studies have substantiated that most of the plants thriving on the QXP possess an abundance of secondary metabolites and robust genetic resources tailored to withstand its severe natural conditions[13,14]. Most studies have focused on sister species within genera, however, due to the long history of species differentiation, population dynamics, and/or breeding systems vary significantly across species, making it rare to investigate the genetic basis of altitude gradient adaptation within the same plant species. Nevertheless, plants native to the deserts of the QXP exhibit remarkable tolerance to multiple stresses such as UV-B radiation, drought, cold, and hypoxia, rendering them optimal pioneer species for restoring desertified lands in this region. Furthermore, a clarified molecular basis of local adaptation in different ecotypes within a plant species can not only forecast the evolutionary trajectory of plant adaptation to future climate changes[15,16], but also provide crucial molecular insights and genetic resources for improving and selecting plant species capable of surviving extreme environments and severe climate fluctuations on the QXP[17]. Meanwhile, due to long-term adaptation to the harsh environment and special climatic conditions of the QXP, indigenous plants have produced many secondary metabolites with medicinal value during their adaptive evolution. Therefore, multi-omics studies among natural populations of plant species are necessary to unravel the molecular metabolic mechanisms underlying the adaptation of desert plants to altitude gradients on the QXP, which can not only reconcile the conflicts between local agricultural development and ecological conservation in these fragile ecosystems, but also provide valuable insights into the indigenous nature of high-altitude medicinal plants.
Agriophyllum squarrosum (L.) Moq., commonly known as sandrice, thrives in the vast deserts and sandy landscapes of arid and semi-arid regions throughout the interior of Asia (
www.efloras.org ). Field investigations underscore its remarkable ecological versatility, thriving at altitudes from about 50 to over 4,000 m, particularly adapting well to the harsh desert conditions of the QXP, where it exhibits strong growth and reproductive success[18,19]. Additionally, sandrice plays a crucial ecological role in reducing wind velocity by at least 90% when withered, and it enriches nutrient-poor soils with carbon and nitrogen, significantly sustaining and restoring fragile desert ecosystems[18,20]. Moreover, despite growing in the infertile sandy soils, sandrice seeds are rich in essential nutrients like amino acids, crude fiber, and polyunsaturated fatty acids, making it an ideal natural functional food[21]. Furthermore, its above-ground parts are abundant in bio-actives, including flavonoids, organic acids, terpenoids, and alkaloids[22], and have been traditionally used in Mongolian medicine for treating kidney inflammation, dyspepsia, fever, and pain relief[23]. Notably, recent studies have indicated that sandrice shows great potential as both an antibiotic substitute and a functional forage crop in antibiotic-free ruminant farming, owing to the abundance of bio-active compounds found in its aerial parts[24,25]. Therefore, exploring the mechanism of alpine adaptation in sandrice would not only help combat desertification in the plateau region, but also enhance our understanding of the factors contributing to the high medicinal quality of its alpine ecotypes.Previous biogeographic studies have underscored notable genetic divergence among sandrice populations across heterogeneous deserts and sandy lands. However, minimal genetic divergence was observed between the alpine group and its adjacent central desert group, despite notable habitat heterogeneity between them[19,26]. Notably, based on metabolomic analysis and common garden experiments, variations were observed in the accumulation of medicinal metabolites with significant pharmacological activity, such as flavonoids, among populations from different altitudinal habitats, even under the same environmental conditions.[27,28]. Recently, cold-stress treatment among ecotypes from different altitude gradients further indicates that flavonoids are crucial for sandrice to defend against low temperatures[29]. These phenomena suggest that flavonoid biosynthesis pathways in sandrice may have been favored through long-term local adaptation to the QXP, and further contributes to its distinctive medicinal properties.
To investigate the apparent paradox of significant differences in secondary metabolite accumulation despite similar genetic backgrounds in sandrice, and to further verify the role of flavonoid biosynthesis pathway in the alpine adaptation of sandrice, this study conducted the first in situ metabolome and transcriptome analyses comparing two ecotypes of sandrice. These ecotypes occupy habitats at different altitudes while sharing the same genetic composition: ETL from the alpine group (2,917 m altitude) and CC from the central desert group (1,530 m altitude). Then, population genetic analysis across 22 natural populations was carried out to determine whether these adaptive functional genes underwent directional evolution along the altitude gradient, compared to neutral genes. This endeavour aims to elucidate the molecular metabolic basis underlying sandrice's adpatation to harsh alpine desert environments, especially the role of metabolites with medicinal value in plant adaptive evolution, which will further provide molecular guidance and genetic resources for the restoration of desertification on the QXP and facilitate molecular marker-assisted breeding to enhance the medicinal quality of this promising plant sepcies.
-
Based on the neutral genetic markers, two wild ecotypes that shared the same genetic composition were collected at the same mature growth period from different altitudinal habitats. One was located in Ertala (ETL), Gonghe county, Qinghai province (36°11'39.48'' N, 100°31'28.26'' E, 2,917 m) on the QXP, while the other one was located in Changcheng county (CC), Gansu province (37°54'10.98'' N, 102°54'4.2'' E, 1,530 m) in the southern edge of the Tengger Desert (Fig. 1a). To minimize variations caused by different growth stages, the ETL ecotype was collected in mid-September 2016, while the CC ecotype was collected in early October. Both the CC and ETL ecotypes were in the early reproductive stage at the time of collection. All collected samples were accurately identified by Prof. Xiao-Fei Ma, Northwest Institute of Eco-Environment and Resources, Chinese Academy of Sciences. Fresh tissues from each ecotype, including leaves, stems and spikes, were promptly flash-frozen in liquid nitrogen and stored at −80 °C in an ultra-low temperature freezer for further extraction of metabolites and RNA.
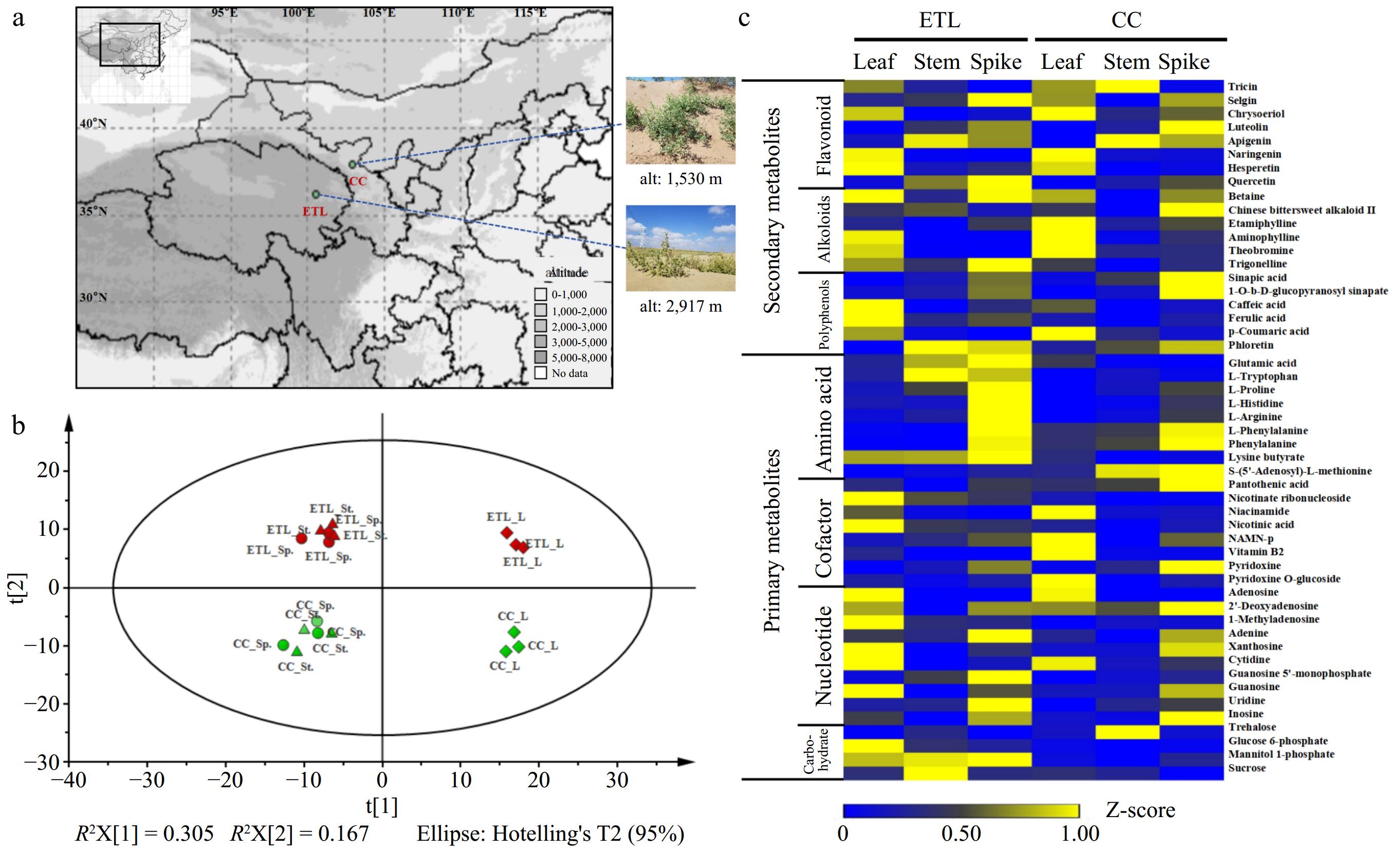
Figure 1.
