








-
Traditionally, plant genetic engineering is defined as a set of techniques and molecular approaches for the transfer of traits or functions between plants or the improvement of an existing function, through the manipulation, modification, and transfer of one or several genes to a host plant[1].
An improvement in plant genetic engineering is genome editing. It can be defined as the precise, accurate, and discrete manipulation of one or more genomic sites, using editing tools based on enzymes or enzyme/RNA complexes. There are several genome editing techniques available to change the structure of genes and their regulation in a timely or extensive manner[2]. Broad or specific genomic editing can provide base deletions, additions, and substitutions in one or more targeted loci and also lead to differential mRNA processing, which leads to changes in gene expression and protein function. This review will detail the different genome editing techniques and explain the differences and advantages of each technique currently used in plant biotechnology[3,4].
-
Plant genome editing methods share six basic steps; in silico design of the genome target site, vector construction, in vitro validation of editing activity using cultured cells, delivery of constructs to host species cells, inducing plant regeneration, and mutant screening and genotyping[5,6].
Recently, the era of genome editing has relied on the development of techniques based on the interaction of DNA with programmed sequence-specific nucleases (SSNs), used for both basic as well as applied plant biology, which revolutionized formerly unstable and non-specific transgene incorporation in the host genome of a crop species[7−9]. SSNs-based gene editing techniques are notably meganucleases (MNs), zinc-finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and clustered regularly interspaced short palindromic repeats/CRISPR-associated protein 9 (CRISPR/Cas9)[3,7].
In common, the techniques that use SSNs produce double-strand breaks (DSBs) in the strands of a targeted DNA locus, since the SSNs are carriers of a sequence-specific DNA binding domain merged with a non-specific nuclease domain, capable of recognizing a specific site sequence and cleaving it. After the double cleavage, there is an activation of the error-prone DNA repair mechanism of non-homologous end joining (NHEJ) or the highly accurate homology-directed recombination for precise genome editing[3,10−12]. Regardless of the chosen technique, SSNs were not only used for simple base insertion/deletion or single gene inactivation, but also significantly enhanced the accuracy of homologous recombination and, thus, allowed precise locus gene replacement[3].
Each of the techniques has methodological peculiarities and intrinsic editing properties, varying with respect to efficiency, off-targets, sensitivity, scope, and expertise. The main genome editing techniques that involve the use of SSNs are described below[3,13,14].
-
MNs were the pioneering enzymes in the editing of the plant genome. Many examples of plant genomes edited by MNs have already been reported in the literature[15]. MNs recognize short specific sites in DNA with 12–40 bp before catalyzing the formation of DSB, making them an excellent delivery system for genome editing[16]. Despite this, MNs only recognize specific target sites and thus are unable to recognize new target sequences different from those naturally recognized by the system[17]. This limits their use to only MNs existing in nature such as I-SceI and I-CreI nucleases[18,19] (Fig. 1).
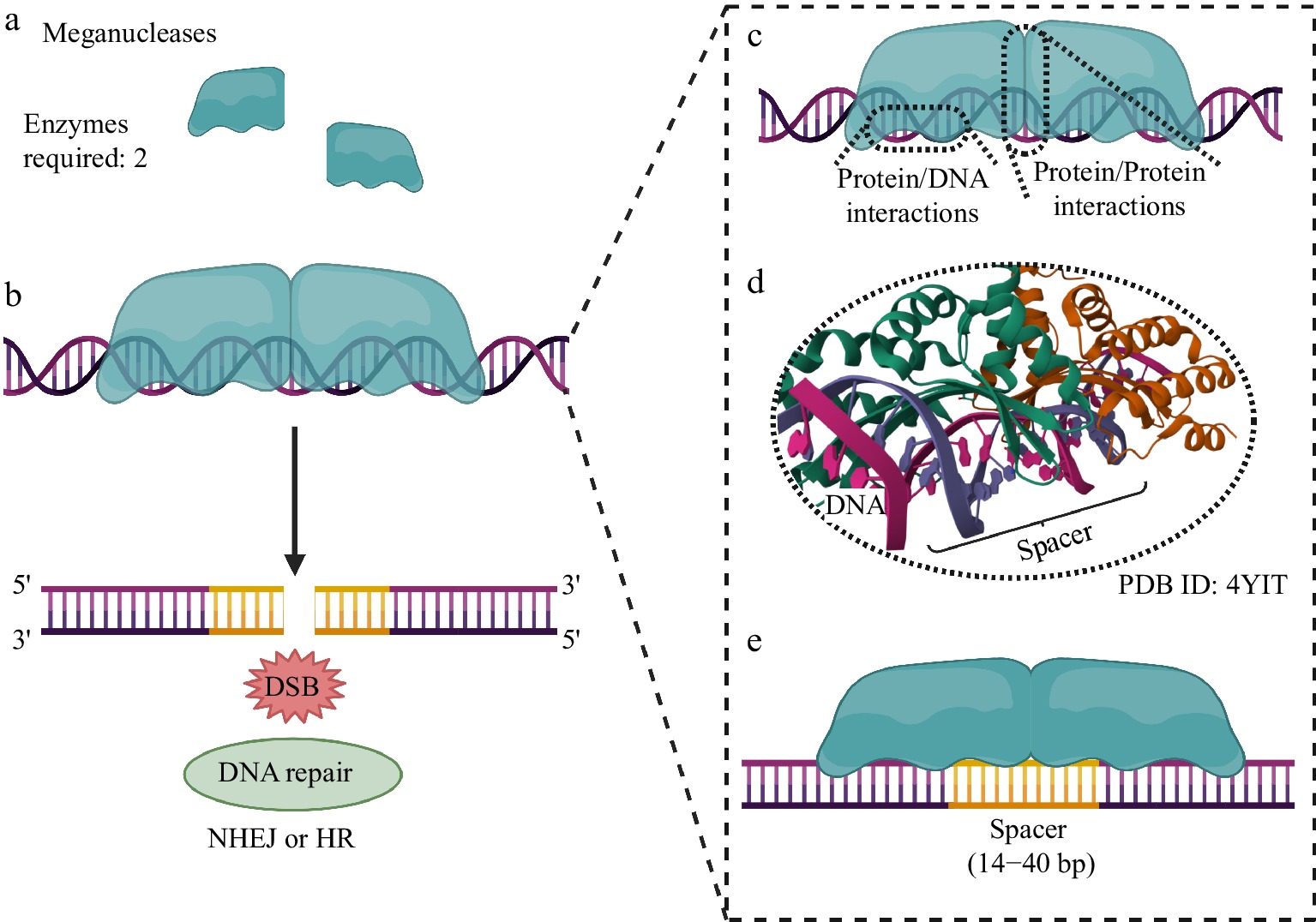
Figure 1.
