-
Ancestral state reconstruction (ASR) refers to inferring characteristics of individuals or populations from their common ancestors, mapping morphological or ecological traits to molecular phylogenetic relationships, and providing insights into biological evolutionary processes[1−3]. The conceptual foundation of ASR is often attributed to Emile Zuckerkandl & Linus Pauling, who were inspired by Frederick Sanger's 1955 method for determining protein primary structure through amino acid sequencing[4]. They proposed that these protein sequences can be used not only to infer phylogenetic relationships but also to reconstruct ancestral protein sequences at the root node of phylogenetic trees[5,6]. ASR is typically employed to link morphology or ecology to molecular phylogenetic studies. ASR can be used to infer ancestral character states of organisms from data such as genetic or protein sequences, morphological features, and ecological traits[7−12]. ASR structures trait evolution[13−15], to help reveal homology, conservation, and trait correlations[16,17], analyze diversification rates[18], link micro- and macro-evolutionary patterns[19], and reassess taxonomic systems[2,20].
The species is the principal unit of evolution[21]. Fungal taxonomy evolves with ongoing species discovery, yet critical groups remain unresolved[22−24]. Fungal taxa have been described at a rate of only 3%–6%[25]. Establishing a natural taxonomic system is imperative to facilitate the scientific comprehension of species diversity[23,25−28]. Traditionally, fungal taxonomy relied primarily on morphological characteristics, such as the shape of the ascomata, ascospore morphology, and the number of septa in ascomycetes[26,29]. Modern fungal taxonomy integrates molecular phylogenetics with morphological, genetic, and ecological data, leading to continuous refinement of classification systems[26,28]. Phylogenetic analysis forms the foundation of evolutionary research by reconstructing the relationships among ancestral lineages. In fungi, molecular phylogenetics has proven particularly valuable for resolving species relationships, and clarifying taxonomic classifications[30−34]. Phylogenetic trees depict evolutionary relationships and serve as a framework for interpreting evolutionary history, classification, character evolution, and divergence times[35,36]. Tree annotation integrates taxonomic traits, biogeography, ancestral reconstructions, niches, and co-phylogenetic associations to enable multidimensional insights into evolutionary dynamics[37,38].
ASR is a distinct analytical approach used to infer the historical states of morphological or molecular traits across a phylogeny. While ASR is a standalone method, its integration into phylogenetic trees through visual annotations—such as colors, symbols, or node labels—transforms it into a key component of tree annotation[2]. ASR is typically performed following phylogenetic tree inferences to estimate ancestral character states, using evolutionary modes. By leveraging genetic information embedded in the phylogeny, it helps reconstruct the sequence and timing of evolutionary transitions[39,40]. Input matrices combine phylogeny (branch lengths) and extant states; however, reconstruction accuracy declines over evolutionary timescales due to information loss[39,41], and ambiguous intermediate states may reduce confidence in inferences[19,42].
ASR has been widely applied in evolutionary studies of both animals and plants. In animals, it has been used to reconstruct Túngara Frog calls[43], identify ancestral morphological traits such as dinosaur cranial structures, and trace the origins of key adaptions[44−46] and infer the evolution of behaviors like sociality[47]. In plants, applications include investigating the origin and evolution of traits such as vascular tissues, seeds, and flowers[48−50], revealing adaptations such as drought and salt tolerance, and photosynthetic pathways (C3, C4, and CAM)[51], and reconstructing biogeographic histories including angiosperm dispersal and tropical rainforest origins[52,53]. In fungal evolution, ASR has focused on ecological niche, geographic distribution, and the evolution of morphological traits (e.g., spore characteristics)[16,54,55]. These traits are generally classified as either continuous or discrete. Continuous traits—such as spore size—vary along a numerical scale and are influenced by multiple genes and environmental factors. In contrast, discrete traits fall into distinct categories, such as spore morphology or colony color, and are often controlled by a few major-effect genes[56−58].
