






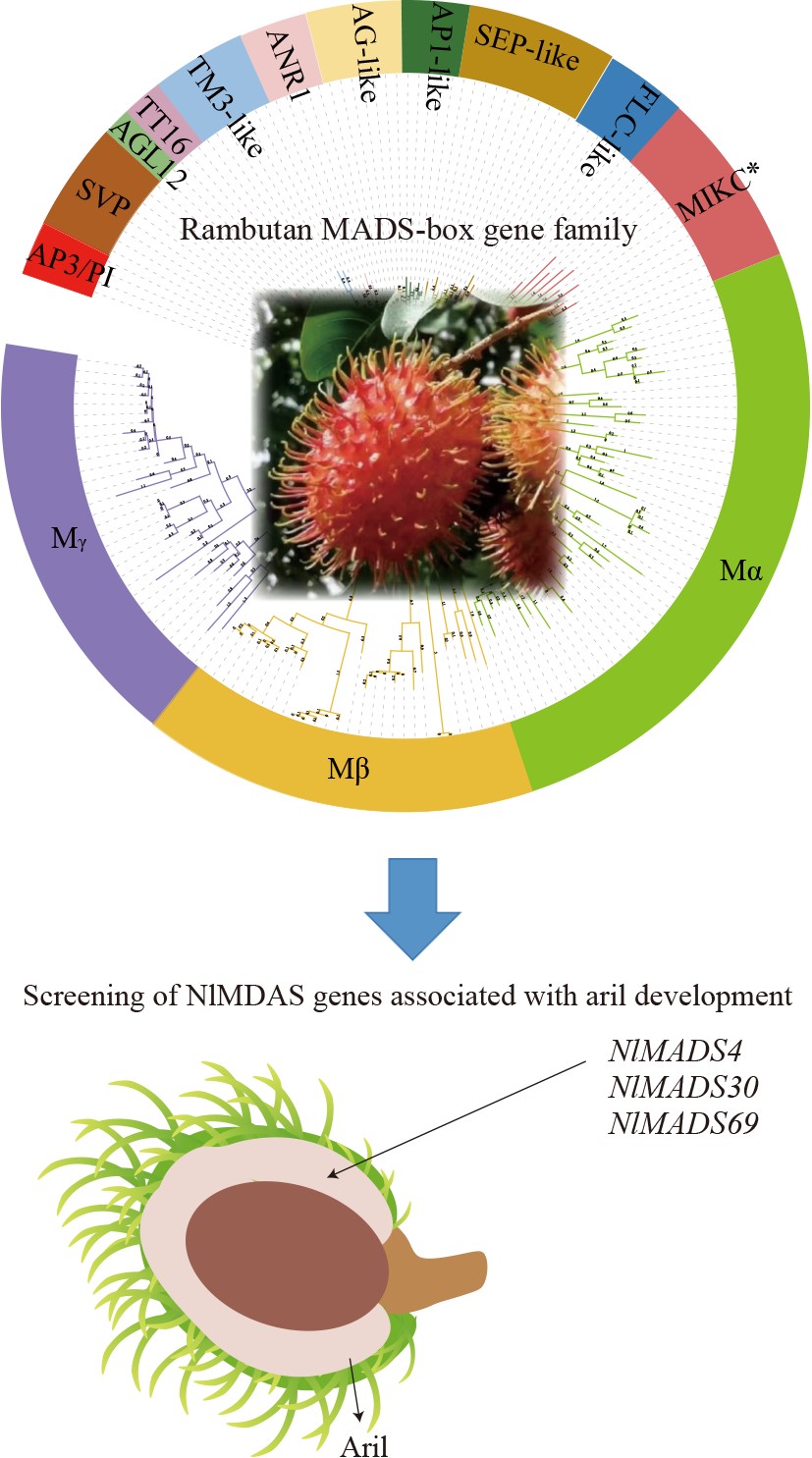



-
Rambutan (Nephelium lappaceum L., 2n = 2x = 32) is an important evergreen fruit tree crop belonging to Sapindaceae family that cultivated in tropical and subtropical regions of the world[1]. Rambutan seeds are coated with fleshy fruits, called 'aril'. However, the molecular mechanisms of flower and aril in rambutan have not got extensive exploration due to the lack of genetic transformation system. Recently, identification and classification of the MADS-box gene family were reported in Arabidopsis[2], pineapple[3,4], rice[5], maize[6], longan[7], lychee[8] and so on, but the MADS-box gene family regulating flower and fruit development in rambutan has not yet been reported.
MADS-box gene family has a crucial influence on plant growth and development. The name 'MADS' derived from four classes of MADS-box genes: 'M' obtained from MINICHRO-MOSEME MAINTENANCE 1 (MCM1) in yeast, 'A' originated from AGAMOUS (AG) in Arabidopsis, 'D' got from DEFICIENS (DEF) in snapdragon, and 'S' stemmed from SERUM RESPONSE FACTOR (SRF) in human[9−12]. MADS-box proteins are composed of two groups, namely type I and type II. In details, Type I can be further divided into Mα-type, Mβ-type, and Mγ-type, while type II is classified into MIKCC-type and MIKC*-type[13]. MIKC-type proteins are mainly distributed in plants, including M (MADS-box) domain, I (Intervening) domain, K (Keratin-like) domain and C (C-terminal) domain[2].
MIKCC-type genes are mainly involved in plant organ identity, especially the regulation of flowers. For example, ABCDE model genes explain how MIKCC subfamily genes regulate floral organ identities. In this model, class A (APETALA1 : AP1) and FRUITFULteL : FUL) control sepals development; class A and class B (PISTILLATA : PI) and APETALA3 : AP3) participate in petals development; class B and class C (AGAMOUS : AG) regulate stamens development; class C determines carpels development; class D (SEEDSTICK : STK) regulates ovules development, while class E (SEPALLATA genes : SEP1, SEP2, SEP3, and SEP4) participate in all models of floral organ development[3,14]. Moreover, there are numerous studies suggest that the MADS-box genes can be involved in the development of fruit-like structures in plants, such as C function genes (AG) and E function genes (SEP)[15−18].
However, there are few studies on aril, especially in rambutan species. In Celastrus orbiculatus Thunb., Aux/IAA, WRKY, ARF and MADS-box genes might be involved in aril development by transcriptomic analysis[19]. Studies on the aril of Ginkgo biloba and Taxus showed that AGAMOUS, AGL6, and the TM8-like gene might be related to aril development[16]. STK and SHP1 transcription factors may regulate aril development in rambutan[1]. Therefore, it was hypothesized that the MADS-box gene family in rambutan might be involved in aril and flower development. Based on this, a preliminary analysis of NlMDAS genes were conducted to better guide the follow-up work.
In the present study, MADS-box gene family members were identified based on the rambutan (cv. Baoyan 7) genome data, 75 MADS-box genes in rambutan were extensively characterized and classified by phylogenetic relationships. Meanwhile, the gene structures, conserved motifs, and cis-element analysis were determined. Moreover, the expression profile of MADS-box genes in different tissues were analyzed and then the GO function was annotated. Finally, the protein interaction regulation network of NlMADS genes in rambutan were predicted. This work provided more useful information about rambutan MADS-box genes and establish a foundation for future study.