Genome editing using MNs. (a) Two MNs are required to generate the DSB at the target genomic site. (b) After DNA cleavage, repair systems based on NHEJ or HR are activated, allowing subsequent modification of the genomic locus for the biotechnological purpose of the experiment. (c) Protein/DNA interactions are established between the MN (through its specific DNA recognition/binding domain and endonuclease domain, for specific DNA cleavage) and the genomic locus to be edited. (d) The endonuclease domain must interact with the genomic spacer region to allow DNA cleavage. (e) Both MNs recognize a long cleavage spacer region, typically 14 to 40 base pairs (bp) long. Created with BioRender.com.
-
ZFNs are one of the main methods for genome editing in crops, especially for gene knockout and site-specific mutagenesis. ZFNs have structural plasticity and a flexible nature, which allows modeling of the recognition and DNA binding domains to virtually any site in the target genome[20]. It typically is comprised of an array of Cys2-His2-Zn motifs forming an approximately 30 amino acid structure with two beta-sheets in opposition of an alpha-helix[21]. The first report on the use of ZFNs occurred in 1996, in which these synthetic chimeric enzymes with their DNA binding domain were fused to the non-specific FokI homodimeric DNA restriction enzyme isolated from Flavobacterium okeanokoites[22]. The dimerization of two ZFN monomers is necessary for the activation of cleavage, thus requiring two ZFNs simultaneously interacting with their respective specific sites on each of the DNA strands. Each monomer with 3 to 6 ZFN modules recognizes sequences on a 9–18 bp DNA strand, targets the spacer region of 5–7 bp, and forms the dimer that catalyzes the formation of a DSB[4,22]. Advances in spacer sequences and FokI orientation have increased site specificity[23]. Therefore, sequence recognition and specificity are determined by the amino acid sequence of each ZF, the number of ZF on each module and the activity of the FokI endonuclease.
The development of this method was a significant breakthrough in genome editing, as the potential to recognize a 36-base sequence greatly improved specificity as compared to Meganucleases, and reduced off-target effects. There are several successful examples of plant genomes edited using ZFN technology, with a template-based approach, which are designed to induce homologous recombination events; as well as non-template approaches, designed to induce indels in specific gene targets. ZFN has been successfully used in the model plants Arabidopsis and tobacco; and crop plants such as soybean, maize, rice, wheat, and tomato[24]. Despite these successful examples, ZFN technology has encountered resistance in the scientific community for several reasons. The Zn-finger protein structure recognizes a 3-base pair DNA sequence, thus overlapping specificities can lead to off-target effects. Moreover, ZF can interact with each other, and these interactions can affect DNA recognition and specificity. ZFNs also show a bias regarding DNA sequence context, in which G-rich sequences are preferably edited in contrast to G-poor regions[25]. Lastly, engineering ZFNs is complex, expensive and technically challenging, limiting its widespread use. Taken together, these characteristics mean that ZFNs are highly dependent on intramolecular interactions and DNA sequence context, therefore limiting the number of good targeting sites[11,26] (Fig. 2).
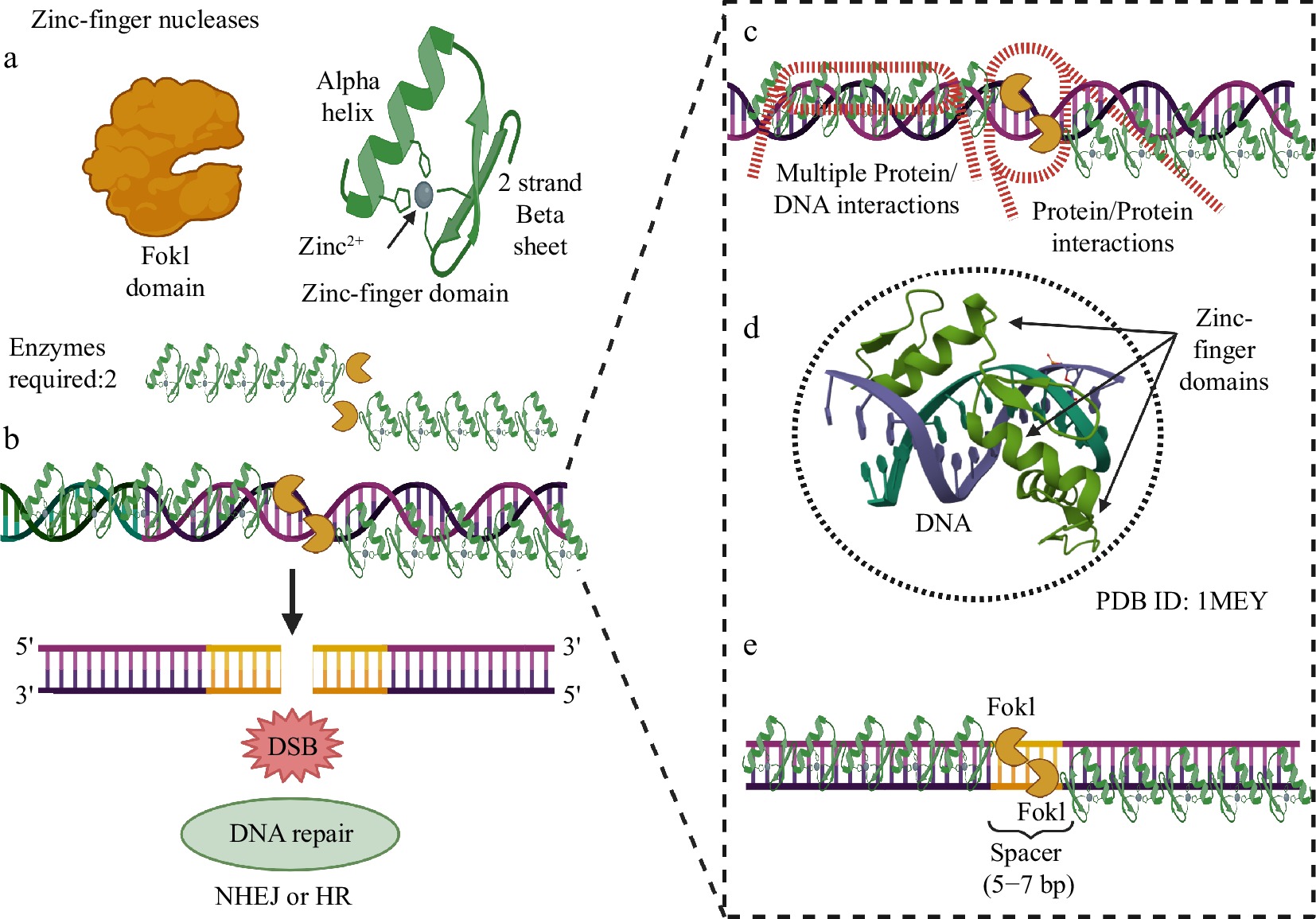
Figure 2.