ASR is a rapidly advancing field that integrates computational methods with diverse biological datasets to infer the evolutionary history of traits. Despite its widespread application in animals and plants, ASR remains underutilized and fragmented in fungal research. To assess the current landscape, a literature search was conducted using the Web of Science database (
www.webofscience.com/wos , 29 April 2025), identifying 2,911 ASR-related articles, of which 1,882 were published in the last decade (2015–present). Among them, 880 studies focused on animals, 562 on plants, 75 on fungi, and 365 on other organisms (Fig. 1).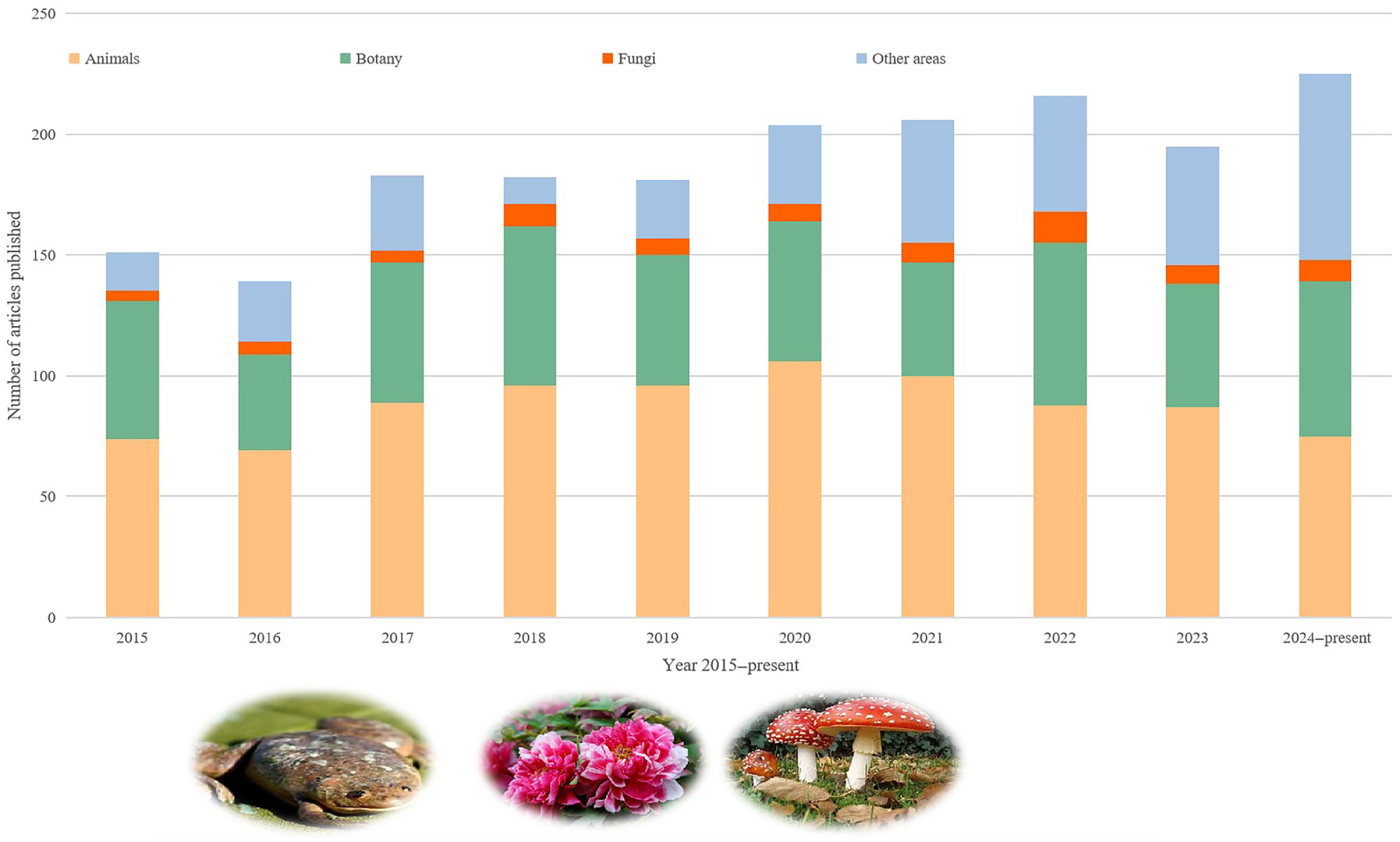
Figure 1.