-
Two bioinformatic approaches were used to identify MADS-box genes in rambutan. First, the MADS-box genes of Arabidopsis and Oryza sativa were used as queries to compare with the whole genome of rambutan. The putative MADS-box genes of rambutan were then examined using BLASTP with an e-value of 1 × 10−3. The TAIR database (
www.arabidopsis.org ) and RGAP database (http://rice.uga.edu ) were utilized for the extraction of MADS-box protein sequences in Arabidopsis and Oryza sativa. In the meantime, HMMER 3.1 was used to identify the MADS-box protein sequences based on the Hidden Markov Model (HMM) profile of the SFR (type I) domain (PF00319) and the MEF2 (type II) domain (PF09047) obtained from the Protein family database (Pfam) database (https://pfam-legacy.xfam.org )[20]. Only proteins with an E-value below 0.01 were included as candidate genes in further analysis. These candidate MADS-box genes were submitted for further verification through the NCBI Conserved Domain Database (www.ncbi.nlm.nih.gov/cdd )[21] and SMART (http://smart.embl-heidelberg.de )[22] to confirm the presence of the MADS-box domain. The ProtParam tool (https://web.expasy.org/protparam )[23] was employed for the analysis of molecular weights and isoelectric points of the proteins. WoLF PSORT (https://wolfpsort.hgc.jp )[24] was used for the prediction of subcellular localization.Phylogenetic, conserved motifs, and gene structure analyses of MADS-box genes in rambutan
-
Multiple homology alignments were performed on amino acid sequences of MADS-box family member in rambutan, Arabidopsis, and rice using MAFFT (
www.ebi.ac.uk/Tools/msa/mafft ) with default parameters. Subsequently, phylogenetic construction was generated using IQ-TREE v1.6.3 (www.iqtree.org ), with ModelFinder as implemented in IQ-TREE[25]. The phylogenetic tree was then visualized via iTOL (http://itol.embl.de )[26]. Gene structure analysis was conducted to elucidate the exon-intron arrangement using the Gene Structure Display Server 2.0 (http://gsds.gao-lab.org )[27]. The MEME online program v5.4.1 (http://meme-suite.org/tools/meme )[28] was employed to identify conserved motifs within rambutan MADS-box proteins, with parameters set as follows: a maximum of 20 motifs and motif widths ranging from 6 to 200.Chromosomal location and collinearity analysis of MADS-box genes in rambutan
-
MADS-box genes were mapped onto chromosomes of rambutan using MapGene2Chromosome v2.1 (MA2C,
http://mg2c.iask.in/mg2c_v2.1) [29]. Gene duplication and collinearity were performed via MCScanX software (https://github.com/wyp1125/MCScanX )[30]. MADS-box genes in rambutan were then compared by BLAST, and genes exhibiting an identity greater than 85% were selectively identified. Subsequently, the non-synonymous substitution rate (Ka)/synonymous substitution rate (Ks) (Ka/Ks value) was computed through TBtools based on these genes[31].Cis-elements analysis of rambutan MADS-box genes promoter sequences
-
The promoter sequences (−2,000 bp) of each rambutan MADS-box genes were extracted from the rambutan genome by TBtools[31]. PlantCARE (
http://bioinformatics.psb.ugent.be/webtools/plantcare/html/ )[32] were used to predict cis-elements. Then Gene Structure Display Server 2.0 (http://gsds.gao-lab.org )[27] was employed to visualize gene structure.GO enrichment analysis and prediction of interaction networks
-
Firstly, all genes of rambutan were annotated by EggNOG (
http://eggnog-mapper.embl.de/ )[33], and then all the GO terms were extracted to make a background file. Secondly, the file of MADS-box genes, the corresponding file of the gene and the GO number and the function annotation file of the GO number was prepared by R packages 'GO.db' and 'dplyr'. Thirdly, the MADS-box genes were enriched into various pathways through the R package 'clusterProfiler'. Finally, these pathways were visualized with the R package 'ggplot'. Protein interaction network for rambutan MADS-box was constructed using STRING version 11.5 (https://string-db.org )[34], and then visualized the STRING output with Cytoscape version 3.5.1[35].Total RNA isolation and real–time quantitative PCR (RT-qPCR) validation
-
The expression patterns of MADS-box genes from different tissues were obtained from rambutan genome RNA-seq dataset. The |log2(FPKM)| value of the expression patterns of NlMADS genes was used to generate the heatmap by using heatmap tools from the Hiplot Pro website (
https://hiplot.com.cn ). The total RNA was extracted from 100 mg of corresponding ground (with liquid nitrogen) rambutan tissues using the RNA prep Pure Plant Plus Kit (TIANGEN Biotech) according to the manufacturer's protocol. The concentration and quality of all RNA were measured using a 1% agarose gel and Nanodrop ND-1000 spectrophotometer (NanoDrop Technologies). The first-strand cDNA was synthesized using Hifair® II 1st Strand cDNA Synthesis SuperMix for qPCR (with gDNA digester plus) (Yeasen Biotech) according to the manufacturer's instructions. The specific primers were designed by PrimerQuest Tool of Integrated DNA Technologies (https://sg.idtdna.com/Primerquest/Home/Index ) and the primer sequences are listed in Supplemental Table S1. For quantification of MADS-box gene expression, NlActin was used as the endogenous control. The RT-qPCR assays were performed in the Bio-Rad CFX96 Real-Time System real-time PCR instrument by using Hieff® qPCR SYBR® Green Master Mix (Yeason Biotech) with three biological replicates. The 2−ΔΔCT method was used to calculate the relative expression levels of each MADS-box gene.Homology analysis of NlMADS4, NlMADS69, and NlMADS30
-
The protein sequences of NlMADS4, NlMADS69, and NlMADS30 were extracted from the genome in rambutan. Homologous genes of other selected species were further analyzed by the BLASTP program on NCBI. Then the alignment results were displayed via DNAMAN, and the conserved domains were predicted in the CD-research program of NCBI.
-
To identify the MADS-box gene family, both HMM profiles and MADS-box protein sequences from Arabidopsis and Oryza sativa were used as queries to perform HMMER and BLASTP, respectively, against the rambutan genome. A total of 75 candidate MADS-box genes were identified in the rambutan genome. Subsequently, these MADS-box genes were designated as NlMADS1 to NlMADS75 based on their positions on the rambutan chromosomes (Supplemental Tables S2−S4). The size of MADS-box proteins ranged from 67 aa to 433 aa. The relative molecular mass (MW) varied from 7.61 to 48.74 kDa, and the isoelectric point (pI) varied from 4.65 to 11.42 (Supplemental Table S2), indicating that MADS-box proteins could perform their functions in different microenvironments. The majority of rambutan MADS-box proteins were predicted to be localized in the nucleus, while four proteins, namely NlMADS26, NlMADS73, NlMADS31, and NlMADS35, were located in the mitochondria. Only one protein, NlMADS33, was predicted to be located in the chloroplast (Supplemental Table S5).
To investigate the evolutionary relationship between rambutan MADS-box genes and known Arabidopsis MADS-box genes, a phylogenetic tree was constructed based on MADS-box protein sequences in rambutan and Arabidopsis. The phylogenetic analysis revealed that the 75 rambutan MADS-box genes were clustered into two types: type I and type II. Specifically, 50 genes belong to type I MADS-box genes, which include 24 Mα-type, 10 Mβ-type, and 16 Mγ-type, while 25 genes were classified as type II MADS-box genes containing 19 MIKCC-type and 6 MIKC*-type (Fig. 1). These 19 MIKCC-type genes were further divided into 10 groups, namely AP3/PI, SVP, AGL12, TT16, TM3-like, ANR1, AG-like, AP1-like, SEP-like, and FLC-like (Fig. 1).
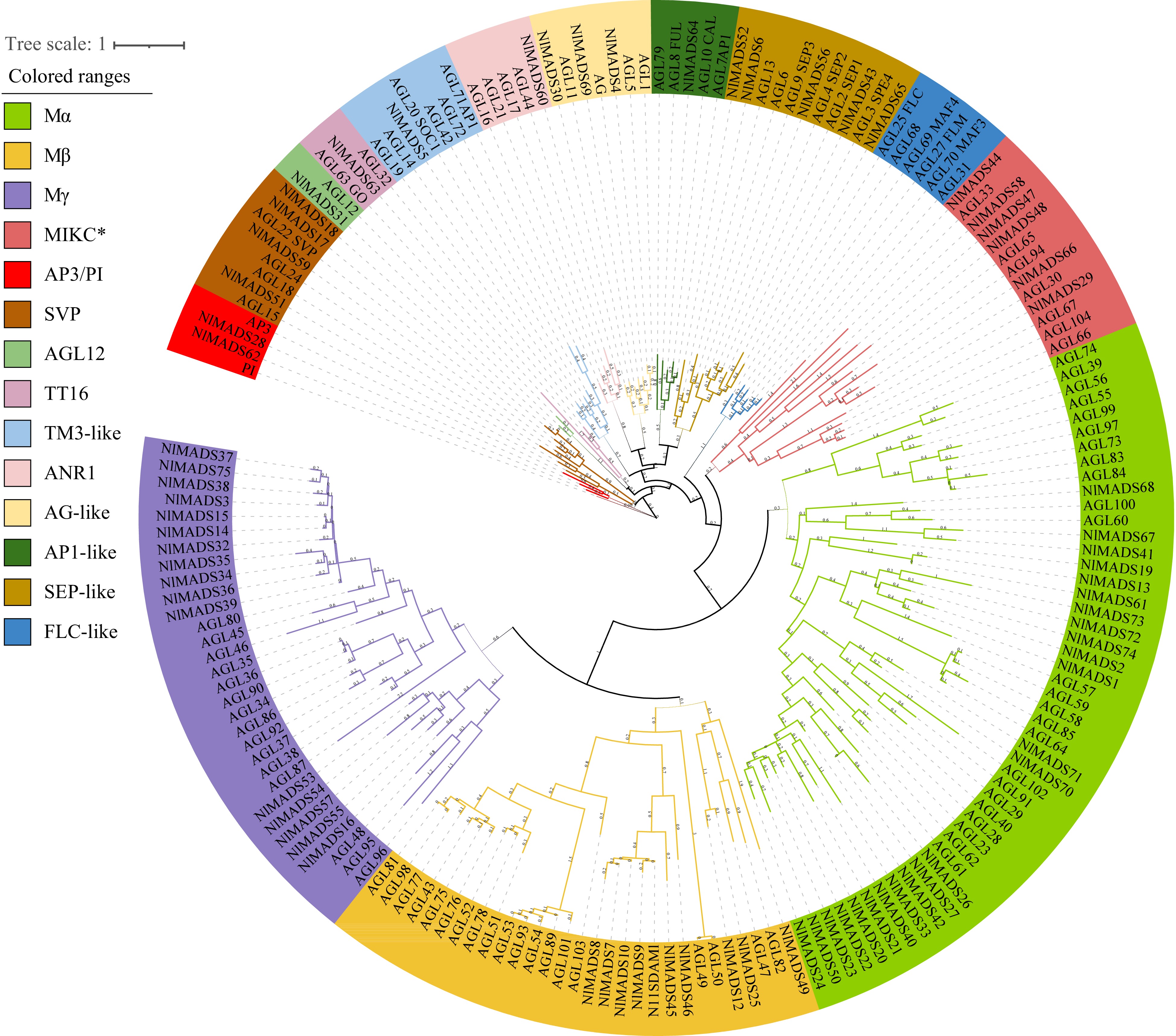
Figure 1.
Phylogenetic analysis of the MADS-box genes from Arabidopsis, rice and rambutan. Different colors represent different subfamilies.