Comparative metabolomic profilings of two altitudinal ecotypes of A. squarrosum. (a) Geographical localization of two altitudinal ecotypes of A. squarrosum provenances. (b) PCA plot of two ecotypes metabolomes revealing the first two principal components with t[1] = 30.5% and t[2] = 16.7%. The meaning of each abbreviation is as follows: L (leaf), St. (stem), Sp. (spike). Each samples have three biological replicates. (c) Heat map showing standardized contents of partial classified metabolites in different tissues of ETL and CC ecotype. These metabolites are classed into two groups according to their chemical characters.
Metabolomic profiling and statistical analysis
-
As the non-targeted approach provides the advantage of discovering important metabolites that might otherwise remain undetected with a targeted approach, to identify the key metabolites and metabolic pathways, particularly the major secondary metabolic pathways, involved in plateau adaptation for sandrice, samples with three biological replicates of each ecotype were prepared for UPLC-MS non-targeted metabonomics using LC-ESI-Q TRAP-MS/MS systems at Metware Biotechnology Co., Ltd (Wuhan, China). The quantification of the metabolites was carried out in MRM mode and the analytical conditions were as the study of Chen et al.[30]. Analyst 1.6.1 and MultiQuant 3.0.2 software were used for data set acquisition, peak recognition, and normalization. Metabolites were annotated by mapping them to the self-built database MWDB (Metware Biotechnology Co., Ltd. Wuhan, China) as well as public databases to identify their chemical structures. Quantitative analysis of metabolites was carried out by a multi-reaction monitoring mode (MRM) on a triple quadrupole mass spectrometer.
To further determine the differentially enriched metabolites (DEMs) between the two ecotypes, PCA (Principal Component Analysis) and PLS-DA (PLS Discriminant Analysis) were performed with SIMCA 13.0 software. DEMs were determined based on relative content, with thresholds set at a variable importance in the projection (VIP) value of ≥ 1 and a fold change of ≥ 2 or ≤ 0.5.
RNA-seq profiling and statistical analysis
-
Total RNA was extracted from each tissue using RNAprep Pure Plant Kit (Tiangen Biotech Co., Ltd, Bejing, China). Double-strand cDNA was constructed following the study of Shi et al.[31]. These fragments were firstly purified with QiaQuick PCR extraction kit (QIAGEN Inc., Valencia, CA, USA) according to the construction, then were end-repaired with A added to the 3' ends, and finally ligated to sequencing adaptors. The ligated cDNA products under size-selected demands were further concentrated by PCR amplification to construct the cDNA libraries. Library preparations were sequenced on an Illumina HiseqTM3000 platform with a 150-bp paired-end mode. Raw sequenced data have been submitted to the NCBI database with the accession number PRJNA659807.
Raw reads containing unknown sequences ('N') exceeding 5% and low-quality reads (with a base quality less than Q20) were eliminated from the dataset. The remaining filtered clean reads were then utilized for de novo assembly with Trinity version 2.4.0[32]. According to the pair-end information, contigs were clustered and assembled into sequences as long as possible after removing redundancies, and then the clustered longest contigs were subsequently amalgamated into the total unigenes. Functional annotation of each unigene was performing BLASTx searches against the public protein and/or nucleotide databases (such as the NCBI Nr, Nt databases, Swiss-Prot protein database, KOG database, GO database, InterPro, and the KEGG database) with an E-value cutoff of 1e-5. Differentially expressed genes (DEGs) between different tissues of the two ecotypes were estimated by DESeq2. A significance threshold was set with a p-value less than 0.05 and an absolute value of fold-change over 2 to determine significant differential expression[33]. Enrichment analysis of DEGs in the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways was performed using KOBAS software (version 2.0.12) and visualized with ggplot2.
Construction of co-expression network
-
To explore the potential regulatory relationships between genes and metabolites, the relative expression of DEGs and the relative contents of DEMs within two interested pathways of phenylpropanoid and flavonoids biosynthesis pathways were firstly standardized respectively using z-score standardization. Then, Pearson correlation coefficients between DEGs and DEMs were calculated by R version 4.2.3. Correlation pairs were selected with an absolute value threshold over 0.9 and p-value lower than 0.05. Finally, the gene-metabolite co-expression network was visualized with Cytoscape-v3.7.2 (available at
www.gnu.org/licenses/lgpl-2.1.html ).Selective pressure analysis of the candidate genes
-
To explore orthologous genes that have potentially undergoing adaptive differentiation between two ecotypes in response to differing altitudinal environments, the transcriptome sequences of each ecotype were first assembled separately by Trinity version 2.4.0[32]. Subsequently, the resulting clean sequences were searched by BLASTp version 2.2.31 under the threshold of E-value < 1e-5, and then were predicted and translated into protein-coding sequences by TransDecoder version 3.0.0. Meanwhile, OrthoMCL-V2.0.9 software was employed to discern potential orthologs and paralogs among the protein sequences derived from each ecotype's transcriptome. Then, pairwise comparisons were further conducted on putative single-copy orthologs to estimate selective pressure, and ParaAT was used to parallelly construct protein-CDS alignments for these orthologs[34]. Finally, the synonymous substitution rates (Ks), non-synonymous rates (Ka) and Ka/Ks value were calculated for each putative single-copy homologous gene between the two ecotypes with KaKs_Calculator 2.0[35] under the YN model of approximate method[36]. Pairs with Ks > 0.1 were excluded to avoid potential paralogs. The positive selection genes (PSGs) with Ka/Ks value higher than 1 were further verified by the codeml program in PAML[37].