Summary of articles on ancestral state reconstruction found on the Web of Science.
This review aims to systematically consolidate recent progress in the application of ASR to fungal taxonomy. The advantages and limitations of commonly used methods and tools are evaluated, and the current state of ASR research is examined across major fungal groups (e.g., Ascomycota and Basidiomycota). By identifying challenges and gaps, future directions to advance the field are proposed. This review provides a foundation for improving ASR approaches in mycology, and offers new insights into the evolutionary history of fungal taxa.
-
ASR serves as a pivotal evolutionary tool, resolving contentious fungal morphological evolution, validating molecular phylogenies through ancestral traits, and elucidating coevolution of ecological-morphological adaptations. These are becoming increasingly prevalent analyses in molecular systematics[16,59]. These methods rely on three fundamental assumptions: constant trait transition rates across phylogenies; correlations between neutral evolutionary variation and phenotypic expression; and available likelihood models with appropriate priors for Bayesian frameworks[60].
Evolutionary diversification exhibits heterogeneous rates driven by distinct mechanisms, including speciation acceleration factors (small-population drift, regulatory mutations, polyploidization), adaptive radiation bursts in brief geological windows, and punctuated equilibrium dynamics[61]. Crucially, large-scale evolutionary patterns manifest predominantly through gradual incremental change—a fundamental premise for evolutionary timescale estimation[62,63]. Despite lineage-specific rate variation (e.g., accelerated molecular clocks in short-generation taxa), conserved linear correlations between biomolecule divergence and post-divergence time underpin molecular clock theory[63,64].
In ASR, taxonomic units located at or near the ancestral position often provide the most critical information. The role of fossils in this process is indispensable, as they contribute at least two significant sources of information that are vital to the reconstruction of ancestral lineages, particularly in revealing the limitations of evolutionary trends previously observed[65]. First, analyzing fossil data enables a comprehensive assessment of the extent and spatial-temporal patterns of character-state transitions across evolutionary clades. Second, temporal constraints embedded in the fossil record allow empirical validation of individual observations used in node reconstructions, thereby refining the accuracy of ASRs[65,66].
ASR methodologies
-
ASR is based on two main principles: the principle of parsimony[67−69], and the principle of stochastic modeling of trait evolution[20,70−72]. The principle of parsimony centers on finding the hypothesis that has the lowest total number of trait change events (e.g., emergence, change of a trait) in the evolutionary tree, with the idea that the 'simplest' explanation is the most likely[68]. The stochastic modeling principle centers on constructing mathematical models (e.g., Brownian motion models) that treat the evolution of traits over time as a stochastic process, and calculating the likelihood of ancestral states based on this model[39,66]. Based on these principles, three main methodological frameworks for ancestry state reconstruction exist: maximum parsimony, maximum likelihood, and the Bayesian inference (Table 1).
Table 1. The main approaches of ASR in mycological research.
Principle Method Parsimony Maximum parsimony Stochastic modeling of trait evolution Maximum likelihood Bayesian inference Maximum parsimony was the earliest ASR approach developed. It infers ancestral states by minimizing the number of evolutionary changes required across a phylogeny, assuming all changes are equally probable[67]. This method is computationally simple and efficient, but it does not incorporate branch lengths or the timing of evolutionary events, which can lead to oversimplified reconstructions and systematic biases[19,73,74].
In contrast, maximum likelihood introduces statistical rigor by modeling character evolution using Continuous-Time Markov Models (CTMMs)[20,75−77]. It calculates the probability of observed data given a particular tree and model, allowing for explicit testing of evolutionary hypotheses. Likelihood-based methods account for branch lengths and use statistical criteria such as the Likelihood Ratio Test (LRT), and Akaike Information Criterion (AIC) to compare competing models[78−81]. In the maximum likelihood estimation framework, AIC is preferentially associated because its goal of directly optimizing prediction accuracy is consistent with the core of the likelihood approach, while BIC focuses on identifying the true model, and AIC is mainly used for small-sample corrections[81]. However, maximum likelihood generates point estimates that may be sensitive to phylogenetic uncertainty—an important consideration in fungal ASR[79,82−84].