Gene structure and conserved motifs of rambutan MADS-box gene family
-
To further investigate the information on rambutan genes, we used MEME to determine gene structure and identify conserved motifs. A total of 20 conserved motifs were discovered and subsequently annotated using CD-search. Motif 1, motif 7, and motif 14 were characterized as MADS domains, while Motif 5 was identified as a K-box domain. With the exception of NlMADS37, all NlMADS genes contained motif 1 or motif 7. Motif 1, as the MADS domain, was found in all MIKCC-type genes, while motif 14 was predominantly present in the MIKCC-type subfamily. Additionally, motif 5 was present in all MIKCC-type genes and in two MIKC*-type genes (NlMADS29 and NlMADS47), but not in type I MADS-box genes. This indicates the importance of motif 5 in MIKCC-type genes (Fig. 2b). It was observed that genes in different subfamilies often possess unique motifs, which are shared within the same group. Motif 19 was exclusively found in the majority of the MIKCC-type subfamily. Most Mα-type genes contained motif 9, motif 11, and motif 12. Members of the Mγ-type group, such as NlMADS57, NlMADS55, and NlMADS53 exhibited motif 20 and motif 15. Conversely, motif 3, motif 4, and motif 13 were only identified in the Mγ-type subfamily. Motif 2 and motif 17 were characteristic of the Mβ-type group (Fig. 2b).

Figure 2.
(a) Phylogenetic relationships, (b) architecture of conserved protein motifs and (c) gene structure in MADS-box genes from rambutan. Conserved motifs were identified based on phylogenetic relationships via MEME program. The different colors represent the 20 conserved motifs. The yellow and green rectangles represent CDs and UTRs respectively, and the blank lines represent introns.
Motif 1, representing the MADS domain, was identified in all MIKCC-type genes, while motif 14 was exclusively found in the majority of the MIKCC-type subfamily. Additionally, motif 5 was present in all MIKCC-type genes and two MIKC*-type genes (NlMADS29 and NlMADS47), but was not observed in type I MADS-box genes. This suggests that motif 5 plays a significant role in MIKCC-type genes (Fig. 2b). Unique motifs were typically identified in genes from different subfamilies, with these motifs being shared within the same group. Motif 19 was exclusively found in the majority of the MIKCC-type subfamily. Most Mα-type genes contained motifs 9, 11, and 12. Members of the Mγ-type group, such as NlMADS57, NlMADS55, and NlMADS53, possessed motifs 20 and 15. In contrast, motifs 3, 4, and 13 were only present in the Mγ-type subfamily. Motifs 2 and 17 were characteristic of the Mβ-type group (Fig. 2b).
The results of the gene structure analysis revealed that NlMADS genes exhibited varying numbers of exons, ranging from 1 to 17 (Fig. 2c). Specifically, genes in the MIKCC-type subfamily typically had 7−10 exons, except NlMADS59, which had 14 exons. Mα-type genes generally had 1−4 exons, while Mβ-type and Mγ-type genes typically had 1−2 exons, except for NlMADS37, which had three exons. Interestingly, the MIKC*-type group displayed a wide range of exon numbers, ranging from 2 to 17. Among the 75 NlMADS genes analyzed, 29 genes had only one exon, all of which belonged to the type I group.
Chromosomal distribution and synteny analysis of rambutan MADS-box gene family
-
The 75 NlMADS genes were distributed unevenly across 16 chromosomes of rambutan (Fig. 3). Type I MADS-box genes were mapped on all the chromosomes due to many members. Out of Type I MADS-box genes, Mα-type genes were assigned to chromosomes 1, 3, 4, 7, 9, 10, 6, 13, 15, and 16. Genes in Mβ-type group were clustered across chromosomes 2, 4, 6, 11, 12 and 16, whereas Mγ group genes were located on chromosomes 3, 4, 10, and 8. There is no regularity in the distribution of Type II MADS-box genes. Notably, chromosome 4 contained the largest number of MADS-box genes, while Chr1 as the largest chromosome only had two MADS-box genes, indicating the number of MADS-box genes on each chromosome are independent of chromosome size.
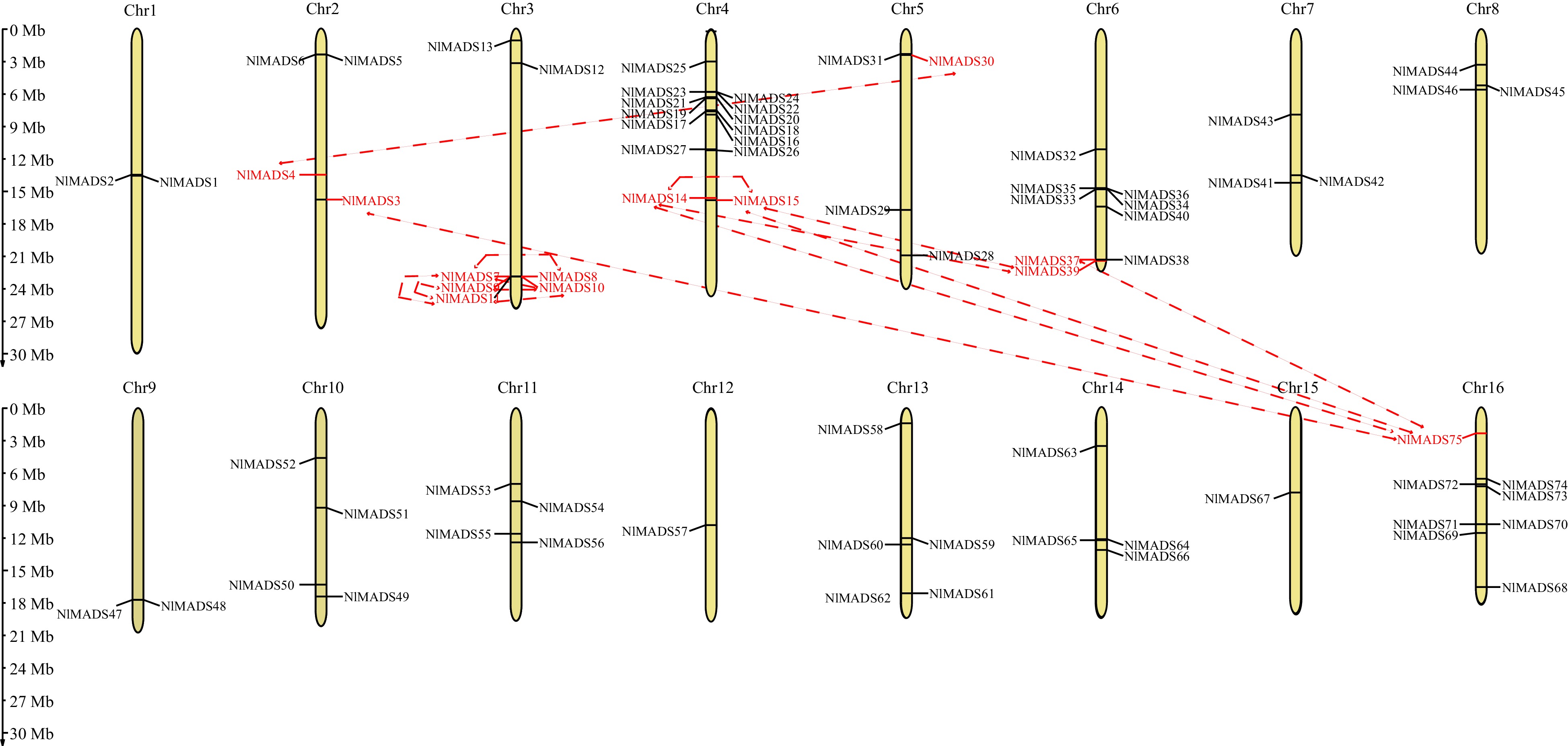
Figure 3.
Distribution of the identified 75 NlMADS genes across the rambutan genome. The dotted lines connected between the two genes represent the duplication events of genes.
The synteny analysis of NlMADS family members in rambutan revealed that these genes were distributed on chromosomes 1, 2, 3, 4, 5, 6, and 16. Among them, seven segmental duplication events were detected in NlMADS genes. A total of seven pairs of tandem duplications were identified, with six pairs located on chromosome 3 (NlMADS7/11, NlMADS7/8, NlMADS7/10, NlMADS9/11, NlMADS9/10, and NlMADS10/11) and one pair on chromosome 4 (NlMADS14/15) (Fig. 4). These findings indicate that segmental duplication and tandem duplication played a significant role in the expansion of MADS-box genes in rambutan. All duplicated gene pairs had Ka/Ks values < 1, indicating they were under purifying selection (Supplemental Table S6). Four pairs of segmental duplications (NlMADS4/30, NlMADS15/39, NlMADS14/39, NlMADS37/75) had Ka/Ks ratios less than 0.3, suggesting their functions were conserved, while the remaining NlMADS gene pairs with Ka/Ks ratios larger than 0.3 indicated potential diverse functions. Additionally, collinearity maps were constructed to analyze the collinearity of MADS-box gene families among rambutan, logan, and lychee (Fig. 5). The results showed that 51 genes in logan were orthologous to 42 genes in rambutan, and 52 genes in lychee were orthologous to 45 genes in rambutan.