Population sampling, DNA isolation, PCR amplification, sequencing, and sequence alignment
-
To further verify whether sandrice has significantly diverged among ecotypes along with different altitude gradients, population analysis was also conducted among populations of sandrice inhabiting different altitudes. A total of 22 sandrice populations with four to six individuals for each population were collected, including the alpine group from the Qaidam Basin with an altitude of 2,000−3,500 m, the middle altitude group from Tengger desert, Ulan Buhe Desert, Kubuqi Desert, and Mu Us sandy land with an altitude of 1,000−2,000 m, and the low altitude group from Otindag sandy land, Horqin sandy land, and Hulun Buir sandy land with altitude of 0−1,000 m (Supplementary Table S1). Meanwhile, a population of A. minus was also collected from Gurbantunggut desert as the outgroup. All the samples were selected apart from > 50 m for each individual. Fresh leaves were dried and preserved in silica gel, and voucher specimens were deposited in the Northwest Institute of Eco-Environment and Resources, Chinese Academy of Sciences.
Total genomic DNA was isolated from the tissue samples with TIANGEN Plant Genomic DNA Kit (Tiangen Biotech Co., Ltd, Bejing, China) following the manufacturer's protocol. All the DNA samples were quantified by Qubit assay HS kit (Life Technologies, Burlington, ON, Canada) with assays read on a Qubit v2.0 (Life Technologies). In total, 24 pairs of primers were designed by PRIMER version 5.0 based on the RNA-seq data (Supplementary Table S2). Among them, genes under positive selection identified by selective pressure analysis, especially those associated with the known stress-resistance pathways of phenylpropanoid and flavonoid biosynthesis pathways[13,38−41], were identified as candidate genes. Conversely, neutral genes annotated with irrelevant functions to stress resistance were considered reference genes (Supplementary Table S2). All these gene fragments were amplified across these 22 populations using 2 × Taq Plus highfidelity PCR MasterMix (Tiangen, Beijing, China) in a Gene-Amp PCR system 9700 DNA Thermal Cycler (PE Applied Biosystems, Norwalk, USA) following the programs listed in Supplementary Table S2. PCR products were purified with TIAN quick Midi Purification Kits (Tiangen, Beijing, China) and then were Sanger-sequenced with both forward and reverse primers by GENEWIZ company (Tianjin, China). Multiple sequences were aligned and adjusted manually by BIOEDIT version 7.2.6.1 software[42]. The structures of all gene fragments were defined by alignment with their corresponding mRNA sequences and their best hits of BLAST on ESTs (Expressed Sequence tags) from NCBI. All these new sequences were deposited in GenBank under accession numbers OM338032-OM338057, OP846852-OP846955.
Population genetics analysis
-
Genetic diversity was estimated for each gene fragment in three groups with different altitudinal gradients by calculating the number of segregating sites (S), nucleotide diversity statistics (θw; π)[43,44], the number of haplotypes (Nh) and haplotype diversity (He)[45] for all sites, silent sites, and nonsynonymous sites with DnaSP version 5.10[46]. Meanwhile, the fixation index (FST) of each loci was also computed among high, middle, and low altitude groups to estimate the genetic divergence degree with AMOVA in the program Arlequin version 3.1.1 with default settings[47].
Furthermore, to detect whether there were any signals of evolutionary adaptation to different altitudinal gradients habitats, neutrality tests were performed for each fragment with several methods, including Tajima's D test , Fu & Li's D* and F* test[43], Fay & Wu's H test[48], DH test[49], McDonald-Kreitman (MK) test[50] and the maximum frequency of derived mutations (MFDM) test[51]. Finally, to understand the evolutionary relationships and patterns of these putative alpine adaptive genes across different altitude populations, genealogical topologies were constructed using the median-joining (MJ) model in NETWORK Version 4.6.1.259[52] for their haplotypes.
-
Based on the non-targeted UPLC-MS/MS metabolic profiling, a total of 506 metabolites were detected. Among these metabolites, 244 metabolites could be matched to known biochemical structures (Supplementary Fig. S1), including 39 flavonoids, 39 amino acids and derivatives, 27 nucleotide and derivatives, 19 polyphenols, 18 vitamins, 16 alkaloids, 11 lipids, seven organic acids, eight coumarins, eight terpnoids, nine carbohydrates, and 43 additional compounds. PCA analysis revealed distinct clustering of metabolites, segregating into ETL and CC groups based on two altitudinal ecotypes and different tissue types (Fig. 1b). Remarkably, metabolite profiles in leaves differed from those in stems and spikes. However, metabolites in stem samples exhibited similarities or equivalences to those in spikes.
Among the 244 metabolites, the most prevalent secondary metabolites were identified as flavonoids, alkaloids, and polyphenols. In comparison with CC, ETL exhibited higher levels of total hesperetin, quercetin, betaine, trigonelline, caffeic acid, and ferulic acid. Conversely, CC displayed greater accumulation of total tricin, chrysoeriol, etamiphylline, theobromine, sinapic acid, and p-coumaric acid (Fig. 1c). Among three tissues of the two ecotypes, the most remarkable and largest number of DEMs were identified as flavonoid and polyphenol compounds, followed by nucleotides and derivatives, as well as lipids. In ETL, the contents of eriodictyol chalcone and ferulic acid O-hexoside significantly accumulated, whereas tricin 7-O-rutinoside showed a notable accumulation in CC. Compared to CC, the leaf of ETL exhibited significant increases in apigenin 7-O-rutinoside and quercetin-3-beta-O-galactoside, while the content of chrysoeriol O-hexoside decreased significantly. In the stem and spike of ETL, hesperetin 5-O-glucoside content was significantly higher, whereas two glycosylated tricins (tricin O-rutinoside and tricin 5-O-hexoside) were significantly reduced. Additionally, kaempferide showed a significant decrease in levels specifically in the stem of ETL (Table 1).
Table 1. List of metabolites in phenylpropanoid and flavonoid biosynthesis pathways in two altitudinal ecotypes of A. squarrosum.
Class Metabolite name ETL vs CC Leaf Stem Spike FC VIP FC VIP FC VIP Flavonoid Naringenin 7-O-glucoside 0.98 0.01 0.61 0.56 0.52 0.56 Hesperetin 5-O-glucoside 1.30 0.27 2.00 1.22 2.38 1.22 Apigenin 7-O-rutinoside 105.03 1.59 2.44 0.66 2.18 0.66 Luteolin 7-O-glucoside 1.44 0.67 1.31 0.43 0.93 0.43 Luteolin O-hexoside 1.77 0.96 1.33 0.78 0.92 0.78 Chrysoeriol O-hexoside 0.39↓ 1.52 0.45 0.94 0.65 0.94 Chrysoeriol C-hexoside 0.42 0.57 0.33 0.72 0.26 0.72 Selgin O-hexoside 1.03 0.54 1.16 0.53 1.07 0.53 Tricin O-rutinoside 0.32 0.89 0.17↓ 1.02 0.32↓ 1.02 Tricin 7-O-rutinoside 0.34↓ 1.09 0.20↓ 1.27 0.40↓ 1.27 Tricin 5-O-hexoside 0.83 0.12 0.43↓ 1.19 0.38↓ 1.19 Eriodictyol chalcone 5.93 1.39 3.43 1.42 2.62 1.42 Quercetin-3-beta-O-galactoside 4.23 1.03 1.19 0.08 1.07 0.08 Kaempferol 3-O-glucoside 1.55 0.96 1.24 0.38 0.95 0.38 Kaempferide 1.23 0.69 0.48↓ 1.42 0.62 1.42 Phenylpropanoids p-Coumaric acid 0.83 1.22 0.69 0.88 0.79 0.88 Caffeic acid 1.74 1.00 1.00 0.22 2.28 0.22 Ferulic acid O-hexoside 3.46 1.26 4.20 1.48 2.75 1.48 The relative abundance of metabolites were displayed in Supplementary Table S1. FC, the fold change of metabolites when ETL compared CC; VIP, variable importance in the projection value. Bold font indicates significantly changed metabolites (FC ≥ 2 or ≤ 0.5,VIP ≥ 0.5). represents increased metabolites in ETL. ↓ represents decreased metabolites in ETL. Transcriptome analysis in two altitudinal ecotypes of sandrice
-
After the removal of sequences with low quality, poly-N, and adaptors, 884,051,398 clean reads were filtered from the 128.96 Gb raw data, with Q30 over 81.13% and the average N50 of 690.38 bp (Supplementary Table S3). Subsequently, these high-quality trimmed clean reads were de novo assembled into contigs and then a joint transcript of 135,686 unigenes. Finally, 69,977 annotated unigenes were identified across all transcripts, each represented in at least one database (Supplementary Table S4). The most abundant KOG terms identified in the unigenes included those related to general function prediction only, signal transduction mechanisms, post-translational modification, protein turnover, and chaperones (Supplementary Fig. S2a). The GO enrichment analysis of the entire transcriptome revealed that the annotated unigenes predominantly participated in metabolic processes and cellular processes (Supplementary Fig. S2b).