Bayesian inference builds upon likelihood approaches by incorporating uncertainty in both phylogenetic trees and model parameters. It estimates the posterior probability of ancestral states through Markov chain Monte Carlo (MCMC) sampling, integrating across different possible trees and evolutionary models[20]. Bayesian methods allow for robust hypothesis testing and provide a probabilistic distribution of ancestral traits, making them especially valuable for modeling complex evolutionary scenarios in fungi.
In summary, maximum likelihood methods are widely used to reconstruct the ancestral morphology of fungi, particularly in groups with high morphological diversity or unresolved taxonomic issues. Bayesian methods are more often applied to ancestral distribution reconstructions, especially for taxa with defined ecological roles or complex biogeographic patterns, and are valuable for exploring species origins and historical dispersal.
Toolkits in ASR
-
While ASR is inherently characterized by uncertainty, this does not imply that its estimates are unreliable or invalid. This uncertainty is mainly reflected in the ambiguity of the reconstruction results, or in the existence of multiple equally parsimonious reconstruction schemes[68]. Thus, any hypothesis testing of ancestral state eigenvalues is highly dependent on the validity of the model fitted.
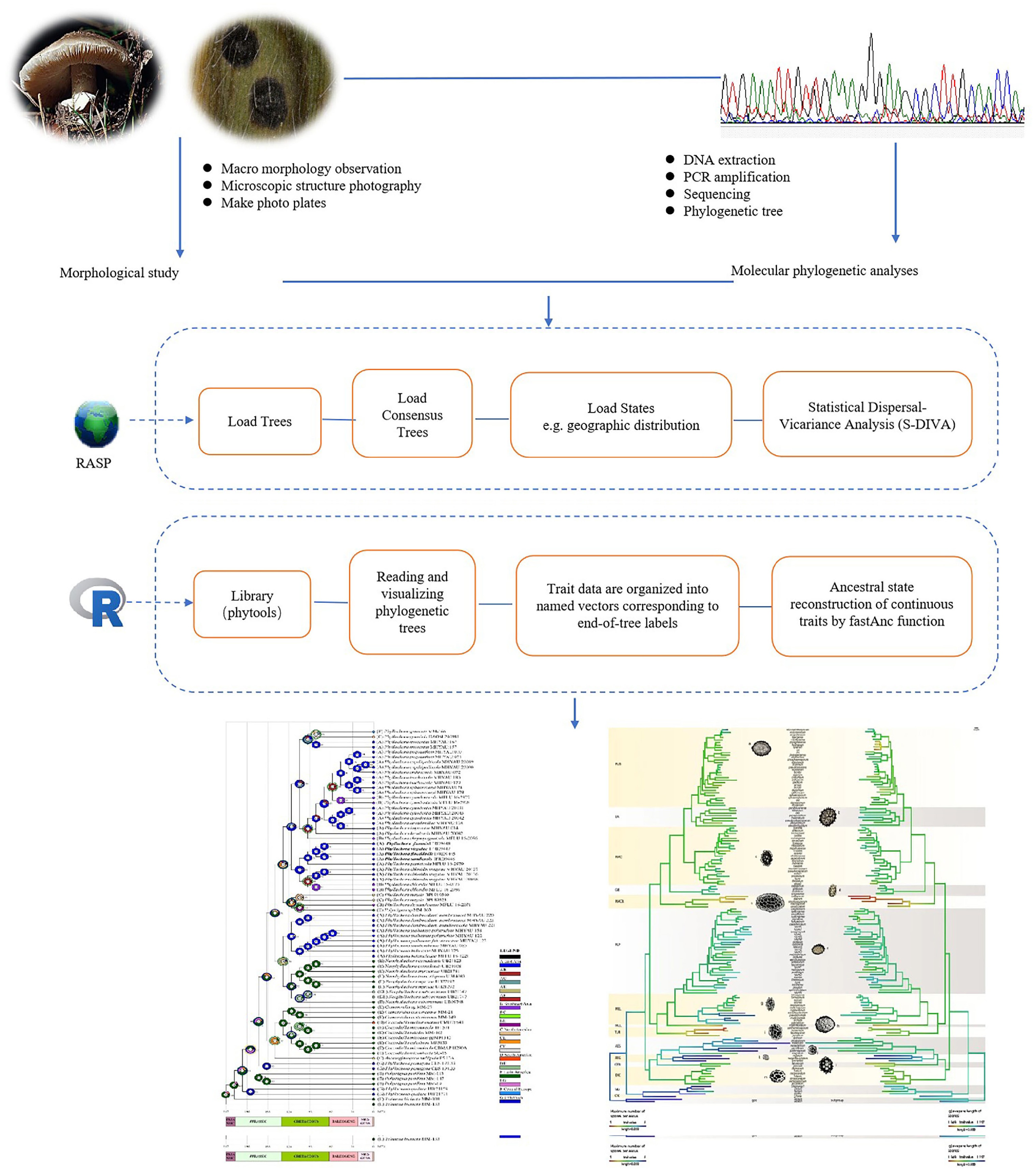
ASR is employed to trace the evolutionary history of biological traits, such as resolving homology vs convergence, inferring extinct ancestral states, or testing taxon monophyly. It relies on a robust phylogenetic framework and appropriate evolutionary models (e.g., maximum likelihood or Bayesian for discrete traits, Brownian motion for continuous traits) to estimate node state probabilities, integrate statistical tests, and assess uncertainty, thereby providing quantitative evidence for trait evolution and informing taxonomic revisions. Commonly used ASR tools include the R package phytools, Reconstruct Ancestral State in Phylogenies (RASP), and Mesquite. This review evaluates the advantages and limitations of these tools and web-based platforms based on published studies (Table 2).