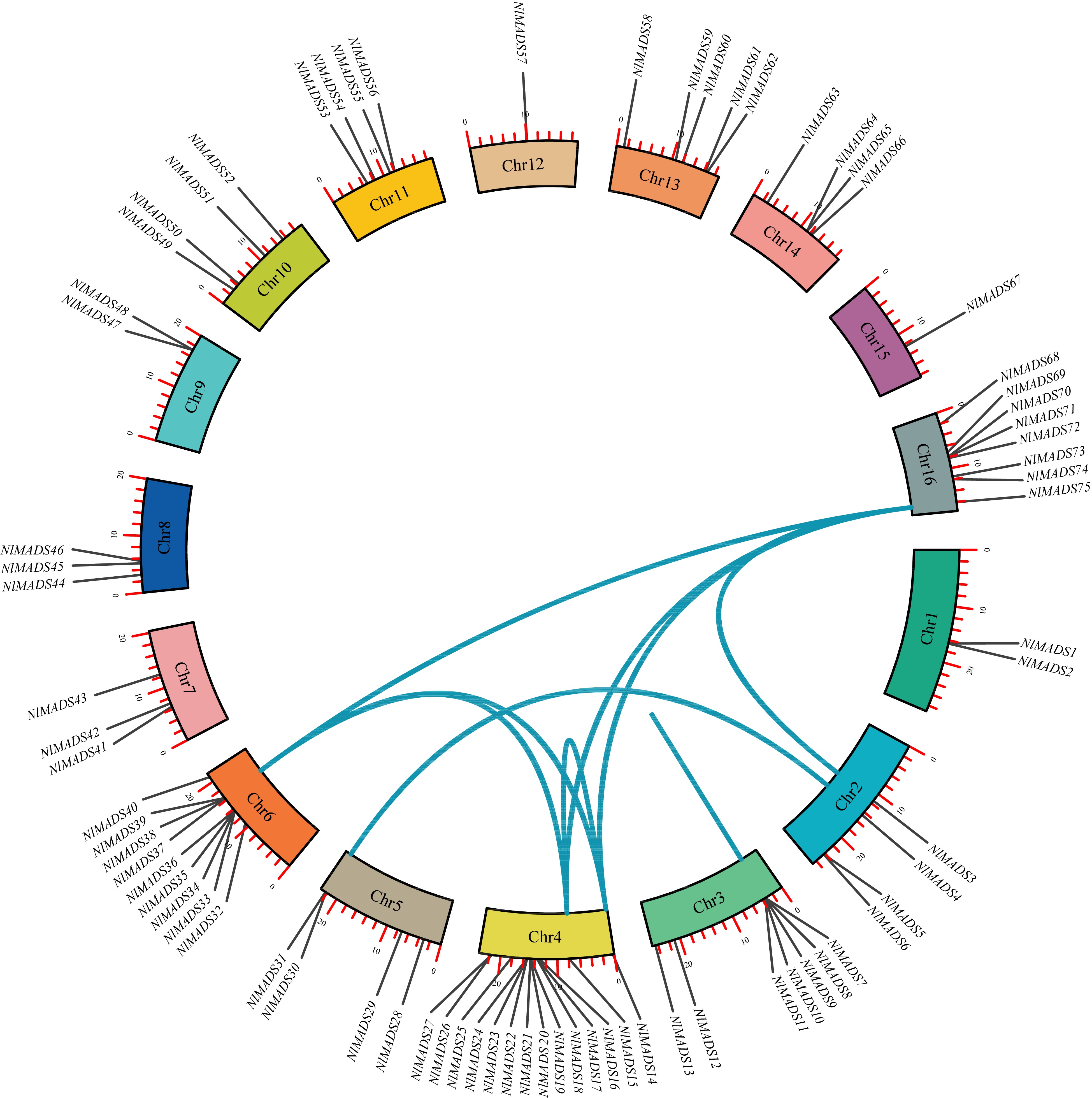
Figure 4.
The synteny analysis of MADS-box family in rambutan. The blue lines connect homologous genes. The different colors represent chromosomes.
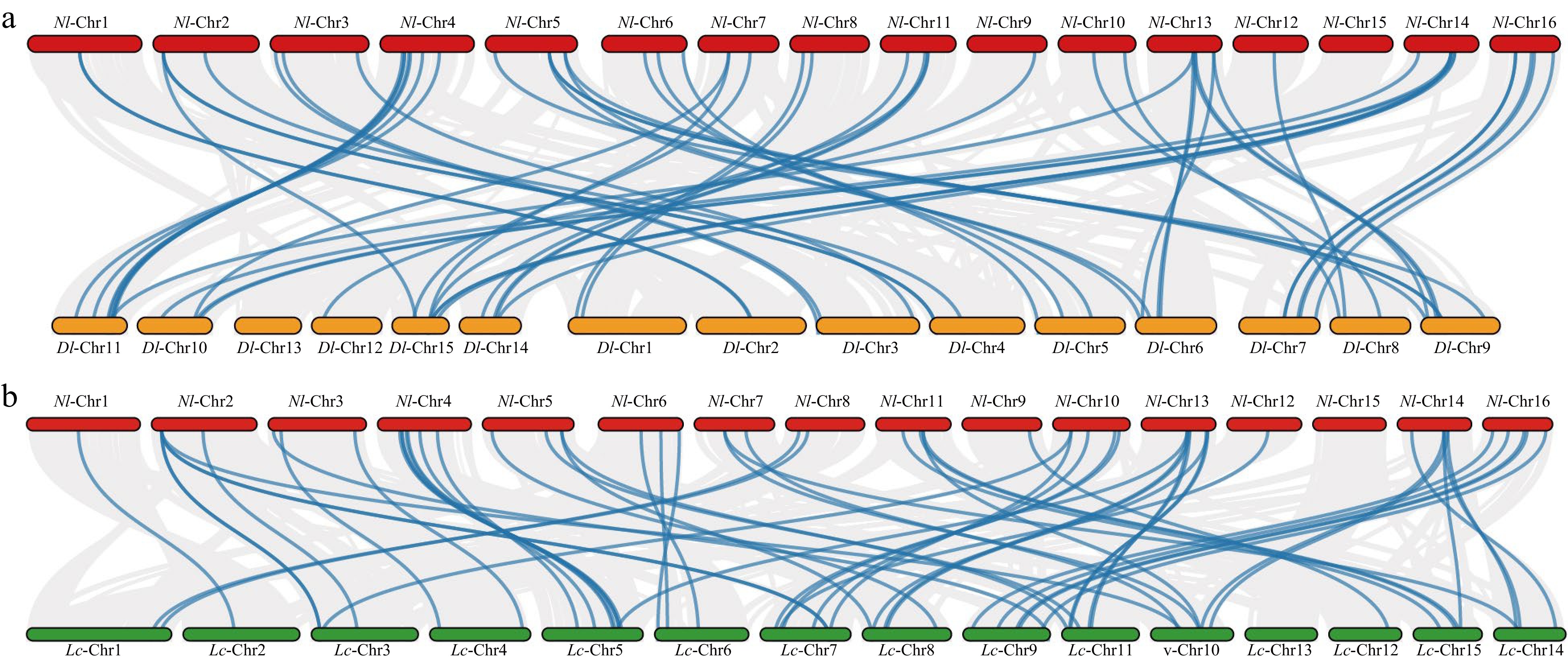
Figure 5.
(a) Synteny analysis of MADS-box family between rambutan and logan. (b) Synteny analysis of MADS-box family between rambutan and lychee. The blue lines connect homologous genes and the gray lines indicate all synteny blocks. The red, yellow and green colors represent the chromosomes of rambutan, logan and lychee, respectively.
Cis-elements analysis in the promoters of MADS-box genes in rambutan
-
The regulation of gene expression is governed by upstream transcription factors that bind to specific cis-elements. To gain further insights into the regulation of NlMADS genes, an analysis of the cis-elements present in the promoter sequences of these genes was conducted. A total of 59 different cis-elements were predicted in the promoters of NlMADS genes, categorized into three groups: growth and development, phytohormone response, and stress response (Supplemental Table S7; Fig. 6). Among these cis-elements, several types were found to be relatively abundant, including light response elements, MeJA response elements, and anaerobic induction elements. We also identified 11 types of hormone-specific cis-elements, such as MeJA-, ABA-, GA-, SA-, and Auxin-responsive elements. Specifically, two types of cis-elements were related to MeJA responsiveness (TGACG motif and CGTCA motif), one related to abscisic acid responsiveness (ABRE), three related to gibberellin responsiveness (TATC-box, P-box, and GARE-motif), two related to salicylic acid responsiveness (TCA element and SARE), and three related to auxin responsiveness (TGA element, AuxRR core, and TGA box). Furthermore, five types of cis-elements associated with multi-environmental responses were identified, namely LTR, GC motif, ARE, TC-rich repeats, and MBS, reflecting responses to low temperature, anoxic conditions, anaerobic induction, defense and stress, and drought, respectively. Additionally, NlMADS62, belonging to the MIKCC-type subfamily exhibited the highest number of cis-elements.