In comparison to the gene expression levels in CC, 16,680 unigenes were up-regulated and 12,773 unigenes were down-regulated in ETL. Additionally, Venn analysis of DEGs revealed that the number of tissue-specific DEGs was higher in the leaf and stem than in the spike (Fig. 2a). The highest number of DEGs with the strongest differential expression level was detected in leaves. KEGG analysis revealed that DEGs of three tissues between the high and middle ecotypes of sandrice were predominantly enriched in pathways such as photosynthesis, starch and sucrose metabolism, flavonoid synthesis, phenylpropanoid synthesis, ribosomal regulation, and carbon metabolism. (Fig. 2b−d).
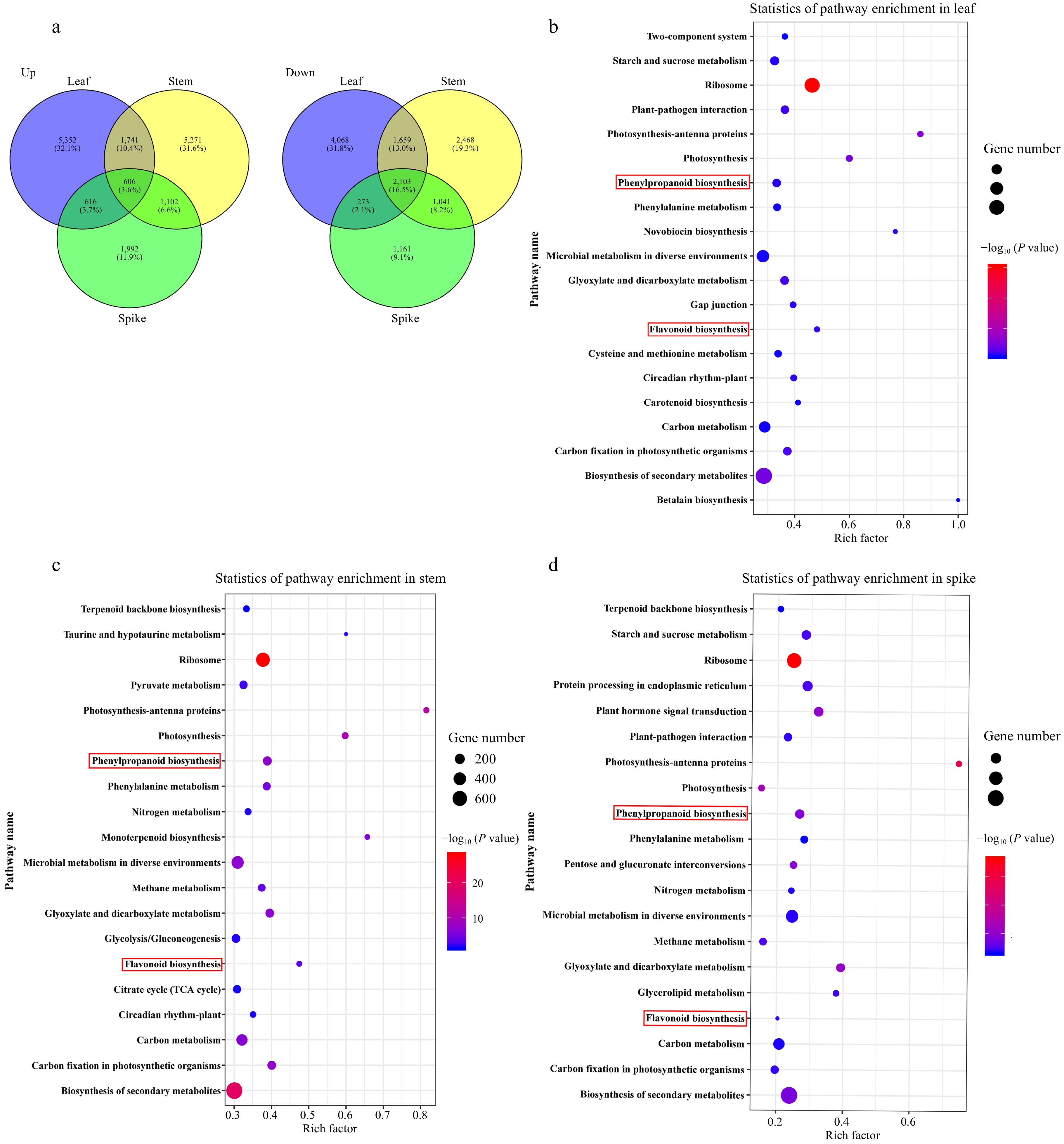
Figure 2.
The state and KEGG pathway enrichment of differentially expressed genes (DEGs) in different tissues from high- and middle-ecotypes of A. squarrosum. (a) Venn diagram indicating the overlapping and unique up-regulated (left) and down-regulated (right). (b)−(d) KEGG pathway enrichment analysis of DEGs in leaf, stem and spike. The number of genes is indicated by the size of the circle and the color of the circle shows significant enrichment through P-value. The top 20 pathways with the minimum P-value are shown in each tissue.
To explore candidate genes involved in high-altitude adaptation, comparative transcriptome analysis was further conducted. A total of 10,275 pairs of single-copy putative orthologous genes were identified after filtering out those with unexpected stop codons. Among them, 127 pairs of orthologous genes showed signs of positive selection with Ka/Ks values greater than 1 (Supplementary Table S5). Although no significantly over-represented KEGG categories were detected, and half of these positively selected genes (PSGs) could not be well-matched to several annotation databases, some PSGs were still putatively annotated to functions related to DNA repair, response to stress, and metabolism (Table 2).
Table 2. Typical differentiated genes for high elevation adaptation.