Table 2. Comparison of the advantages and disadvantages of the software and online websites for constructing ASR.
Software Advantages Disadvantages Beast Framework for testing evolutionary hypotheses that does not require a single tree topology to be defined. It consumes computational resources. Fossil-based phylogenetic positions inferred from morphological data still need to be considered in conjunction with other evidence (e.g. palaeontological evidence). Diversitree Focuses on statistical modeling of species diversity and lineage differentiation, which can be used to explore patterns of species formation and extinction. Performs better with large amounts of data. Mesquite This is professional evolutionary biology software optimized for the analysis of comparative organism data, and is ideal for small datasets and educational applications. Processing large data sets is inefficient and the functionality is relatively simple, making it unsuitable for complex analysis. Multi-state Allowing the system and its components to assume more than two performance levels can provide a more realistic and accurate representation. Involves the estimation of a larger number of rate parameters. PHYLIP Open source and free, supports a wide range of phylogenetic analysis methods. The interface is older, less intuitive, and the functionality is relatively basic. R Powerful and flexible. It cannot be used to develop web-type or internet-type applications. RASP Supports multiple analysis methods and allows users to select models according to data characteristics, offering greater flexibility. Running Bayesian methods or complex models on large datasets or phylogenetic trees is time-consuming and requires high-spec hardware. To demonstrate the applicability of ASR in mycology, two representative case studies are presented. For discrete traits, Li et al.[54] employed RASP 4.2[85] to infer ancestral geographic distributions of Phyllachora species. For continuous traits, Cseh et al.[59] utilized the phytools package[86] in R4.3.2 to reconstruct ancestral spore traits in the genus Tuber. Building on these examples, we propose a standardized ASR workflow (Fig. 2) that integrates both discrete and continuous trait analysis.