Main aspects of genome editing with ZFNs. (a) Functional domains of ZFNs: the FokI cleavage domain and the DNA-specific recognition and binding domain; Zinc-finger domain (containing an alpha-helix and a beta-sheet with two antiparallel beta-strands, in addition to a Zn2+ cation. (b) Two ZFNs are required for the cleavage of the genomic locus, with simultaneous double activation of the FokI domains, which need to interact after touching each other. The provoked DSB activates DNA repair systems (NHEJ or HR). (c) Multiple DNA/protein interactions and between FokI domains allow two compatible ZFNs to catalyze two phosphodiester bond cleavage reactions within the spacer region. (d) Tandem Zinc-finger domains are essential for the specificity of recognition and binding to DNA at the genomic editing locus. (e) The spacer region capable of presenting an editable site for the FokI domains is short, between 5 and 7 bp. Created with BioRender.com.
-
Another widely used technique for editing the plant genome is TALENs. TALENs are artificial restriction enzymes that combine the FokI nuclease domain with the DNA-binding domain of transcription activator-like effector (TALEs) TALEs have been reported after studies with the phytopathogenic bacteria of the genus Xanthomonas, and capable of infecting numerous crops[11]. TALEs are proteins, capable of penetrating the nucleus of cells infected by Xanthomonas and targeting specific DNA sequences to modify the expression of endogenous genes of the host plant, to stimulate cell growth and the biosynthesis of useful metabolites for the pathogen[27]. The ability to target and modify specific DNA sequences sparked an interest in modifying these proteins for gene editing. TALE proteins are comprised of an array of highly conserved repeat sequences of 34 amino acids in length, although repeats of 33 and 35 are also present. Each 34 AA repeat is conserved, except at positions 12 and 13. These positions vary within the repeats and are called repeat-variable di-residues (RVD). Each RVD within the 34 amino acid sequence recognizes one nucleotide specifically. This code is relatively simple. The most common RVD within TAL effectors are HD, NG, NI, and NN, which recognize the nucleotides C, T, A, and G, respectively[28,29]. TALENs are engineered to acquire in tandem modules consisting of specific pairs of amino acid residues, called repeat variable di-residues (RVDs), which cooperatively link to the editing site, configuring themselves in a specific binding domain to the target sequence in the DNA, endowed with 33-35 amino acid residues. In addition, each TALEN receives at its C-terminal a FokI nuclease domain to promote a DSB at the editing site[11,30]. As with ZFNs, two TALENS are needed, simultaneously associated with each of the DNA strands, in convergent orientations, since the FokI domains activate each other to generate a DSB. TALENs require a spacer region of 12–21bp for FokI dimerization and cleavage[30].
Compared to MNs and ZFNs, TALENs have the advantage of allowing virtually any possible combination of their DNA recognition module arrangements, making these SSNs versatile tools for gene editing. Another interesting aspect of TALENs is their greater specificity in recognizing target sites in DNA when compared to other nucleases. Typically, a TALEN has an extension of 15–20 RVDs and a TALEN is often able to associate with target sites of 30 or more bp in the DNA recognition domain. Since the DNA interaction site with the TALEN RVDs is larger, there is a greater specificity of interaction between the editing system and the site immediately before the cleavage point[31]. The genes encoding TALENs can be integrated directly into host cells or by vectors carrying the sequences[32] (Fig. 3).
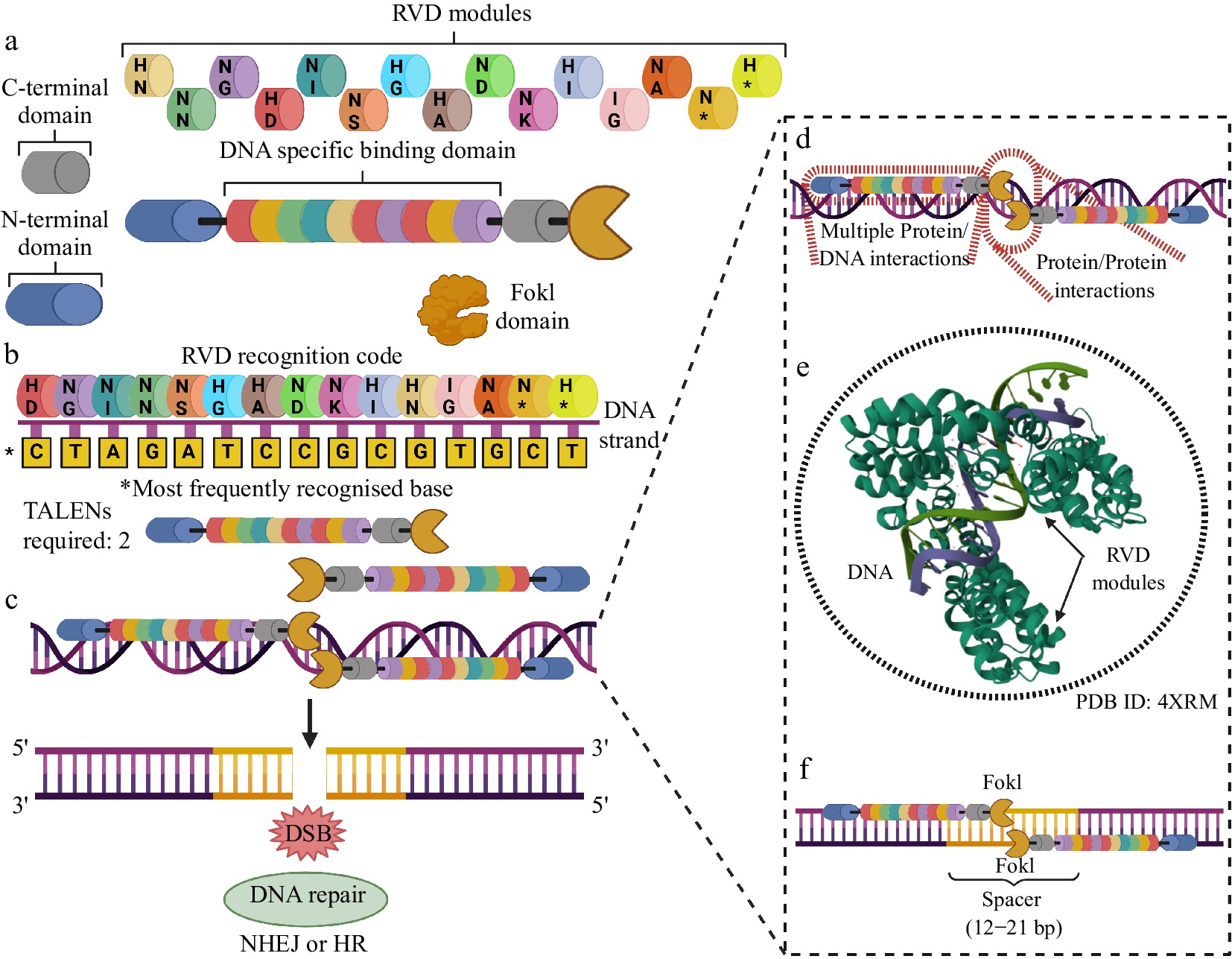
Figure 3.