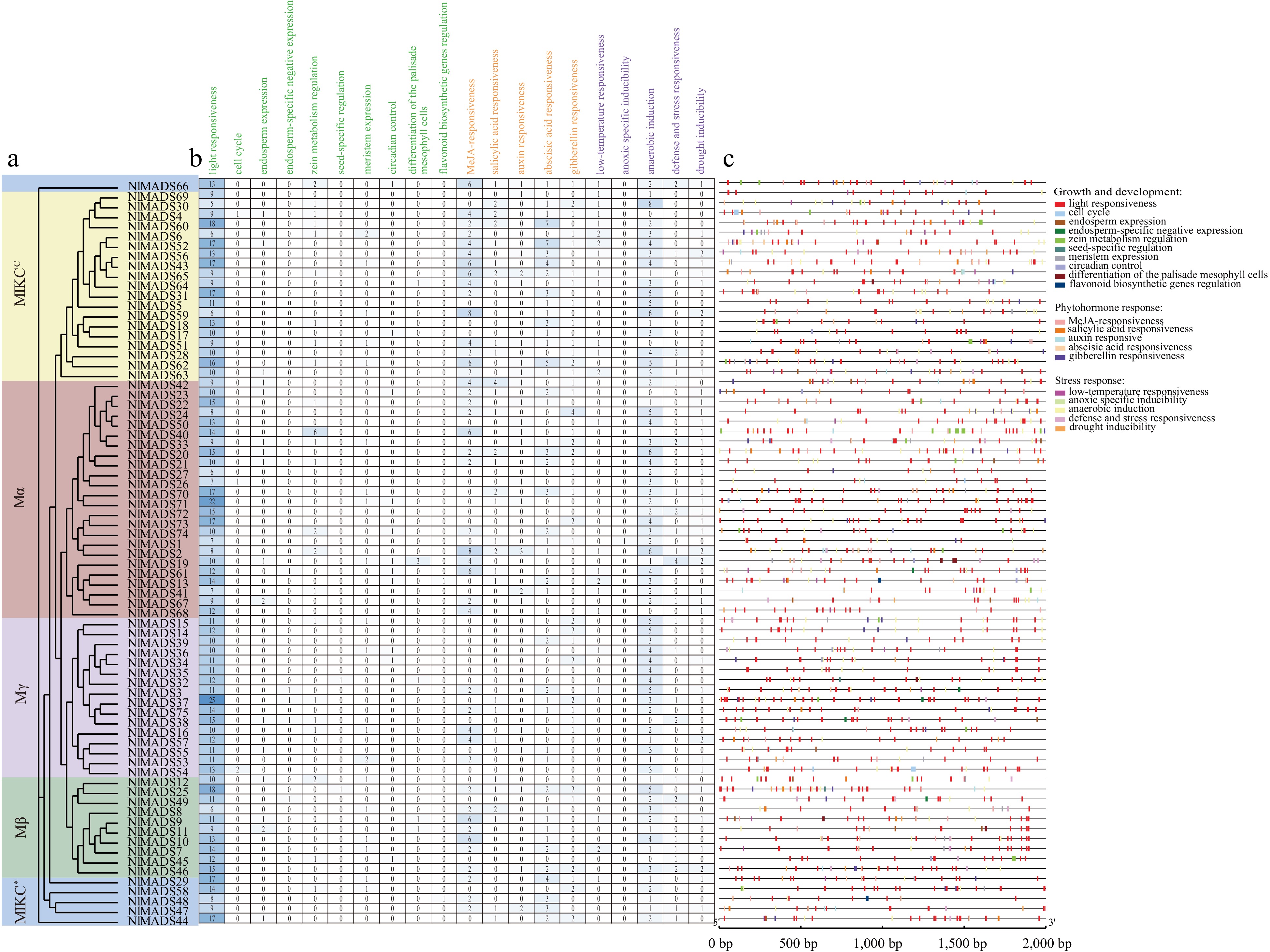
Figure 6.
Cis-elements analysis in rambutan promoters. (a) Phylogenetic relationships. (b) The number of cis-elements. (c) The distribution of cis-elements of promoter sequences (2,000 bp) of NlMADS genes.
Functional annotation analyses of NlMADS gene family
-
To further elucidate the function of the NlMADS family, these genes were annotated and enriched using Gene Ontology (GO). We identified that 75 NlMADS genes were significantly enriched in 52 biological processes (BP), four molecular functions (MF), and three cellular components (CC). The GO term enrichment analysis revealed that the majority of genes were enriched in the BP GO term (Supplemental Table S8) and were involved in organ development, particularly in flower and embryonic development, such as floral organ development (GO:0048437), embryo sac development (GO:0009553), and plant ovule development (GO:0048481). Additionally, fourteen genes were enriched in the regulation of hormone levels (GO:0010817) and regulation of auxin polar transport (GO:2000012), suggesting their potential involvement in auxin regulation (Fig. 7).
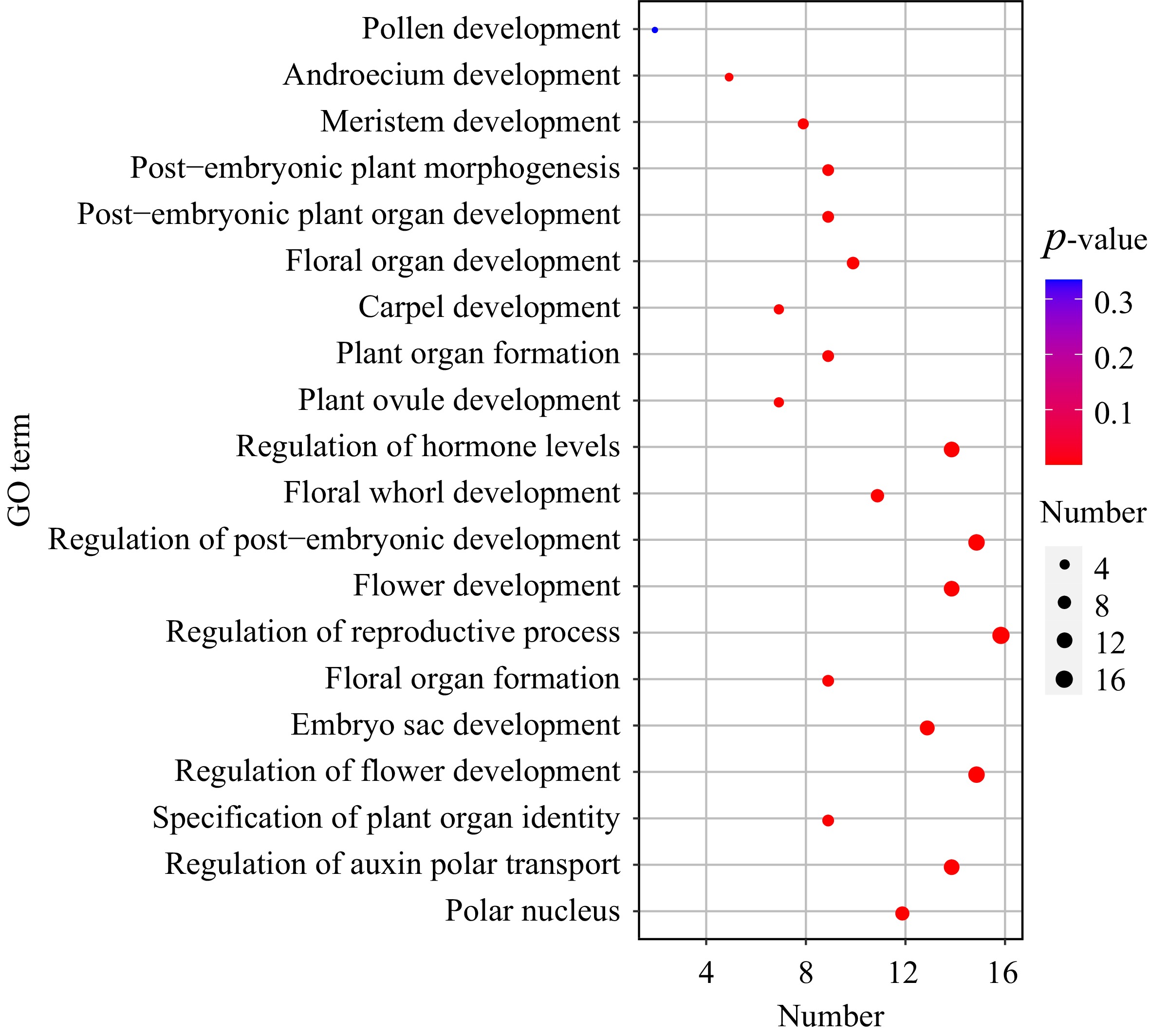
Figure 7.
GO enrichment analysis of NlMADS genes in rambutan.
Tissue-specific expression patterns and protein–protein interaction network of NlMADS family members
-
To explore the expression patterns of rambutan MADS-box genes across various tissues, we analyzed 19 RNA-seq libraries representing different tissues and developmental stages of rambutan using publicly available RNA-seq data from the rambutan genome. Genes with FPKM values below 3 were considered as not expressed. Among the 75 NlMADS genes, 44 genes were filtered out, and a heat map illustrating the expression levels of the remaining 31 genes was generated (Fig. 8; Supplemental Table S9). The expression profiling of NlMADS genes indicated that 11 genes (with FPKM values exceeding 100) exhibited high expression levels in diverse tissues. Notably, all 11 of these highly expressed genes belonged to the MIKCC-type subgroup (Fig. 8). Among these highly expressed genes, seven showed significant expression in flowers, including NlMADS6, 28, 43, 56, 62, 64, and 69. Additionally, NlMADS5 exhibited high expression exclusively in leaves and stems. Furthermore, NlMADS4 and NlMADS30 displayed specific expression patterns during aril development, with up-regulation observed in the early stages of development.