Examples of ASR in mycological research
-
Increasingly applied to Ascomycota, ASR has emerged as a key evolutionary tool, exemplified by tracing yeast enzyme evolution. By reactivating ancestral dehydrogenases in fragmented AAD homologs, Yang et al. provided functional evidence supporting ancestral lignin degradation roles[9]. Furthermore, ASR uniting with molecular dating analysis has been applied to the study of the species origin and geographic distribution patterns of different taxa of fungi. For instance, Réblová et al. hypothesized that the genus Codinaea originated in the temperate zone and transitioned to the tropics, with ancestral distributions in Eurasia and the Americas and regional derivation areas in Africa and Australia[87]. In the study of new species of the genus Phyllachora in the southwest of China, the ancestral distribution of the species of the genus was analyzed by the RASP based on the global distribution of extant species of the genus, and the species of Phyllachora originated in Latin America at about 194 Mya. The biogeographic distribution of the species is influenced by historical events. In addition, the migration of Phyllachora species from Latin America to Southeast Asia occurred during the Jurassic period[54]. Rathnayaka et al. hypothesized that the order Botryosphaeriales originated during the Cretaceous period, at which point the Botryosphaeriaceae and Phyllostictaceae diverged from each other during the Mesozoic Era[55]. ASR has been instrumental in reconstructing ascomycetes' morphological evolution, revealing through ASR that the Pezizomycotina lineage—including major subclades such as Leotiomyceta, Sordariomycetes, and Dothideomyceta—possessed an open-type fruiting body (apothecium) with an exposed hymenium as the ancestral reproductive structure[15]. For instance, Chroodiscus is inferred to have originated in the Late Cretaceous (~80 Mya) on the Gondwanan landmass (e.g., South America/Australia). Its subsequent radiation into tropical and subtropical regions occurred via continental fragmentation and long-distance dispersal. At the same time, island isolation (e.g., Pacific Islands) facilitated the formation of localized endemic species[88]. ASR by Divakar et al., through the maximum likelihood method, revealed that the ancestor of the Parmeliaceae is a kind of foliose lichen[13]. Similarly, Spribille et al. applied ASR to trace lichen symbiosis origins, revealing its recurrent evolution in fungi, mainly Ascomycota, with some Basidiomycota with convergent symbiotic traits. Molecular clock analyses dated lichenized lineages to the Devonian (approximately 400 million years ago), aligning with land plant radiation, highlighting their pivotal role in early terrestrial ecosystem formation[89]. Meanwhile, ASR has also been used to study the evolution of decomposition capacity and the ecological functions of saprophytic ascomycetes. Floudas et al. reconstructed the evolutionary history of ligninolytic enzymes (e.g., lignin peroxidase LiP, manganese peroxidase MnP) by analyzing the genomic data of 31 seeded cysticolous fungi and found that their origins can be traced back to the Paleozoic era[90].
In Basidiomycota research, ASR serves as a powerful tool to unravel evolutionary trajectories of wood-decaying fungi, including decay modes, mating systems, and ecological adaptations. Hibbett's ASR analysis of 130 Homobasidiomycetes species, using nuclear/mitochondrial DNA and parsimony methods, inferred ancestral white-rot decay. Findings indicate recurrent evolution of brown-rot decay and bipolar mating systems, while coniferous substrate decomposition emerged as a widespread polymorphic trait[91]. Furthermore, ASR has been successfully applied to resolve the evolution of morphological traits in Agarics taxa. Caboň et al. used multi-locus phylogenetic reconstruction in their study of Rubrinae to distinguish R. firmula, R. rubra, R. rutila, and R. veternosa in a well-supported Rubrinae branch. These species have traditionally been categorised into two distinct subclasses based on morphology. However, ASRs demonstrated that their common ancestor possessed yellow spores and was related to Angiosperms. It is noteworthy that the possession of dark yellow or ochre spores characterises all species within the Rubrinae branch[8]. In the study of Coniferiporia, Wang et al. revealed that the ancestral fungus initially parasitized pine and cypress fungi before shifting approximately 17.01 Mya to infect another plant originating in eastern Eurasia, subsequently extending its range into western North America, Central Asia, and eastern Europe[92]. Their subsequent work on Hymenochaetales evolution reconstructed that the most recent common ancestor of this