Unigene ID ETL_Leaf
FPKMCC_leaf
FPKMETL_spike
FPKMCC_spike
FPKMETL_stem
FPKMCC_stem
FPKMLa Ka Ks Ka/Ks Gene annotation Possible functions and biological process TR86494|c0_g1_i1 0 0 1.71 2.76 0.20 0.34 1044 0.00787 0.00365 2.157 Shikimate O-hydroxycinnamoyltransferase (HCT) Phenylpropanoid biosynthesis;
Flavonoid biosynthesisTR80792|c3_g1_i2 51.14 25.12 6.13 1.94 29.94 4.86 1176 0.00780 0.00423 1.845 Flavonol synthase (FLS) Phenylpropanoid biosynthesis;
Flavonoid biosynthesisTR45359|c0_g2_i1 29.58 5.35 1.73 0.27 7.19 1.72 489 0.0164 0.00899 1.828 Caffeoyl-CoA O-methyltransferase (CCoAOMT) Phenylpropanoid biosynthesis;
Flavonoid biosynthesisTR934|c0_g1_i1 0.94 0.84 2.68 2.48 2.49 1.38 1143 0.00288 0.00272 1.056 A/G-specific adenine DNA glycosylase (ANG ) Base excision repair TR74739|c0_g2_i1 2.13 3.45 3.52 3.52 3.50 2.85 1791 0.00514 0.00318 1.615 Uracil-DNA glycosylase (UNG) Base excision repair; Immune diseases TR39350|c0_g4_i1 1.59 3.59 1.53 2.28 9.18 6.21 1434 0.00358 0.00330 1.087 Vegetative cell wall protein gp1(GP1) Defense response TR39755|c0_g1_i1 7.79 2.74 5.65 4.64 7.37 3.86 1089 0.00505 0.00345 1.463 Elongator complex protein 4 (ELP4) Response to oxidative stress, abscisic acid signaling pathway TR40190|c7_g5_i1 25.76 28.38 23.06 20.04 31.05 27.46 300 0.02602 0.00831 3.130 Calcium-dependent protein kinase 1 (CDPK1) Plant-pathogen interaction; Response to drought, cold stress; Salicylic acid biosynthesis FPKM, gene expression level; La, the length of candidate genes (bp); Ka, nonsynonymous substitution rate; Ks, synonymous substitution rate; Ka/Ks, selective strength; p-value, the value computed by Fisher exact test when calculating Ka/Ks For example, two genes (TR934|c0_g1_i1, TR74739|c0_g2_i1), which encoded A/G-specific adenine DNA glycosylase (ANG) and uracil-DNA glycosylase (UNG) participated in DNA base excision repair; TR39350|c0_g4_i1 encoding Vegetative cell wall protein gp1 (GP1) took part in defense response; TR39755|c0_g1_i1 encoding a subunit of elongator complex (ELP4), mediated ABA signaling pathway and manifested oxidative stress resistance. TR40190|c7_g5_i1, which encoded putative calcium-dependent protein kinase family protein (CDPK1), played a vital role in pathogen resistance abiotic stress and salicylic acid biosynthesis. Besides, three genes encoding shikimate O-hydroxycinnamoyltransferase (HCT), flavonol synthase (FLS), and caffeoyl-CoA O-methyltransferase (CCoAOMT), which are involved in phenylpropanoid and flavonoid pathway, were suggested to be under strong positive selection between the two altitudinal ecotypes (Table 2).
Correlation analysis of DEMs and DEGs in two altitudinal ecotypes
-
The integrated analysis between DEMs and DEGs in the phenylpropanoid and flavonoid pathway revealed a significant relationship between the accumulation of metabolisms and the expression of key genes. For example, PAL, CHS, CHI, F3H, and FLS showed significant up-regulation in the high-altitude ecotype ETL compared to CC, consisting of significantly elevated enrichment of corresponding downstream flavonoid and phenylpropanoid contents in ETL (Fig. 3a). Subsequently, as illustrated in Fig. 3b, correlation analysis between gene expression in flavonoid and phenylpropane metabolic pathways and the enrichment of metabolites further revealed that the expression of the COMT1 is positively correlated with the accumulation of most metabolites in the flavonoid and phenylpropanoid metabolic pathways, except for luteolin-O-hexoside and quercetin 3-O-glucoside, which exhibit negative correlations. In terms of differential accumulation of metabolites between the two altitude ecotypes, the level of naringenin 5-O-glucoside directly correlated with the differential expression of CHI, CHS, and F3H. Meanwhile, the accumulation of chrysin 5-O-hexoside was positively correlated with the expression of CHS and COMT. The high expression of these genes would reduce the availability of the substrate naringenin for chrysin synthesis, thereby prompting the production of products from alternative pathways (Fig. 3a). Differences in the accumulation of ferulic acid O-hexoside correlated directly with the differential expression of PAL. Furthermore, the content of quercetin 3-O-glucoside was negatively correlated with the expression levels of CHI, CHS, 4CL, and F3H genes in the high-altitude ecotype of sandrice, leading to reduced quercetin synthesis compared to the mid-altitude ecotype.
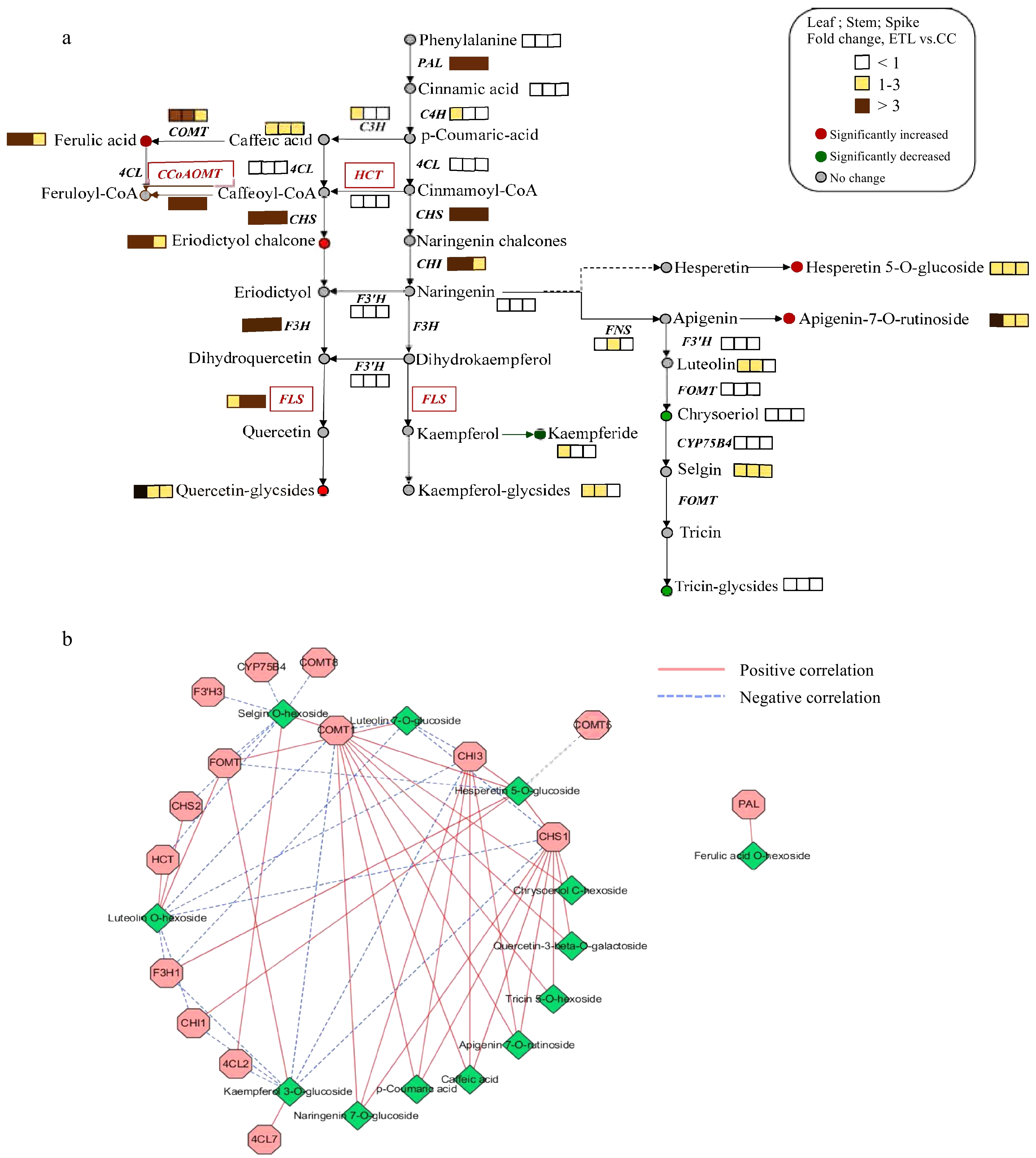
Figure 3.
Phenylpropanoid and flavonoid pathways in A. squarrosum. (a) The schematic representation of gene and metabolite changes in phenylpropanoid and flavonoid pathways. The dotted line represented unreported pathway. (b) The interacted network constituted from genes and metabolites co-expression in phenylpropanoid and flavonoid pathways.