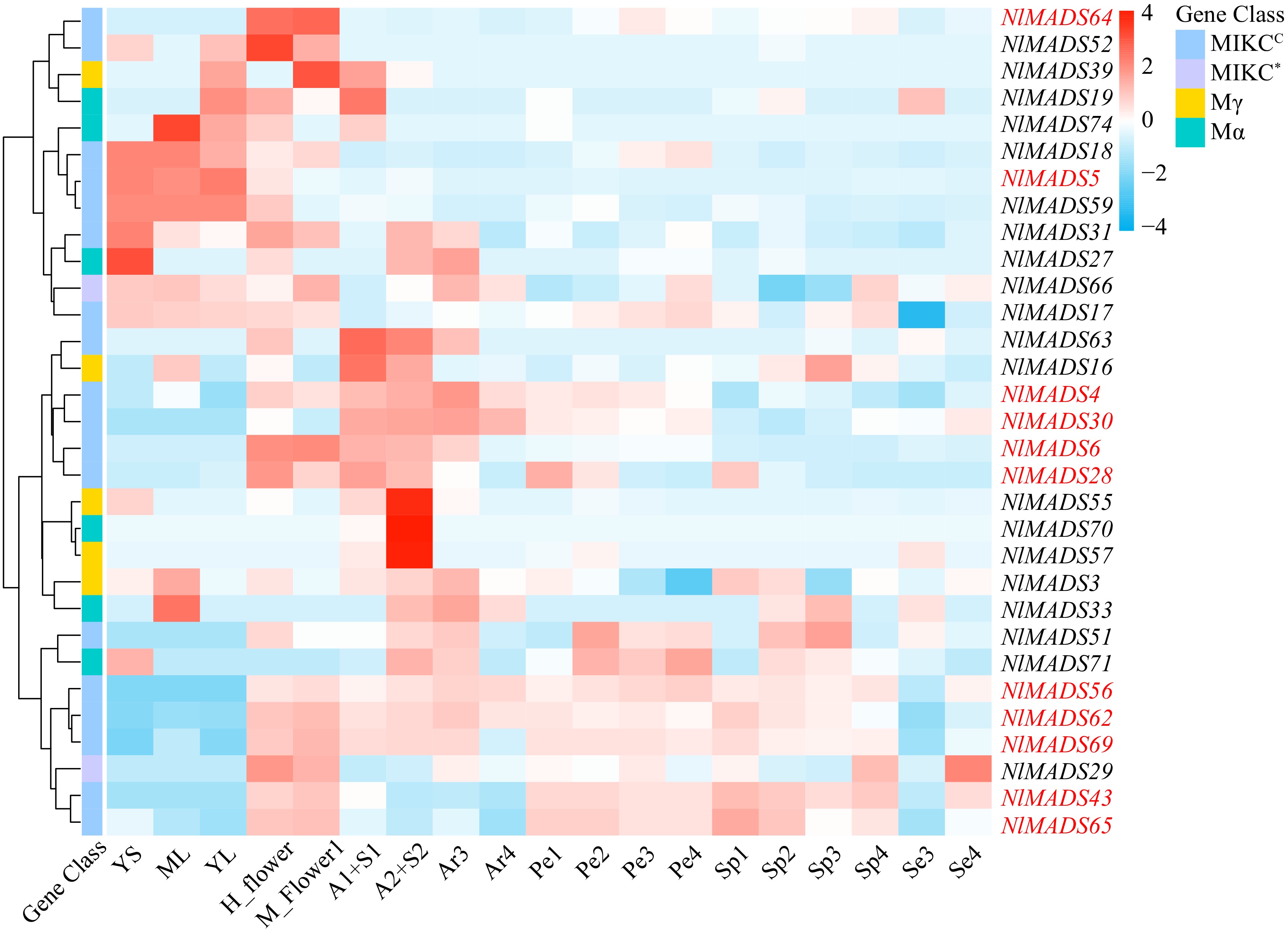
Figure 8.
The expression profile of NlMADS genes. Thirty one genes (FPKM > 3) were selected for expression visualization. Red-marked genes (FPKM > 100) are expressed at higher levels at some stages. YS, ML, YL, H_flower and M_flower represented young stems, mature leaves, young leaves, hermaphroditic flowers, male flowers respectively. A1+S1 represented the sample mixed with aril stage1 and seed stage1, and A2+S2 indicated the sample mixed with aril stage 2 and seed stage 2. Se3 and Se4 represented seed development. Ar3 and Ar4 indicated the development of the aril from thin to thick. Pe1, Pe2, Pe3, and Pe4 represented pericarp development, and Sp1, Sp2, Sp3, and Sp4, represented spine development.
To investigate the expression patterns of NlMADS genes in different tissues, 19 RNA-seq libraries were selected from different tissues and developmental stages of rambutan based on public RNA-Seq data of the rambutan genome. Genes with an FPKM value of less than 3 were considered not expressed. Out of 75 NlMADS genes, 44 genes were filtered out, and a heat map displaying the expression levels of the remaining 31 genes was produced (Fig. 8; Supplemental Table S9). The expression profile of NlMADS genes revealed that 11 genes (FPKM > 100) were highly expressed in different tissues. It is worth noting that all 11 of these highly expressed genes belong to the MIKCC-type group (Fig. 8). Among these highly expressed genes, seven are highly expressed in flowers, including NlMADS6, 28, 43, 56, 62, 64, and 69. In addition, NlMADS5 was highly expressed exclusively in leaves and stems. NlMADS4 and NlMADS30 were specifically expressed during the aril development process and were up-regulated in the early stages of its development. NlMADS62 and NlMADS69 exhibited high expression levels during rambutan fruit development, particularly in the aril, pericarp, and spine. NlMADS43 and NlMADS56 are potential regulators of rambutan fruit development, excluding the aril. NlMADS65 displayed high expression only in the initial stage of spine development. To validate the reliability of the transcriptome data, 13 NlMADS genes were chosen for RT-qPCR analysis, and the results were consistent with the transcriptome data (Fig. 9). MIKCC-type proteins with FPKM values exceeding 10 were utilized to construct regulatory networks. Nineteen genes were selected to predict protein interactions and annotate their functions in Arabidopsis using the STRING program to unravel potential roles and metabolic pathways (Fig. 10; Supplemental Table S10). The analysis revealed numerous close interactions among these MADS-box proteins (Fig. 10a; Supplemental Table S11). Notably, within these predicted interactions, NlMADS4 (SHP1), NlMADS69 (AG), and NlMADS30 (STK) exhibited early upregulation during aril development, displaying consistent expression patterns (Fig. 8). Consequently, these three genes may interact to regulate aril development (Fig. 10b).
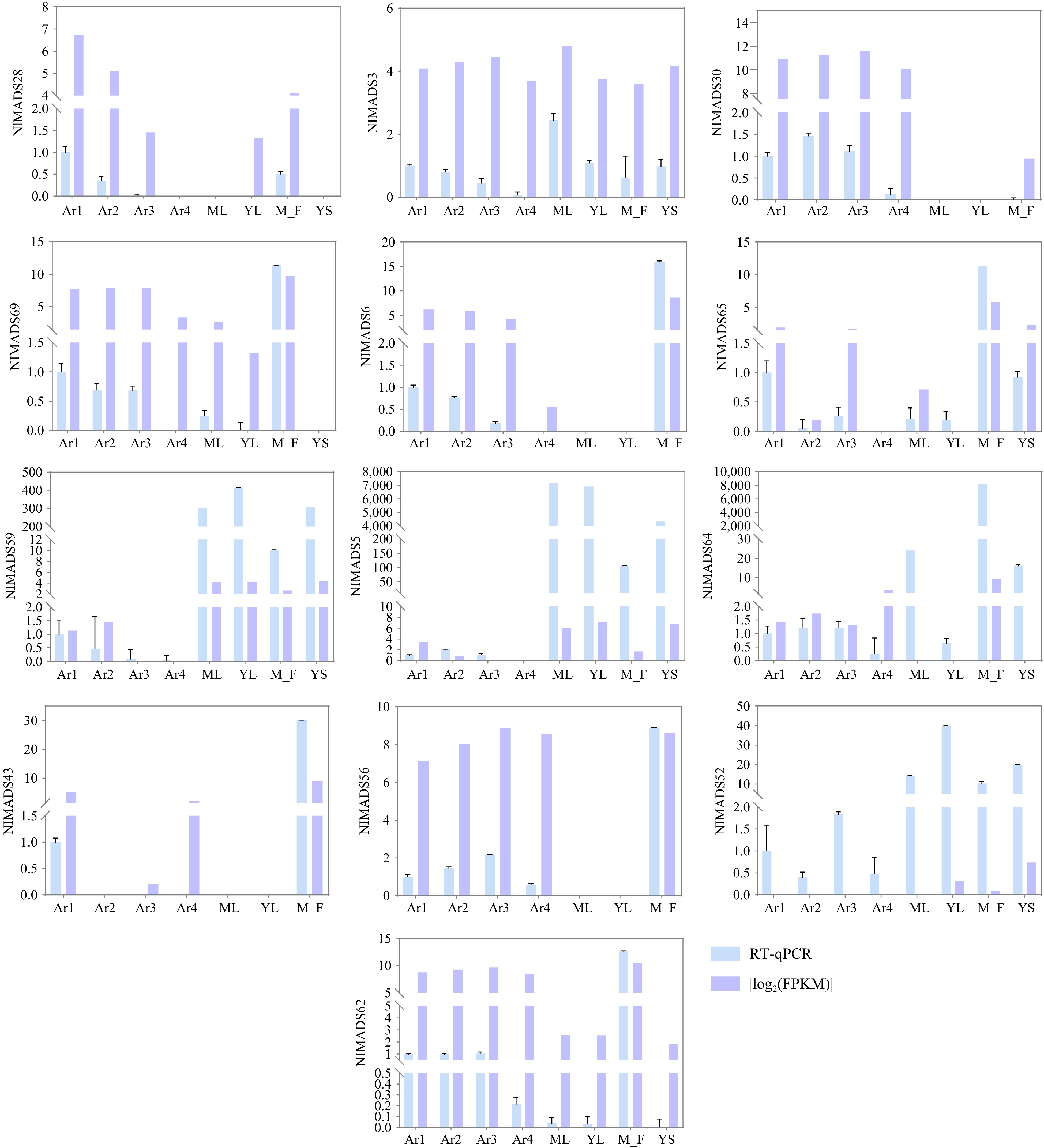
Figure 9.