Most important aspects of using TALENS for genome editing. (a) TALENS have components varying in length and structural complexity. Each TALEN has two stabilization domains (N- and C-terminal), a FokI cleavage domain, and an extensive DNA-specific recognition and binding domain, made up of RVD modules with pairs of amino acid residues organized in tandem. Each combination of residues in an RVD pair can recognize and specifically bind to a base along a DNA strand. (b) The RVD DNA recognition code indicates which pair of amino acid residues are most likely to recognize and bind to a given DNA base. The same pair can bind to more than one base but with different probabilities. The figure shows the pairs with their respective most frequent bases. (c) Two TALENs with opposite orientations are needed to produce a DSB. (d) TALEN/DNA interactions are extensive, which allows for a higher level of editing precision of this technology when compared to MNs and ZFNs. (e) The RVD modules present sequential binding cooperation to the genomic editing locus, which increases the specificity of DNA recognition. (f) A spacer of 12 to 21 bp is required for the mutual activation of the FokI domains and the precise cleavage of the DNA. Created with BioRender.com.
Compared to ZFNs, TALENs have been more widely used to edit plant genomes. Many plants have already been subject to genome editing by TALENs such as Arabidopsis, tobacco, and grasses, such as rice and Brachypodium. However, there are major drawbacks to the use of TALENS as a genomic editing tool. The large size[11,32] (~950 to ~1,900 amino acids) of the proteins and their repetitive modular nature make it difficult for molecular manipulation and more complex to design and assemble tandem repeats for specific site recognition and editing. Additionally, some level of off-targets are still observed, although less than MNs and ZFNs.
-
MNs, ZFNs, and TALENs constitute the first generation of SSNs for the editing of plant genomes. The CRISPR/Cas9 system has started the second generation of editing tools, an integrated set of molecules for genomic manipulation that consumes less time, requires simpler and shorter protocols, is easier to design, are more robust, and is more cost effective[3,11].
The two main molecular components of the CRISPR/Cas9 system are single guide RNA (sgRNA), which targets a specific DNA sequence, and independent Cas9 protein, which can catalyze the simultaneous cleavage of the two DNA strands at a target site without the need for dimerization, as required in ZFNs and TALENS. Therefore, sgRNA sequence design is all that is needed to access any editing site in the target genome, making CRISPR/Cas9 a much more friendly and easy-to-handle tool than TALENs and ZFNs[4] (Fig. 4).
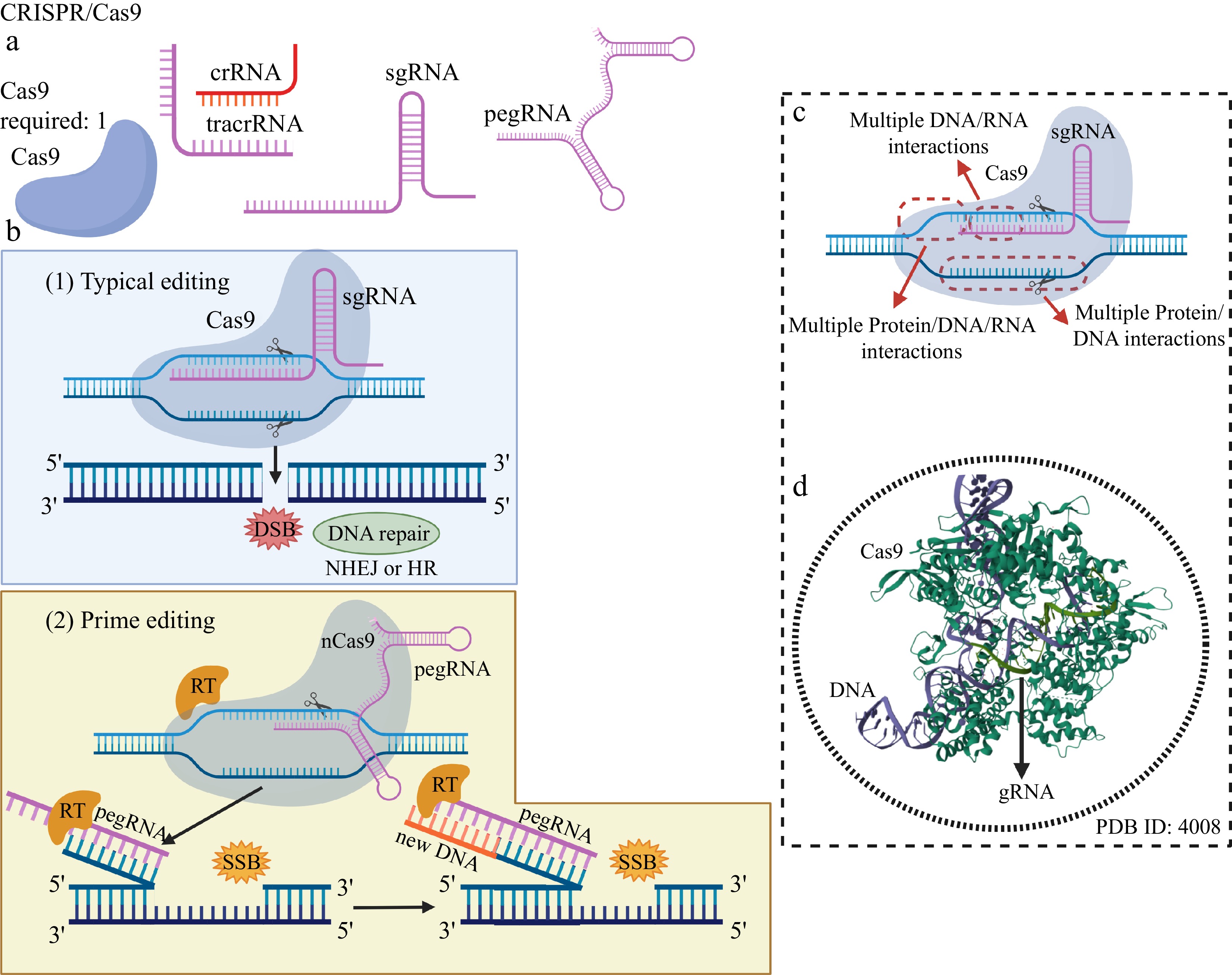
Figure 4.