Population genetics and divergence among populations across varying altitude gradients
-
Population genetics analysis revealed that putative alpine adaptive genes exhibited higher levels of nucleotide diversity compared to each reference locus, as well as the haplotype diversity (Supplementary Table S6). Interestingly, further analysis of genetic diversity across different altitude gradient groups revealed discernible differences, despite the statistical insignificance (Supplementary Fig. S3). Specifically, gene segments at higher altitudes exhibited notably lower nucleotide diversity. Notably, certain gene segments in high-altitude populations, such as PAL, C3H, FOMT, FNS, F3H, CYP75B4, and CHS, displayed nucleotide diversity as low as zero (Supplementary Fig. S3).
Fixation index (FST) values were also estimated to assess genetic differentiation among high, middle, and low altitude populations with population genetic data from candidate genes and reference loci, supplemented by SNPs obtained through restriction site-associated DNA sequencing (RAD-seq), which provided a representation of genome-wide variation. Significant genetic differentiation was observed among the high-, middle-, and low-altitude populations (Fig. 4). Specifically, between the high and middle altitude populations, several candidate genes showed notable levels of FST: UNG (FST = 0.410), CDPK1 (FST = 0.228), FLS (FST = 0.144), GP1 (FST = 0.104), and CCoAOMT (FST = 0.099), which were all higher than the average genome-wide FST level (FST = 0.051), indicating significant genetic divergence. Within the phenylpropanoid and flavonoids pathway, F3'H (FST = 0.525), and CHI (FST = 0.300) exhibited the highest levels of genetic differentiation between the high and middle populations.
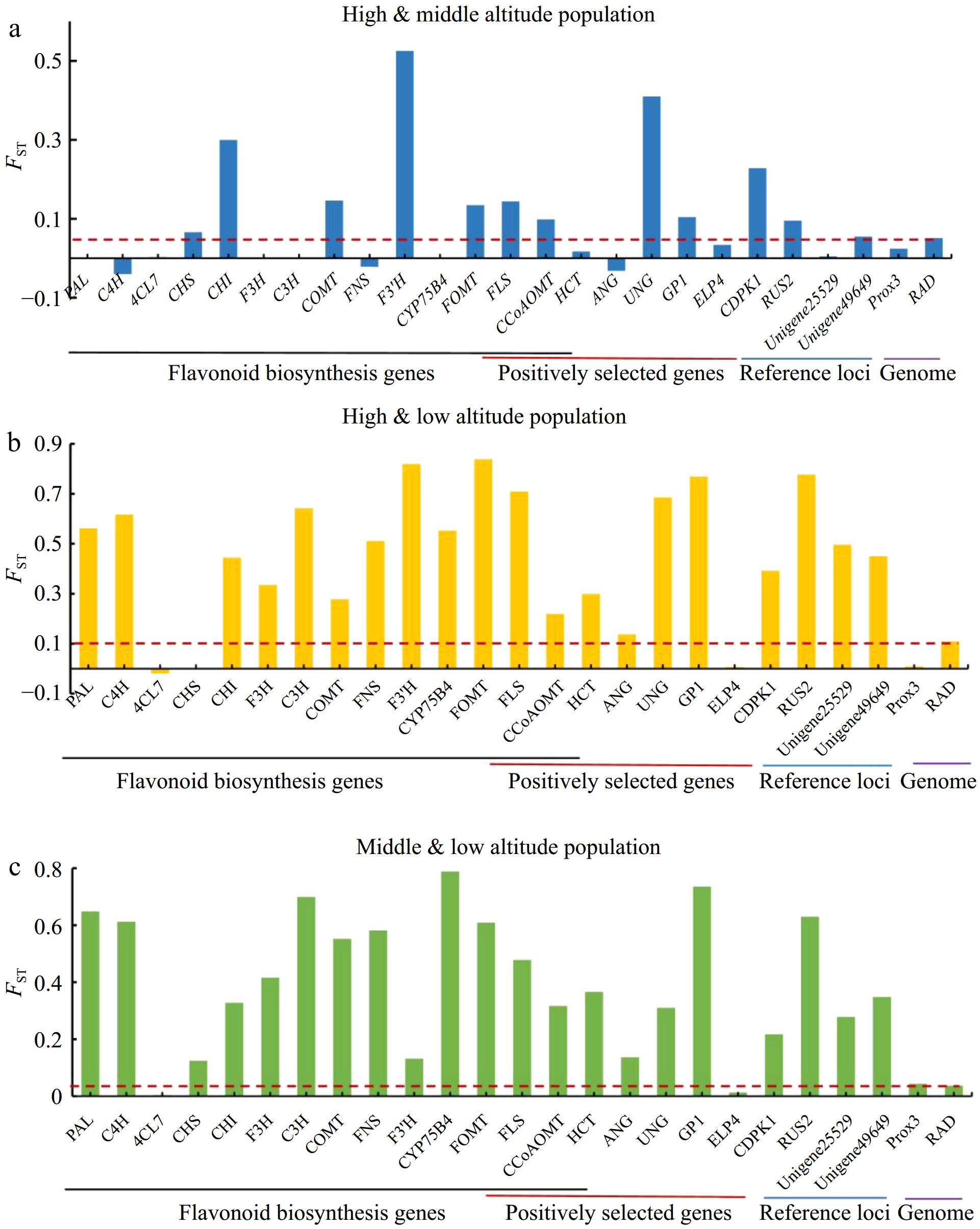
Figure 4.
Genetic differentiation among different altitudinal populations in A. squarrosum. (a) High- and middle-altitude populations. (b) High- and low-altitude populations. (c) Middle- and low-altitude populations. The red dashed line represents the average genome-wide FST level.
Neutrality tests were further conducted for all candidate genes and reference loci among 22 populations of sandrice along with altitudinal clines. Among these, four PSGs (ELP4, GP1, FLS, and HCT) were fixed in the high-altitude group with only one allele, as well as nine genes involved in phenylpropanoid and flavonoids pathways, such as FLS, HCT, PAL, C3H, FOMT, FNS, F3H, CYP75B4, and CHS (Supplementary Table S6). Furthermore, as indicated by Tajima's D, Fu & Li's D*, F* values, UNG showed robust signal of positive selection in the high-altitude populations (Supplementary Table S7). Interestingly, for PSG CCoAOMT, involved in the phenylpropanoid and flavonoids pathway, Tajima's D and F* were significantly greater than zero. The MK test of CCoAOMT was also significant, suggesting that it was under balancing selection at the population level. However, the previous Ka/Ks value indicated positive selection for this gene (Table 2). Furthermore, haplotype network and topology analysis of CCoAOMT across different altitude populations showed that H1 might be the ancestral haplotype, while haplotypes H3 and H4 were specific to the high-altitude populations (Fig. 5a). Combined with distinct expression patterns of its alleles in high and middle altitude ecotypes, as revealed by transcriptome data featuring a non-synonymous mutation (Fig. 5b), CCoAOMT appears to have been selected due to ecological differentiation.