Relative expression levels of NlMADS genes by RT-qPCR. Blue rectangles represent relative expression from RT-qPCR. Purple rectangles were |log2(FPKM)|.
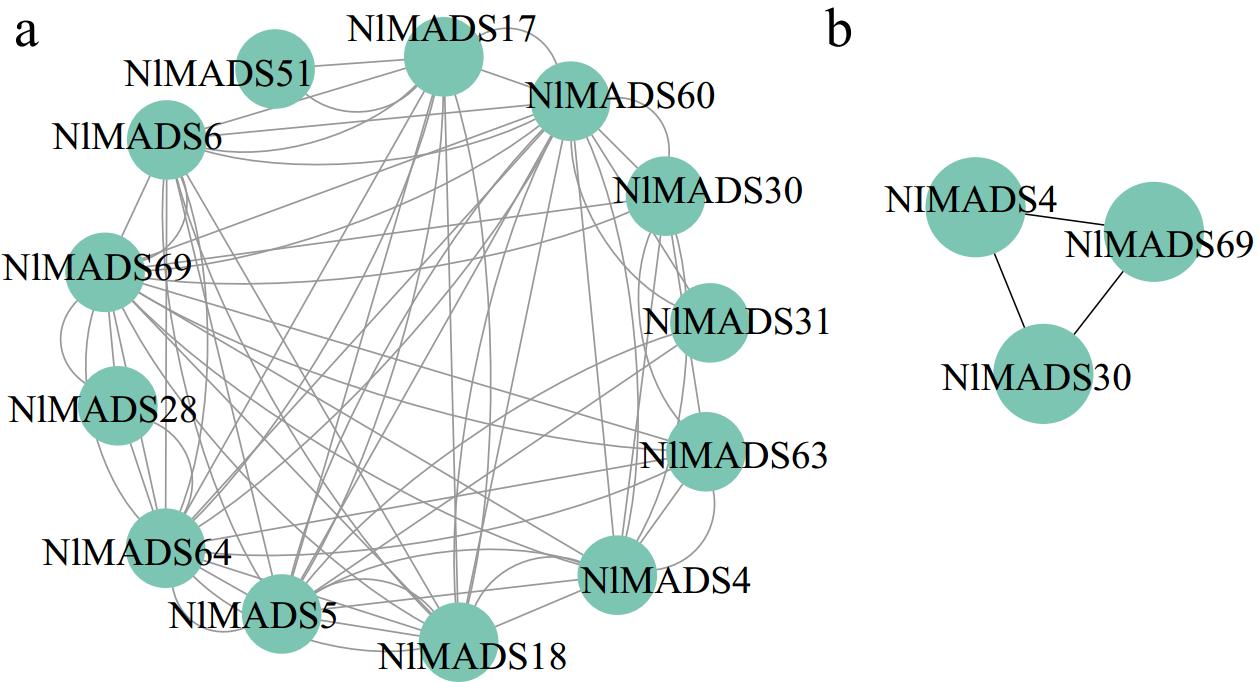
Figure 10.
Protein–protein interaction network of NlMADS MIKCC-type proteins based on their orthologs in A. thaliana.
Protein sequences alignment of NlMADS69, NlMADS4, and NlMADS30
-
To further understand the potential functions of NlMADS69, NlMADS4, and NlMADS30, they were compared by blastp with the genome sequences of lychee, Carica papaya, Solanum lycopersicum, Prunus persica, and Arabidopsis, respectively. Homologous proteins for NlMADS69 were identified, including LIAG (LITCHI028933.m1), CpAG (XP_021898389.1), SIAG (NP_001266181.1), PpAG (ACL31234.1), and AtAG (AT4G18960.1). For NlMADS30, the selected homologous genes were LIAGL11 (LITCHI011705.m1), CpAGL11 (XP_021908122.1), SIAGL11 (XP_010312975.1), PpSTK (XP_007223946.1), and AtSTK (AT4G09960.4). The homologs of NlMADS4 included LISHP1 (LITCHI026995.m1), CpSHP1 (XP_021898390.1), PpSHP1 (XP_007217264.1), and AtSHP1 (AT3G58780.4).
Both the MADS and K-box domains are present in these genes and their homologs, with the MADS domain exhibiting greater conservation than the K-box domain (Supplemental Fig. S1). The alignment of homologs of NlMADS69 revealed inconsistencies in single-site amino acids of conserved domains in plants with fleshy fruits compared to Arabidopsis, such as C-I, K-R, KG-RS, and S-T. Furthermore, a site in the K-box domain of the NlMADS69 homologous alignment displayed differences between plants with and without aril and the K-box domain of NlMADS69 exhibited segment-deletion sequences compared to other homologous genes. Similar conditions were observed for NlMADS30 and NlMADS4. Multiple amino acid sites exhibited differences in fleshy fruits and silique, as well as in plants with and without aril. These variations in amino acid sites within the conserved domains of NlMADS69, NlMADS30, and NlMADS4 may contribute to differences in fleshy fruit development and aril presence compared to other plants.
-
The MADS-box gene family has undergone extensive examination in various plant species, including Arabidopsis, rice, longan, lychee, and sugarcane[2,5,7,8,36], as advancements in sequencing technologies have led to the sequencing of an increasing number of plant genomes. Previous investigations have highlighted the diverse roles played by MADS-box transcription factors in plant growth, development, and responses to environmental stresses. However, limited information exists regarding the characterization of MADS-box genes in rambutan. Therefore, conducting a comprehensive analysis of NlMADS gene family members and their expression patterns across various tissues and developmental stages would prove invaluable for furthering our understanding of the molecular mechanisms governing rambutan growth and development, as well as for exploring their potential applications in rambutan breeding efforts.
In this study, 75 NlMADS genes were identified in rambutan for the first time. These genes were classified into two types: Type I, comprising Mα (24), Mβ (10), and Mγ (16) genes, and Type II, consisting of MIKC* (6) and MIKCC (19) genes. The MIKCC-type genes were further divided into 10 subfamilies based on phylogenetic relationships to shed light on the functional evolution of NlMADS genes. Although the number of NlMADS genes in rambutan is lower than in certain species like Arabidopsis (107), longan (114), lychee (94), and sugarcane (182), it is higher than in many other species such as pineapple (48) and cucumber (43)[4,37]. This disparity may be due to events like genome or gene duplication, which can lead to neofunctionalization, subfunctionalization, or nonfunctionalization[38]. Notably, the number of Type I genes surpassed that of Type II, aligning with observations in other species such as Arabidopsis (Type I: 68, Type II: 39), longan (Type I: 63, Type II: 51), and lychee (Type I: 56, Type II: 37).
However, gene structure analysis revealed that Type II genes possess a higher number of exons and introns compared to Type I genes, which aligns with findings in Arabidopsis, rice, longan, and lychee[2,5,7,8]. In rambutan, Type I NlMADS genes typically exhibit a simple structure with one or two exons, while Type II NlMADS genes are more complex, containing 2 to 17 exons. Introns play crucial roles in various stages of mRNA processing, including transcription regulation, genome organization, and alternative splicing, thereby influencing gene expression and contributing to the maintenance and regulation of gene function[39]. Introns experience weak selection pressure, potentially leading to rapid evolution in genes lacking introns, whereas genes containing larger or more introns may contribute to the acquisition of new functions during evolution[40]. Consequently, the regulatory mechanisms and functions of Type II genes may exhibit greater variability and complexity compared to Type I genes.
Gene duplications play a crucial role in the evolution of genomes and genetic systems[41]. The analysis of duplicated NlMADS genes in the rambutan genome revealed 14 pairs with Ka/Ks ratios < 1, indicating the importance of purifying selection during NlMADS gene duplication. Cis-acting elements play essential roles in plant development and physiology by regulating gene expression, and their divergence often contributes to evolutionary changes[42]. In this study, the promoter sequences of the NlMADS gene were analyzed to predict several cis-acting elements involved in plant growth and development, phytohormone response, and stress response. This finding is consistent with previous reports in the literature[7,8], indicating a certain level of conservation of cis-acting regulatory elements among different species. Several studies have highlighted the significance of hormones such as MeJA and abscisic acid in plant adaptation to biotic and abiotic stresses[43]. In the study of cis-acting elements, investigating hormone-related cis-regulatory elements (CREs) is crucial. Hormone signaling pathways, mediated by CREs play a pivotal role in regulating various aspects of plant growth and development. For instance, the abscisic acid (ABA) responsive element (ABRE) is critical for the regulation of genes involved in seed dormancy and stress responses[44]. Cis-elements such as the TCA-element (related to SA) and the TGACG motif (related to JA) are important for the activation of defense genes involved in plants' immune responses[45]. By studying the hormone-related CREs, we will have a better understanding of the regulatory networks that govern plant physiology and adaptation to their environment. Taken together, the diverse cis-elements discovered upstream of these NlMADS genes suggest their involvement in plant responses and various biological processes. These elements likely play crucial roles in ensuring the normal growth and development of rambutan, as well as enhancing its ability to adapt to diverse natural environments.