Genome editing using the classic CRISPR/Cas9 system and Prime editing. (a) Editing elements typically present in the CRISPR/Cas9 system: the DNA cleavage enzyme Cas9, the recognition RNAs of the genomic editing locus: crRNA, tracrRNA, a single guide RNA (sgRNA) and a prime editing guide RNA (pegRNA) – in the case of Prime editing. (b) In typical editing, a sgRNA internal to Cas9, containing a sequence of bases that are complementary and homologous to the bases of a DNA strand at the genomic editing locus, is used to access the editing region, which is doubly cleaved by a single Cas9 with both internal endonuclease domains, resulting in a DSB and activation of DNA repair systems. In the Prime editing system, a modified Cas9 (nCas9), linked to a reverse transcriptase (RT) and carrying only one active DNA cleavage domain, is guided by a peg RNA to the genomic editing locus. After pairing the bases of an extensive region of the pegRNA with one of the DNA strands of the editing locus, nCas9 catalyzes the production of a single-strand break (SSB) by a single nick in the DNA. RT reads the editing sequence present in a region of the pegRNA and synthesizes a DNA strand containing the editing, which is attached to the end of the nicked DNA. (c) Multiple and extensive interactions between Cas9, sgRNA and these with both DNA strands at the genomic editing locus confer a high level of specificity, precision and editing accuracy. (d) The main cause of the high specificity of DNA editing provided by the CRISPR system is the multiple interactions between nucleic acids (sgRNA and DNA), governed by complementarity and homology, which are not observed in MNs, ZFNs, and TALENs. Created with BioRender.com.
Over the past few years, the CRISPR/Cas9 system has proven to be the most powerful technology for editing the plant genome. CRISPR/Cas9 editing technology is derived from the prokaryotic adaptive immune system type II that protects bacteria against phage attacks. The system was discovered in bacteria adapted and included in the toolbox of engineered nucleases. The mechanism has three stages of operation: spacer acquisition (adaptation), biogenesis (expression), and target degradation (interference)[3,12].
In 1987, the CRISPR/Cas system was identified in the E. coli genome, with many Cas genes organized in a Cas operon (coding the Cas enzyme and ribonucleases), and a CRISPR array of homologous palindromic repeated units are interspaced by repeated spacer[3]. The successive array of repeats, the CRISPR sequences, are regularly spaced repeats with an average length of 32 bp with variations from 21 to 47 bp in different organisms[3,11]. Every CRISPR short regular repeated sequence has a unique set of nucleotides that are extremely conserved in specific species and can be transcribed into short RNAs, called 'repeats'. Between two repeats, a short mobile sequence called 'spacers' occur, which are inserted in these repetitive sites when the cell is invaded by viruses[33].
The spacers function as a bacterial defense activation and alarm system that triggers CRISPR/Cas to neutralize sequences from the invading organism. During the adaptation phase, a new spacer is acquired by the CRISPR-array in the bacterial genome, derived from the invader's genetic material. It is followed by the expression stage, in which the activation of CRISPR genes leads to the biosynthesis of the precursor CRISPR-RNA (pre-crRNA) and the expression of the Cas genes that lead to the production of Cas proteins[34]. In the second stage of CRISPR immunity, mature crRNAs are produced after processing the precursor CRISPR-RNA by a distinct subunit of a multiprotein Cas complex; by a single, multidomain Cas protein, or by non-Cas RNase III. At this stage, the protospacer adjacent motif (PAM), a sequence of conserved dinucleotides of the virus, and absent in the bacterium's genome. PAM can be targeted by a Cas/crRNA complex, initiating the recognition of the protospacer, followed by sequential DNA unwinding from PAM-proximal region to PAM-distal region, leading to the formation of a crRNA-DNA heteroduplex and activation of Cas nuclease activity[3,11,34−36].
In the CRISPR/Cas9 system, maturation of the crRNA requires an extra class of RNA, called trans-activating crRNA (tracrRNA), in addition to RNAse. The major difference from the type II CRISPR/Cas system to types I and III is the presence of a crRNA/tracrRNA duplex formed by the hybridization of crRNA and tracrRNA and a single large Cas9 endonuclease protein. The hybrid structure formed by the pre-crRNA and tracrRNA allows the recognition of the complex by RNAse III and the maturation of pre-crRNA into crRNA. The crRNA/tracrRNA duplex formed then binds to Cas9 and guides the editing complex to the genomic target site, signaled by spacers derived from crRNA, leading to the formation of DSB by Cas9[3,11,34].
The comparison between different plant genome editing tools, their properties, and applications is shown in Table 1.
Table 1. Technical, biotechnological aspects, and possibilities of using ZFNs, TALENs, and CRISPR/Cas9 system for the genomic manipulation of plants.
Parameter ZFNs TALENS CRISPR/Cas9 system Design complexity ++ ++ + Versatility in editing + ++ +++ Simultaneous editing in multiple sites + + +++ Large-scale library delivery + + +++ Specificity + ++ ++++ Efficiency ++ ++ +++ Cost + +++ + Homologous recombination frequency + + + Mutation frequency after non-homologous end joining (NHEJ) + + ++ Use as epigenetic modifiers ++ +++ ++++ Use as gene-knockout in model organisms − − +++ − absent, + low, ++ moderate, +++ high, ++++ very high. Modified from Khan[11]. The most common way of using the CRISPR/Cas system for editing the plant genome is based on the dual promoter method. The strategy consists of using two promoters to individually control the expression of the most important elements of the CRISPR/Cas system: the sgRNA and the enzyme Cas[37]. The Cas gene is normally cloned under the control of an RNA polymerase II (RNA Pol II) promoter and the gene encoding the sgRNA is controlled by an RNA polymerase III (RNA Pol III) promoter[38]. Eventually, both genes may be under the control of two strong RNA Pol II promoters, able to regulate temporal and spatial expression more accurately[39]. A simplification of this strategy also works, with the simultaneous control of the sgRNA gene and the Cas gene under the same Pol II promoter[38,39].