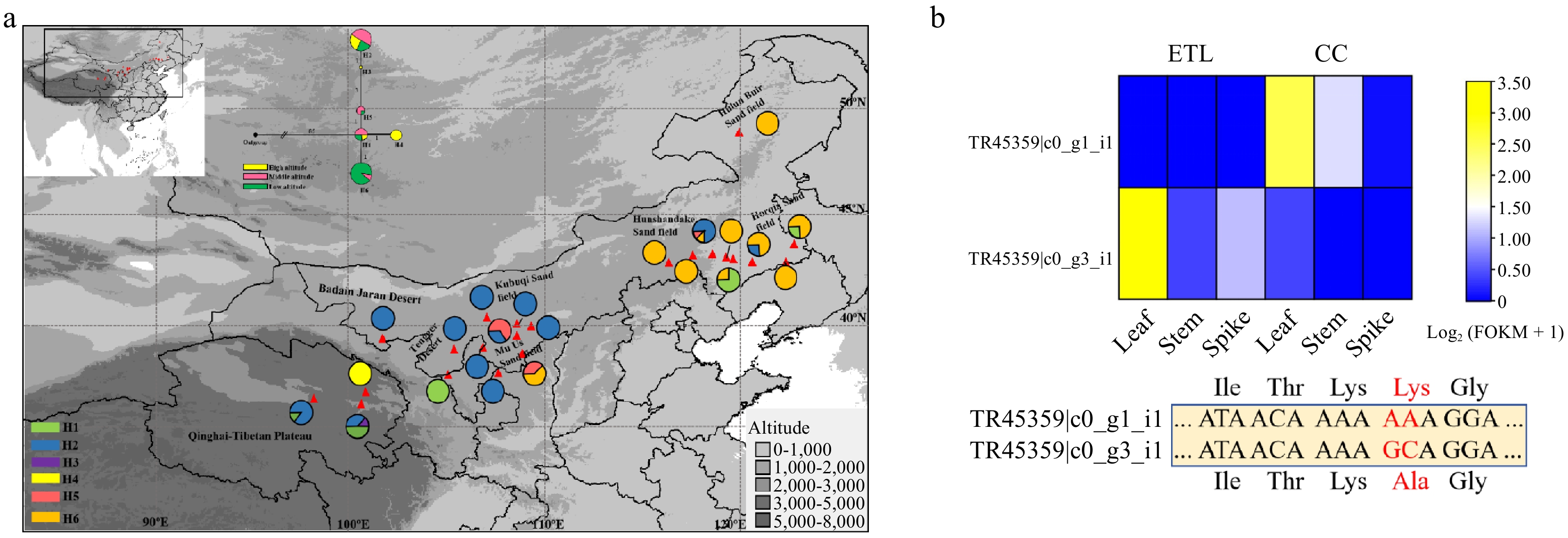
Figure 5.
Haplotype distribution of CCoAOMT in A. squarrosum populations and the expression of two alleles of this gene based on transcriptome data. (a) Haplotype topology of CCoAOMT. (b) Expression levels and mutation sites of the two CCoAOMT alleles in high-altitude and middle-altitude ecotypes of A. squarrosum.
-
The QXP, adjacent to arid Central Asia, offers diverse habitats for biomes, however, it is highly sensitive to ongoing climate changes, particularly extensive desertification of alpine meadows[3]. Vegetation colonizing these desertified areas has evolved adaptive traits to cope with extreme environmental conditions resulting from climate change scenarios, and are also a priority candidate for vegetation reconstruction in alpine desertified lands[17]. However, few studies have explored the molecular basis of adaptation in plant species native to alpine desert ecosystems, especially concerning ecotypes across altitude gradients. As a pioneering species in vegetation restoration of desertified lands, sandrice provides compelling evidence that long-term local adaptation to multiple stresses drives adaptive divergence. This adaptation could significantly impact the success of ecological restoration and development in alpine grassland ecosystems threatened by desertification. Meanwhile, it also provides a solid foundation for improving and developing the medicinal value of sandrice.
The role of flavonoids and phenylpropanoids in sandrice's adaptation to alpine environments
-
Metabolites, particularly secondary metabolites, are pivotal for shaping species-specific traits and are integral to how plants respond to challenging environmental conditions[53]. Previous research indicates that in adapting to alpine conditions, plants have evolved to produce a rich array of secondary metabolites, including phenylpropanoids, flavonoids, ascorbic acid, etc. Besides the outstanding pharmaceutical values, these compounds are also pivotal in helping plants resist environmental stressors, providing numerous advantages such as antioxidative properties, the scavenging of reactive oxygen species (ROS), UV-B radiation absorption, enhanced cold tolerance, and the maintenance of osmotic balance[54]. Phenolics such as flavonoids and phenylpropanoids have even been demonstrated to exhibit species-specific distribution patterns that accumulate along altitudinal gradients in certain plant species[13,41,55]. In this study, compared to the mid-altitude ecotype of CC, significant enrichments of secondary metabolites were observed in the alpine ecotype of ETL, particularly in the phenylpropanoid and flavonoid biosynthesis pathways, such as hesperetin, betaine, quercetin, apigenin, caffeic acid, ferulic acid, etc (Fig. 2c). Interestingly, among these DEMs, apigenin, nobiletin, and ferulic acid were primarily found in glycoside forms (Table 1), which demonstrated potent antioxidant activity by effectively scavenging ROS to safeguard cellular functions and biotic stressor resistance[55,56]. Significant accumulations of flavonoid glycosides have also been found in qingke (Hordeum vulgare L. var. nudum), a crop that has been cultivated and exposed to long-term and intense UV-B radiation on the QXP[10]. Previous studies based on common garden experiments have demonstrated flavonoid accumulation in sandrice is positively correlated with temperature and UV-B radiation, but negatively affected by precipitation and sunshine duration[27,28]. Thus, for the alpine ecotype of sandrice, the significant accumulation of metabolisms, especially the flavonoids, and phenylpropanoids, would hypothetically be induced for adaptation to the long-term harsh alpine desert environment stressors, which further contribute to the high medicinal quality of alpine ecotype.
Alpine adaptive evolution of phenylpropanoid and flavonoid biosynthesis pathways in sandrice
-
Besides detecting a substantial number of DEMs within the phenylpropanoid and flavonoid biosynthesis pathways between mid-altitude and alpine ecotypes, a significant clustering of DEGs associated with the phenylpropanoid and flavonoid biosynthesis pathways was also observed between these two ecotypes (Fig. 2). Correlation analysis of DEMs and DEGs further demonstrated that the differential expression of constitutive genes in the phenylpropanoid and flavonoid biosynthesis pathways would contribute to the divergent enrichments of the downstream DEMs in sandrice. For instance, it has been observed that the transcriptional up-regulation of flavonoid biosynthesis genes (such as PAL, CHS, CHI, FLS, F3H, etc.) significantly enhances the accumulation of hesperetin, apigenin, and quercetin (Fig. 3a, b). These metabolites have been documented to boost plant resistance against UV-B radiation, drought, extreme temperatures, pathogens, and oxidative damage[55,56].
Meanwhile, Ka/Ks analysis between the two altitudinal ecotypes identified that DEGs involved in the phenylpropanoid and flavonoid biosynthesis pathways, such as FLS, CCoAOMT, and HCT, experienced significant positive selection (Table 2). Furthermore, population genetic studies also identified alleles of nine genes (FLS, HCT, PAL, C3H, FOMT, FNS, F3H, CYP75B4, and CHS) out of the 15 tested genes from the phenylpropanoid and flavonoid biosynthesis pathways were fixed in the high-altitude populations (Supplementary Table S6). Previous studies based on RAD-seq and several neutral genetic markers have elucidated that there is no significant genetic differentiation between the high- and middle-altitude populations. This indicates that the high-altitude populations and mid-altitude populations share the same origination and dispersal patterns[19,26]. Thus, the fixation of alleles for these genes in the high-altitude populations might have occurred after populations' colonization on the QXP. Previous simulation and population genetic analyses in wild barley have revealed that advantageous alleles could be selectively preserved and tend to become fixed within the populations under strong environmental pressures[57]. To survive the harsh desert conditions of the QXP, advantageous alleles in the genes related to the phenylpropanoid and flavonoid biosynthesis pathways might be selectively retained. Then, under the environment's directional selection, these alleles could progressively replace other alleles and eventually establish themselves within the high-altitude populations.