order likely possessed crust-like, resupinate basidiomes and a simple, smooth to toothed hymenophore configuration. Their study also showed that this ancestor grew a saprotroph on both angiosperms and gymnosperms[18]. Meanwhile, ASR has been extensively applied to investigate host specificity, pathogenic mechanisms, and genomic evolution in plant pathogenic basidiomycetes. Aime et al. conducted ASR on rust fungi (order Pucciniales), and proposed that the diversification of major lineages was closely linked to shifts in host plant groups. For example, plant diversification in the Poaceae and Fabaceae families drives adaptive radiation, and there are genome evolution features such as transposon burst, horizontal gene transfer (HGT), and so on[93]. Kijpornyongpan et al. hypothesized that the common ancestor of the fungal taxa of the subphylum Ustilaginomycotina has the pathogenic properties of the smut fungi, and that the pathogenic ancestor may have carried a family of genes (e.g., secreted effector proteins, cell-wall degrading enzymes, and genes for the synthesis of secondary metabolites) that are involved in the infestation of plant hosts. The retention of these genes has been observed in certain extant pathogenic species, while they appear to be degraded or lost in non-pathogenic species. The role of horizontal gene transfer (HGT) or events of gene family expansion/contraction in the evolution of pathogenicity is a key area of research. The present studies provide ample evidence to demonstrate the importance of ASR in the study of the evolution of fungi in the basidiomycetes species[94].
In summary, the maximum likelihood method is the more commonly used approach in the reconstruction of the ancestry of morphological features of fungal species. This method is particularly suitable for taxa with high morphological diversity or significant taxonomic controversies, and can be applied to analyze both micro- and macro-morphological characteristics. For example, the ancestral reproductive structure of Pezizomycotina and its infrageneric taxa in the Ascomycota is the exposed hymenium (apothecium)[15], and a morphologically based taxonomic reconstruction of the Agaricomycetes in the Basidiomycota[16]. Conversely, the Bayesian method (e.g., BBM in RASP) is more prevalent in the context of ancestral distribution reconstruction of fungal species. This approach is particularly suitable for taxa with well-defined ecological functions or complex distribution patterns, focusing on the occurrence of events such as the origin or historical spread of fungal species. Examples of taxa that are particularly well-suited to this approach include lichenized fungi in the Ascomycota[88,89], and Agaricomycetes in the Basidiomycota[92].
-
Recent advancements in molecular techniques and evolutionary biology have significantly propelled ASR research in fungal systematics. However, achieving a comprehensive understanding of fungal evolutionary history and the timing of critical evolutionary events necessitates integrating multidisciplinary approaches. Key limitations include: (1) restricted species sampling in fungal phylogenetics, undermining the representativeness and accuracy of phylogenetic trees; (2) absence of dedicated ASR models specifically tailored for fungal taxa, with current frameworks predominantly adapted from animal or plant studies; (3) oversimplified assumptions in reconstructing fungal biogeography, apart from the limited sample size involved, often neglecting ecological complexity (e.g., climate shifts, tectonic movements, host plant distributions).
The main problems and challenges are summarized as follows: (1) Data quality issues: morphological or molecular data for many species are missing or incomplete, leading to inaccurate reconstruction results; samples of extant species may not represent the true diversity of ancestry, especially for extinct species. (2) Limitations of model selection: a significant number of reconstruction methods rely on simplified evolutionary models (e.g., Markov models), which may fail to capture complex evolutionary processes. The model that is selected may not be suitable for the evolutionary pattern of a particular trait (e.g., continuous traits vs discrete traits). The estimation of model parameters (e.g., evolutionary rate) may be subject to error, which may affect the reconstruction results. (3) A failure to consider environmental and ecological factors: numerous methodologies for reconstruction neglect the repercussions of environmental transformation on trait evolution. Interactions between species (e.g., predation, competition) have the potential to influence trait evolution; however, these factors are typically excluded from the model.