CRISPR/Cas system technology has already been used to generate new crops with different characteristics, such as soybean seeds with a high oleic acid content[40], potatoes with reduced browning after cutting[41], rice plants with high-tillering and a dwarf habit[42], tomato plants resistant to bacterial diseases[43], and corn plants that are more tolerant to drought[44], among other examples. Considering the short time since its discovery, the evolution of its application is astonishing, changing the scale on which genetic engineering applied to crop improvement is capable of evolving. In short, several economically important characteristics have been incorporated into various cultivated plants[1]. These characteristics include better yield, greater nutritional value, greater stress tolerance, and greater resistance to pests and herbicides. Plant species such as rice, wheat, corn, tomato, apple, and the legume model Lotus, among others, were all successfully edited[45,46].
-
Cas proteins are classified according to their phylogeny, structure, and function and are used as classification parameters for CRISPR/Cas systems. The classes of CRISPR/Cas systems are defined based on the enzyme(s) required for the pre-crRNA processing and interference stages. If a single Cas is used for these purposes, then the CRISPR/Cas system belongs to class 2. If several enzymes are required, then it belongs to class 1[47]. Within each class, there may be subdivisions into types of systems, each defined by its type of signature protein (Table 2).
Table 2. Some examples of classes/types and signature/standard proteins of CRISPR/cas systems for editing the plant genome.
Standard Cas nucleases Class Type Signature
Cas proteinMain features Ref. 2 II Cas9 Most popular Cas for editing the
plant genome recognizes PAM NGG
Requires a crRNA-tracrRNA duplex
or a single-guide RNA for target recognition[48,49] V Cas12a More specific than wild-type SpCas9 Requires a crRNA for target recognition, but the crRNA is
short (~42 nt) Targets T-rich regions generates a DSB distal from the PAM site, with staggered ends[50−57] Cas12j
(CasΦ)Recognizes PAM at the 3' end of target sequences and initiate staggered cleravages on both target and non-target DNA strands Is smaller than other Cas proteins, with 700 to 800 residues [58,59] Based on Zhang et al.[3] Streptococcus pyogenes Cas9 (SpCas9) is the most used CRISPR/Cas. Cas9 is considered the standard enzyme of the CRISPR type II system since it recognizes a simple PAM (NGG) and was adapted for genome editing from a wide range of plants[60].
The discovery of new signature Cas and Cas9 variants/orthologs constitutes some of the greatest technical advances in improving the CRISPR/Cas9 system for editing the plant genome, since the different Cas proteins recognize different PAMs, which increases the versatility of choosing modules for editing within the CRISPR/Cas toolbox[4].
In addition to a new Cas signature, modifications to Cas9's PAM-interacting (PI) domain are also an excellent alternative for expanding editing possibilities[3,4]. Variants of Cas9 with the mutated PI can recognize other PAMs and even more than one PAM (Table 3).
Table 3. Some examples of Cas9 engineered/orthologs and other Cas mutated for new possibilities for editing plant genomes.
Type Name Modification PAM Ref. Engineered Cas9 variants Cas9 nickase (nCas9) Point mutations D10A in the RuvC domain or H840A in the HNH domain
only cleaves the targeting or non-targeting strand, respectivelyNGG [61] Dead Cas9 (dCas9) Simultaneously promotes point mutations D10A in the RuvC domain or
H840A in the HNH domain nuclease activity is abolishedNGG, NGN, NNG, and NNN [62] SpCas9 VQR Point mutations 1135V/R1335Q/T1337R NGA [63] SpCas9 EQR Point mutations D1135E, R1335Q, and T1337R NGAG [63] SpCas9 VRER Point mutations D1135V, G1218R, R1335E, and T1337R NGCG [63] xCas9 Point mutations A262T/R324L/S409I/E480K/E543D/M694I/E1219V NG, GAA and GAT [64] SpCas9-NG Point mutations R1335V/L1111R/D1135V/G1218R/E1219F/A1322R/T1337R NG, GAA and GAT [65] SpRY Point mutations A61R/L1111R/N1317R/A1322R/R1333P NGN [66] iSpyMacCas9 Point mutations R221K and N394K NAAR [67] Cas9 orthologs Staphylococcus aureus
Cas9 (SaCas9 KKH)Point mutations E782K/N968K/R1015H NNGRRT [68] Streptococcus thermophilus Cas9 (St1Cas9) − NNAGAA [69] Brevibacillus laterosporus Cas9 (BlatCas9) − NNNNCND [70] Streptococcus macacae Cas9 (SmacCas9) − NAA [71] Lactobacillus rhamnosus Cas9 (LrCas9) − NGAAA [72] Faecalibaculum rodentium Cas9 (FrCas9) Point mutations E796A, H1010A, and D1013A NNTA [73] Cas12 orthologs AsCas12a − TTTV [74] AsCas12a RR Point mutations S542R/K607R TYCV and CCCC [75] AsCas12a RVR Point mutations S542R/K548V/N552R TATV [75] enAsCas12a Point mutations E174R/S542R/K548R VTTV, TTTT, TTCN, and TATV [76] LbCas12a − TTTV [74] LbCas12a RR Point mutations G532R/K595R TYCV and CCCC LbCas12a RVR Point mutations G532R/K538V/Y542R TATV [75] FnCas12a − TTV, TTTV, and KYTV [77] FnCas12a RR Point mutations N607R/K671R TYCV and TCTV [77] FnCas12a RVR Point mutations N607R/K613V/N617R TWTV [77] N can be any purine or pyrimidine base (A, G, C, or T). Based on Zhang et al.[3] -
Much progress has taken place since the first report on plant genome editing in 2013. The expansion of the CRISPR/Cas toolbox, with the discovery/engineering of new Cas enzymes, the redesign of sgRNAs, and the development of editing platforms have led to accurate editing of the plant genome, generating targeted mutagenesis, base editing, transcriptional regulation, and activation of DNA repair pathways[3,11] (Fig. 5).