Interestingly, Ka/Ks analysis revealed that CCoAOMT, another critical gene in the biosynthesis of flavonoids and phenylpropanoids, is under diversifying selection between two altitudinal ecotypes (Table 2). Additionally, signals of balancing selection on this gene were also observed among the high-altitude populations, and the mid-altitude populations as well (Supplementary Table S7). CCoAOMT is a key enzyme that catalyzes the methylation of caffeoyl-CoA to feruloyl-CoA. This reaction is crucial in the production of monolignols, the fundamental building blocks of lignin. Lignin is vital for maintaining the structural integrity of plants and enhancing their resistance to pathogens, and also play a significant role in enabling plants to withstand abiotic stresses such as cold and drought[58,59]. In recent years, increasing research underscores that balancing selection, by preserving genetic diversity, is a fundamental evolutionary mechanism significantly contributing to the adaptability and survival of species in changing environments, ensuring populations can cope with new challenges and thrive across diverse ecological niches[26,60]. This result suggested that the balancing selection of genes involved in the phenylpropanoid and flavonoid biosynthesis pathways plays a pivotal role in enabling sandrice to endure and prosper under the varied extreme conditions of desert environments, including the harsher environment of alpine deserts.
In general, to survive and thrive in the diverse and extreme desert environment for sandrice, functional genes such as those involved in phenylpropanoid and flavonoid biosynthesis pathways experienced balanced selection among populations with different ecological niches, which could preserve high genetic diversity to ensure populations that can cope with diverse challenges. However, populations of sandrice inhabiting the alpine deserts endure even harsher conditions compared to those in other northern deserts. Under such environmental stress and selective pressure, a greater number of functional genes of phenylpropanoid and flavonoid biosynthesis pathways undergo directional selection, resulting in a shift in the population's genetic makeup toward certain favorable alleles. In some cases, this process leads to the presence of only one advantageous allele for specific functional genes within the population, influencing gene expression and subsequently regulating the high accumulation of downstream flavonoids and phenylpropanoids to enable the population to thrive in the harsh environments of the QXP deserts. As a result of long-term local adaptation, the accumulation of flavonoids and phenylpropanoids in sandrice has significantly diverged among ecotypes from different altitudes, even within the same common garden[27,28].
In addition to the phenylpropanoid and flavonoid biosynthesis pathways, pathways of photosynthesis, starch and sucrose metabolism, flavonoid synthesis, phenylpropanoid synthesis, ribosomal regulation, and carbon metabolism, and several genes related to chronic hypoxia, oxidative stress, DNA damage repair, and stress response regulation, such as ANG, UNG, PRX3, ELP4, CDPK1, and GP1, are also suggested to play crucial roles in sandrice's adaptation to the harsh desert environment of QXP (Fig. 2, Table 2). To gain a comprehensive understanding of sandrice's adaptation mechanisms to alpine deserts and the formation of medicinal components in sandrice under high-altitude environmental factors, further genetic evidence is needed to validate the molecular functions and regulatory relationships among these adaptive alleles and pathways, the expression of functional genes, and the subsequent synthesis and accumulation of those anti-stress metabolites.
This research was supported by the National Natural Science Foundation of China (Grant No. 32271695); Lanzhou Youth Science and Technology Talent Innovation Project (Grant No. 2023-QN-140); Chinese Academy of Sciences Strategic Biological Resources Program (Grant No. KFJ-BRP-007-015); Gansu Province to Guide the Development of Scientific and Technological Innovation Special Fund Competitive Project (Grant No. Y939BD1001), and National Natural Science Foundation of China (NSFC, Grant Nos 32171608 and 32201378).
-
The authors confirm contribution to the paper as follows: study conception and design: Ma XF, Yan X, Yin X; data collection: Yin X, Qian C, Fan X, Zhou S; analysis and interpretation of results: Yin X, Qian C, Fang T, Yang J, Chang Y; draft manuscript preparation: Qian C, Yin X, Yan X, Chang Y. All authors reviewed the results and approved the final version of the manuscript.
-
All the raw sequenced data was submitted to GenBank (www.ncbi.nlm.nih.gov) (accession numbers: PRJNA659807; OM338032-OM338057, OP846852-OP846955).
-
The authors declare that they have no conflict of interest.
-
# Authors contributed equally: Chaoju Qian, Xiaoyue Yin
- Supplementary Table S1 Geographical distribution information of A. squarrosum populations used in this study.
- Supplementary Table S2 Primers information for population genetic analysis.
- Supplementary Table S3 The summary of assembly from transcriptome sequencing data in A. squarrosum.
- Supplementary Table S4 Summary of gene annotation against the seven databases.
- Supplementary Table S5 The list of positively selected genes in A. squarrosum.
- Supplementary Table S6 The nucleotide and haplotype diversity of flavonoid biosynthesis genes, positively selected genes and the reference loci in A. squarrosum.
- Supplementary Table S7 The neutralist tests of flavonoid biosynthesis genes, candidate positively genes and reference loci in A. squarrosum.
- Supplementary Fig. S1 Results of non-targeted metabolomics detection in the aboveground tissues of A. squarrosum. (a)The distribution of the known and unknown structural metabolites; (b) The classes and number of metabolites.
- Supplementary Fig. S2 Gene annotations of unigenes of A. squarrosum. (a) Histogram presentation of EuKaryotic orthologous groups (KOG) classification. (b) GO function classification of unigenes.
- Supplementary Fig. S3 The nucleotide diversity of flavonoid biosynthesis genes, positively selected genes and the reference loci in different altitude groups of A. squarrosum. (a) Watterson’s parameter; (b) Nucleotide diversity; (c) Haplotypic diversity.
- Copyright: © 2025 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Qian C, Yin X, Yan X, Fan X, Zhou S, et al. 2025. Adaptive phenylpropanoid and flavonoid biosynthesis pathways of Agriophyllum squarrosum on the Qinghai-Xizang Plateau. Medicinal Plant Biology 4: e008 doi: 10.48130/mpb-0025-0005 

Adaptive phenylpropanoid and flavonoid biosynthesis pathways of Agriophyllum squarrosum on the Qinghai-Xizang Plateau
- Received: 18 November 2024
- Revised: 09 January 2025
- Accepted: 09 February 2025
- Published online: 24 March 2025
Abstract: With climate change and human activities, the Qinghai-Xizang Plateau (QXP) faces increasing risk of desertification. High-altitude desert plants exhibit remarkable resilience, making them ideal for restoring desertified lands on the QXP. Sandrice, a medicinal herb, disperses widely across Asian deserts including the QXP. To elucidate the molecular mechanism of sandrice adaptation to the QXP, in situ metabolome and transcriptome analyses were conducted between high and mid-altitude ecotypes. Comparison analysis revealed that up-regulated genes in the high-altitude ecotype were primarily involved in phenylpropanoid and flavonoid biosynthesis pathways, leading to higher accumulation of these medicinal metabolites in the high-altitude ecotype. Additionally, Ka/Ks analysis indicated significant divergence in DEGs such as FLS, CCoAOMT and HCT between the two ecotypes. Population genetic analysis across altitude gradients showed that FST values for genes in phenylpropanoid and flavonoid biosynthesis pathways were higher than genome-wide FST values. Notably, nine out of 15 genes in these pathways, including FLS and HCT, were fixed in all the high-altitude populations, as a consequence of strong directional selection by the alpine desert environment, which supports phenylpropanoids and flavonoids play critical roles for sandrice adapting to alpine desert environments. Moreover, balancing selection could also facilitate sandrice's spread across diverse desert conditions, whose signal was witnessed in CCoAOMT within the QXP populations. This study bridges our understanding from medicinal metabolites to the genetic basis of alpine ecotypes adapted to harsh environments on the QXP, providing valuable molecular insights and genetic resources for ecosystem restoration and the indigenous nature of high-altitude medicinal plants.