Previous studies have demonstrated considerable variability in the expression of MADS-box family genes, both across species and within species. For example, in pineapple, the majority of MADS-box family genes show heightened expression levels in flowers, while certain genes display differential expression patterns between photosynthetic and non-photosynthetic leaf tissues[4]. Similarly, in longan, MADS-box family genes such as DlAP1, DlFUL, and DlMADS109 exhibit diverse expression levels across various tested tissues[7]. Analysis of Carica papaya, encompassing 152 samples unveiled distinct expression profiles for different subclasses of MADS-box family genes across various tissues[46]. Through expression analysis of rambutan MADS-box family genes, we aimed to decipher the molecular mechanisms underlying biological development. Our findings revealed that NlMADS genes are primarily active during flower and aril development. Notably, most genes in the Type I and MIKC* subgroups showed negligible expression, leaving their functions elusive. It is plausible that they are expressed solely in specific cells or under particular conditions. Conversely, certain NlMADS genes displayed tissue-specific expression, particularly those belonging to the MIKCC-type.
The ABCDE model stands as the foremost and classical model elucidating flower development in plants, with MADS-box family genes assuming pivotal roles, extensively studied across various model plants. In this model, A-class genes (AP1, CAL, FUL, and AGL79) dictate floral organ identity, fostering petal and sepal development while delineating floral meristem identity[47]. B-class genes (AP3 and PI) intricately specify petal and stamen structures. C/D class genes (AG, SHP1, SHP2, and STK) primarily orchestrates stamen, carpel, ovule, and fruit development[48−50]. Furthermore, E-class genes (SEP) exhibit partially redundant functions throughout flower development. Broadly, in Arabidopsis and other hermaphrodite species, sepals are determined by A- and E-class genes, petals by A-, B-, and E-class genes, stamens by B-, C-, and E-class genes, and carpels by C-, D-, and E-class genes[51]. Our investigation unveiled several NlMADS genes, namely NlMADS6 (NlAGL6), NlMADS28 (NlAP3), NlMADS43 (NlAGL4), NlMADS56 (NlAGL2), NlMADS62 (NlAP3), NlMADS64 (NlAP1), and NlMADS69 (NlAG), preferentially expressed in flowers. This finding underscores their potential contributions to flower differentiation and development, in line with the roles delineated in the ABCDE model.
Additionally, NlMADS4 (NlSHP1), NlMADS30 (NlSTK), NlMADS62 (NlAP3), NlMADS63 (NlTT16), NlMADS69 (NlAG), and NlMADS56 (NlAGL2) demonstrated notably elevated expression levels in the aril, with upregulation observed during the initial three stages of aril development. This pattern suggests their potential involvement in fleshy fruit development, particularly in aril formation. The AGAMOUS (AG), SEEDSTICK (STK), SHATTERPROOF (SHP1), and SHP2 genes, members of the AG clade, primarily contribute to the development of reproductive organs, fruit ripening, and seed dispersal[50,52]. In Arabidopsis, these genes function redundantly and are indispensable for specifying ovule integument identity and its subsequent development[53,54]. Notably, the rice STK ortholog, OsMADS13, exhibits confined expression within ovules and plays a crucial role in determining floral meristem fate, facilitating the transformation of ovules into carpelloid structures[55].
Previous research on the rambutan genome has underscored the significant involvement of D-class genes, such as STK and SHP1, in aril development, likely attributable to the aril's developmental origin from the ovule stalk[1]. Moreover, transcription factors within the ABCDE model of flower development genes also govern aril development in Ginkgo and Taxus. This encompasses B-class genes (APETALA3 and PISTILLATA), C-class genes (AGAMOUS), D-class genes (SEEDSTICK and SHATTERPROOF), and E-class genes (AGL6 and SEPALLATA), all contributing to the formation of fleshy structures[16,56]. Additionally, prior research has indicated that orthologs of AGL20, such as AtAGL20, OsMADS50, and GmSOC1-like, regulate flowering time in Arabidopsis, rice, and soybean, respectively[57−59]. However, the present findings reveal that NlMADS5 (NlAGL20) exhibits high expression exclusively in rambutan leaves and stems, with no detectable expression in reproductive organs, suggesting that NlMADS5 may have distinct roles in rambutan development compared to other plant species. In summary, these discoveries underscore the significance of MIKCC-type genes in rambutan flower and aril development. Furthermore, the identified NlMADS genes may serve crucial functions in aril development, presenting themselves as potential candidates for further functional characterization. Analysis of the protein-protein interaction network of the NlMADS genes unveiled various interactions among them, indicating their potential collective regulation of rambutan development and stress response through the formation of heterologous complexes. Particularly, NlSHP1 (NlMADS4), NlAG (NlMADS69), and NlSTK (NlMADS30) emerge as key nodes in the protein interaction network, underscoring once again their crucial role in regulating rambutan development.
In conclusion, this study provides the first comprehensive genome-wide characterization of the MADS-box gene family in rambutan. These findings shed light on the potential functional roles of NlMADS genes in regulating the rambutan development process.
-
The authors confirm contribution to the paper as follows: study conception and design: Zhang W; data collection: Dong F, Wan S; Analysis and interpretation of results: Dong F, Wan S; draft manuscript preparation: Dong F, Wan S, Zhang W. All authors reviewed the results and approved the final version of the manuscript.
-
The data that support the findings of this study are available in the NCBI GeneBank repository. These data were derived from the following resources available in the public domain: PRJNA728838.
The research was supported by the Project of Sanya Yazhou Bay Science and Technology City (Grant No: SCKJ-JYRC-2023-21).
-
The authors declare that they have no conflict of interest.
-
accompanies this paper at (https://www.maxapress.com/article/doi/10.48130/tp-0024-0026)
-
Received 5 May 2024; Accepted 14 June 2024; Published online 16 August 2024
-
The MADS-box gene family of rambutan were analyzed..
Expression pattern analysis was used to predict whether NlMADS genes could regulate the aril.
NlMADS4, NlMADS30, and NlMADS69 were screened as the candidate genes for aril development.
-
# Authors contributed equally: Fei Dong, Suyan Wan
- Supplemental Table S1 List of RT-qPCR primers used in this study.
- Supplemental Table S2 Basic information of MADS-box genes in rambutan.
- Supplemental Table S3 The information of NlMADS genes position.
- Supplemental Table S4 Chromosome Lengths of rambutan.
- Supplemental Table S5 Subcellular localization of NlMADS genes.
- Supplemental Table S6 The calculation of Ks, Ka and Ka/Ks of NlMADS genes.
- Supplemental Table S7 Cis-regulatory elements of NlMADS genes.
- Supplemental Table S8 The information of GO enrichment analysis about NlMADS genes.
- Supplemental Table S9 FPKM value of NlMADS genes in different tissues of rambutan.
- Supplemental Table S10 Homologous alignment MIKCC-type genes in rambutan and Arabidopsis genes by String.
- Supplemental Table S11 The interactions of MADS-box proteins.
- Supplemental Fig. S1 The homologous alignment of MADS domain and K-box domain of NlMADS69, NlMADS30, and NlMADS4. The green rectangles represented genes in the plants with aril. The red boxes represented the different amino acid sites between the plant with fleshy fruit and silique. The yellow boxes represented the different amino acid sites between the plants with aril and without aril.
- Copyright: © 2024 by the author(s). Published by Maximum Academic Press on behalf of Hainan University. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Dong F, Wan S, Zhang W. 2024. Genome-wide identification, characterization, evolution, and expression pattern analyses of MADS-box gene family in rambutan. Tropical Plants 3: e026 doi: 10.48130/tp-0024-0026 

Genome-wide identification, characterization, evolution, and expression pattern analyses of MADS-box gene family in rambutan
- Received: 05 May 2024
- Revised: 11 June 2024
- Accepted: 14 June 2024
- Published online: 16 August 2024
Abstract: Rambutan (Nephelium lappaceum L.) is a popular tropical fruit with a unique flavor and an economically valuable plant within the Sapindaceae family. MADS-box transcription factors are widely present in all eukaryotes and affect the morphogenesis, growth, and development of various plant organs. In this study, 75 MADS-box genes in rambutan were successfully characterized. Among them, 50 were identified as type I, including 24 Mα-type, 10 Mβ-type, and 16 Mγ-type genes. Twenty five were identified as type II, including 19 MIKCC and six MIKC* type genes. These NlMADS genes were randomly located on 16 chromosomes based on chromosomal mapping. Synteny analysis indicated the occurrence of seven pairs of tandem duplication and seven pairs of segmental duplication events. Prediction of cis-acting elements demonstrated the involvement of MADS-box genes in plant growth and development, hormone response, and stress response. RNA-seq data showed that most rambutan MADS-box genes were highly expressed in flowers and aril development, particularly within the MIKCC-type group. Notably, NlMADS4, NlMADS30, and NlMADS69 showed specific high expression during aril development, suggesting their critical role in this process. Furthermore, MIKCC-type MADS-box members displayed higher expression levels across different tissues, indicating their importance during plant growth and development. Protein-predicted regulatory networks suggested potential close interactions between MIKCC-type proteins in rambutan. The GO term enrichment analysis showed that the majority of genes might be involved in floral organ development (GO:0048437), embryo sac development (GO:0009553), and plant ovule development (GO:0048481). This paper conducted a comprehensive analysis of the rambutan MADS-box family for the first time, which will provide valuable insights into the molecular mechanisms governing flower and aril development in rambutan and other Sapindaceae species with aril structures.
-
Key words:
- Rambutan /
- MADS-box gene family /
- Genome-wide /
- Expression pattern /
- Aril development