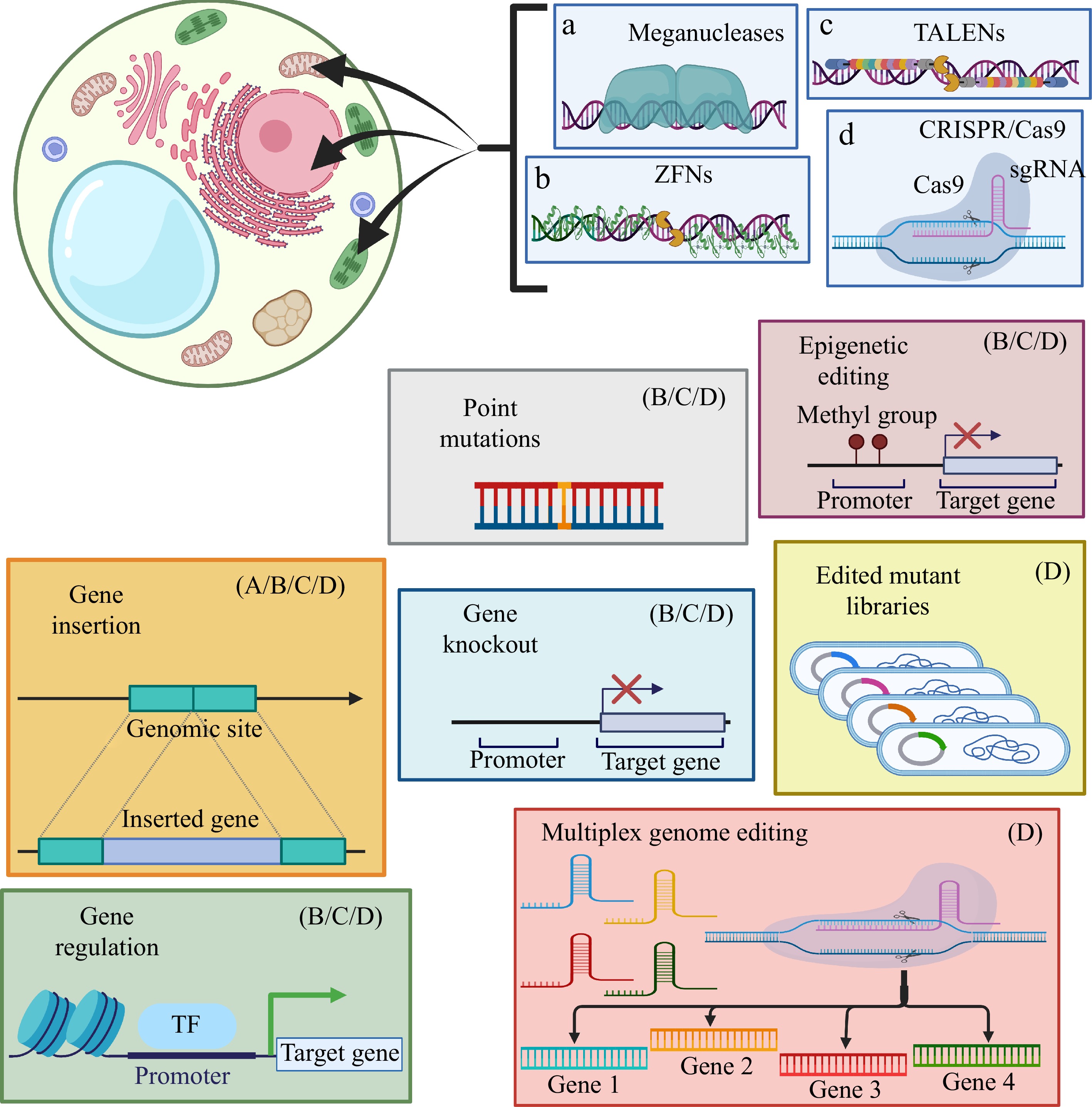
Figure 5.
Some recurring biotechnological approaches to genome editing in plants. Diagram shows the different methodological applications of the main plant genome editing tools, as well as their limitations of use. Created with BioRender.com.
After the production of a DSB by the Cas enzyme, the insertion of a DNA fragment (coding or not), as well as the replacement of a genetic element, can occur through two repair pathways: typical/canonical NHEJ or homology-directed repair (HDR). Typically, NHEJ and alternative microhomology-mediated end joining (MMEJ) lead to gene knockout and HDR is used for gene replacement[78].
NHEJ is the main pathway for DNA repair after the occurrence of a DSB. The mechanism leads to the production of insertions/deletions (indels) by the loss of bases at the ends of the strips that flank the cut, until obtaining homologous internal sequences, which are exposed for recombination after the removal of bases at the superfluous ends[79]. This explains the fact that this repair method is error-prone, leading to loss of gene function. NHEJ pathways can be inhibited genetically or enzymatically[80]. Frequently, inhibition of NHEJ pathways can lead to an increase in HDR pathways in plants[3].
The editing of the plastid genome, cannot occur by NHEJ. In this organelle, the enzyme repair machinery by NHEJ is not present. This is in contrast to the HDR pathway, which can occur with or without the use of SSNs, which frequently act as editing enhancers[81].
When using NHEJ, great care is required in the design of sgRNA to better achieve the experimental objectives, since the NHEJ-based editing is strongly influenced by the target sequence features and the broken ends generated by SSNs. If the sgRNA is precisely designed to recognize the target site, it is possible to predict the expected indels[3,11].
HDR is a secondary DNA repair pathway, but it is a major approach to editing plant genomes. Although HDR is not the most active repair pathway, it helps achieve precise genomic editing in plants[3,11]. HDR allows editing by gene targeting (GT), the most efficient means for the introduction of exogenous genes and for modifying the content of native plant genes. Accurate editing of the plant genome by HDR has been widely reported using Cas9 and Cas12a. More and more technical efforts are being made to improve genomic editing by replacement and targeted gene insertions via HDR[82].
In the early years after the discovery of the CRISPR/Cas9 system, GT in higher plants was technically limited, until the advent of SSNs revolutionized the method, making it more accessible and easier to handle. After the production of a DSB by SSNs, the exogenous DNA inserts can be linked in a directed way in the donor template to repair the DSB, provided that the flanking sequences of the fragment of interest are homologous to the edges of the DSB flanks[83].