-
Fungi are a group of organisms that are widespread throughout the Earth, with diverse morphological characteristics, and ecological functions. They play an important role in maintaining ecosystem balance and biodiversity[95,96]. Nevertheless, due to the extensive variety of fungi and the similarity of morphological features among species of the same taxonomic category, traditional classification systems have certain limitations in the context of fungal taxonomic studies. The introduction of ASR provides a novel approach and valuable method for fungal taxonomic studies. By inferring ancestral traits, ASR helps to unravel the evolutionary history of fungi and their taxonomic characteristics, revealing the origin and evolution of key features such as morphological characteristics, metabolic pathways[16,97]. ASR has enhanced understanding of the evolution of fungal ecological functions, such as the origin of the lignin-degrading capacity of fungal species, and provides a theoretical basis for understanding fungal ecological roles[90]; it can also be used to study the co-evolutionary relationship between fungi and their hosts, revealing changes in the host range of pathogenic fungi and the evolution of virulence factors as well as the origins of their pathogenicity mechanisms, and transmission pathways[98]. Furthermore, ASR can be used to help infer the geographic origin and dispersal pathways of fungi and reveal their biogeographic distribution patterns and historical dispersal events[54,92]. It can also be used to calibrate phylogenetic trees, enabling more accurate estimates of divergence times in conjunction with fossil records or molecular clock analyses[99,100].
-
As evolutionary biology advances, ASR is expected to play an increasingly prominent and in-depth role in fungal taxonomy. Integrating multi-omics data, innovative algorithms, and ecological functional inference will enable the precise resolution of key evolutionary innovations. Future breakthroughs will depend on the development of fungal-specific algorithms and the construction of global evolutionary databases, facilitating cross-boundary interaction with host evolutionary histories. These advances will help bridge scales from genes to ecosystems, further advancing the application in fungal taxonomy.
This review summarizes current ASR methodologies and applications in mycology, highlighting its potential to shift fungal evolutionary research from descriptive event cataloging toward mechanism-driven analysis, thereby refining classification systems and deepening our understanding of fungal evolution.
We are grateful to the National Natural Science Foundation of China (Grant No. 32170024), the Yunnan Province Ten Thousand Plan of Youth Top Talent Project (Grant No. YNWR-QNBJ-2018-267), and the Yunnan Fundamental Research Projects (Grant No. 202401AT070017) for the financial support for the present study.
-
The authors confirm their contributions to the paper as follows: study conception and design: Wang YT, Wu HX; data collection: Wang YT; analysis and interpretation of results: Wang YT, Wu HX; draft manuscript preparation: Wang YT, Wu HX. All authors reviewed the results and approved the final version of the manuscript.
-
All data generated or analyzed during this study are included in this published article.
-
The authors declare that they have no conflict of interest.
- Copyright: © 2025 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Wang YT, Wu HX, Chomnunti P. 2025. The role of ancestral state reconstruction in mycological researches: methods and insights. Studies in Fungi 10: e022 doi: 10.48130/sif-0025-0023 

The role of ancestral state reconstruction in mycological researches: methods and insights
- Received: 01 June 2025
- Revised: 12 August 2025
- Accepted: 01 September 2025
- Published online: 20 October 2025
Abstract: Clarifying the phylogenetic relationships of fungi by integrating genetic evolution and morphological characteristics is central to modern fungal taxonomy. Ancestral state reconstruction (ASR) is a key phylogenetic tool that applies statistical models to infer the evolution and timing of ancestral morphological traits using genetic data. By mapping traits onto phylogenies, ASR helps clarify the evolutionary transitions and origin of traits. This paper reviews the theoretical foundations of ASR and surveys commonly used methods and software in fungal systematics. It also highlights the practical applications of ASR in fungal taxonomy, particularly in reconstructing morphological evolution and testing molecular phylogenetic hypotheses. The findings highlight ASR as an indispensable tool for analyzing fungal phylogenies, addressing taxonomic controversies, and supporting evolutionary research. This review presents relevant insights into the application of ASR in fungal classification, providing methodological support, and a scientific basis for the classification and evolutionary studies of important fungal taxa. The future direction of the field is to integrate multi-omics, innovative algorithms, and ecological function inference to accurately analyze key events in evolutionary innovation.
-
Key words:
- Annotation of phylogenetic tree /
- Evolution /
- Taxonomy /
- Fungi diversity