The efficiency of editing plant genomes by the HDR pathway can be increased by the adoption of simple molecular strategies. For instance, the use of longer homology arms in the exogenous fragment and in the flanks of the DSB increases the probability of gene replacement by HDR. Other strategies such as increasing donor availability by enriching the mass of exogenous DNA delivered to the cell, or by adding the target sequences themselves on the flanks of the donor fragment, can also increase HDR efficiency, especially if the delivery is made by Agrobacterium. Genetic transformation by biolistics can also be used, provided that free dsDNA or ssDNA donors are delivered alongside the CRISPR components[78]. A geminivirus-based replicon system can be used to previously replicate the donor fragment[18,84]. Furthermore, the inclusion of a nuclear localization signal (NLS) peptide to the 5′-end of the donor fragment facilitates the transport of the donor sequence into the nucleus, thus increasing efficiency[85]. In other cases, HDR efficiency can be increased by recruiting biotinylated donor DNA interacting with streptavidin – biotin and aptamer -linked-SpCas9 or avidin-linked SpCas9[86,60]; by fusing HUH endonucleases and Cas9[87]; by the use of nCas9, since nicks tend not to lead to NHEJ activation; and, finally, using RNA donors[88].
Another interesting approach to editing is base editing. This consists of the permanent conversion of one nucleotide target to another, without the formation of a DSB or the presence of a fragment of donor DNA[89]. Base editing uses two types of editors: cytosine base editors (CBEs) and adenine base editors (ABEs). The first CBEs were composed of the rat cytidine deaminase apolipoprotein B mRNA editing enzyme (rAPOBEC1) fused to dCas9[90,91]. Deamination of cytosine (C) leads to the C-to-T transition[90,53].
The second generation of CBEs was produced to improve editing efficiency. The new CBEs consist of first-generation CBEs fused to the uracil DNA glycosylase inhibitor (UGI), capable of inhibiting the uracil DNA glycosylase (UDG) enzyme, which is a protein that hinders first-generation CBEs in the conversion of C-to-U[89]. The third and fourth generations of CBEs use a mutated Cas9 (D10A) to induce a mismatch repair (MMR) on the editing site[89]. The indel formation during the base editing method can also be minimized by the fusion of CBEs with the bacteriophage Mu protein Gam, capable of protecting DSBs against degradation by precise binding to dsDNA extremities[92].
The NHEJ, HDR, and base editing approaches allow for numerous applications in editing plant genomes, which can vary in terms of the number and extent of changes in target DNA in addition to the type of delivery[3,11]. Table 4 shows the main types of manipulation at the DNA level by CRISPR/Cas and other technologies, as well as the tools used for editing.
Table 4. Main approaches available for editing the plant genome and the types of alterations brought about by them.
Modification Approach Main features knock-in HDR after SSN
actionGene activation enhanced by
long ssDNA donorsknock-out NHEJ or HDR after
the action of SSNsLower in polyploid than diploids
plant speciesPoint mutations Base editing Depends on CBEs and ABEs Epigenome editing CRISPR-directed methyltransferases
and demethylasesEditing epigenetic marks for
transcription regulationGene replacement NHEJ or HDR
(preferred) after
the action of SSNsHomology-dependency among
DSB flanks and donor template
flanksMultiplexed
genome
editingCRISPR array Long transcripts with multiplexed
sgRNAs for multiple
simultaneous editings
Expression or delivery of sgRNAs
through ribonucleoproteins
(RNPs) or particle bombardmentGene
transcriptional
regulationCRISPR array Cas9 fusion with various
transcription effectors can be
used to repress or activate
transcription.Chromosomal
rearrangementCRISPR array Target highly endogenous
retrotransposons and LINEs -
Although the CRISPR/Cas9-based system of producing mutations has revolutionized plant breeding, indels are generated randomly, with no control or specificity. This is due to the double-strand break (DSB) activity of Cas9 and the DNA repair mechanism of non-homologous end joining (NHEJ) this triggers[89,90]. Homologous recombination repair can occur after DSB formation, however, it is not frequent and for genome editing, requires a donor DNA. Therefore, a new genome editing system with base editing precision and no need for donor DNA was developed to address these issues. Base-editing utilizes nucleotide deaminases fused to an engineered Cas9 with no double-strand break activity (dCas9) or with only single-stranded nick activity (nCas9)[89,90]. These versions of Cas9 still require an RNA guide sequence (sgRNA) making it possible to target specific regions of the genome. The R-loop structure created by sgRNA/Cas9 exposes nucleotides to deaminase activity[93,94]. The alternative nCas9 nicks single-stranded DNA on the non-edited strand and triggers the mismatch repair system which uses the edited strand for repair[95]. The edited base is further maintained by DNA replication. Initially, this system was developed with cytosine deaminase from rats, which converts cytosine to uracil by removing its amine group[89]. Although this system showed base editing capability, it was not efficient. The low frequency is explained by the cellular response to G-U mismatches, which triggers a base excision repair mechanism that through the activity of a uracil UDP glycosylase, removes U from these mismatches, thus working against this conversion. To overcome this, recent versions of CBE incorporated uracil glycosylase inhibitors (UGI) into the system, which greatly improved editing efficiency[89,90]. This system is known as BE3 and has been the basis for base editing in crop plants[96,97]. Other cytosine deaminases from different sources, such as the fish species Petromyzon marinus (PmCDA1) and different versions of the human cytosine deaminase (APOBEC3) have also been tested showing different efficiency rates depending on plant species[97,98].
BE3 has some technical limitations due to both the activity of cytodine deaminases and the Cas9 used. Although the R-loop structure exposes cytosines to the activity of deaminases, not all cytosines contained within the target sequence are equally exposed and this has implications that can limit or expand its use[97]. Typically, the cytosines positioned from nucleotide 4 to 13 (counting from 5' to the 3' PAM sequence of the protospacer) are more likely to be edited, depending on the Cas enzyme used. This is known as the activity window and can be narrowed or widened depending on the application[95,97]. For instance, for a single nucleotide conversion aimed at altering a protein coding sequence, having several cytosines edited in the activity window is an undesired effect, whereas if the goal is large-scale mutations for variability or to modify cis-regulatory elements, a large activity window can be desirable[97]. Protein engineering and naturally occurring orthologs from different Streptococcus species have enabled scientists to create different versions of Cas nucleases, expanding the scope of target sequences available for editing and thus providing better-suited editing platforms depending on the desired application. This includes PAM sequence requirements. Cas proteins with alternative PAM requirements and even Cas proteins that do not require PAM sequences have been tested[99−103]. However, a balance needs to be considered when choosing Cas PAM sequences, as more relaxed PAM sequence requirements increase off-target effects[97].
CBEs limit editing to changing a C-G pair to a T-A pair, the opposite modification is theoretically possible by using an adenine deaminase. Interestingly, the naturally occurring adenine deaminases do not function on DNA. Through protein engineering of a tRNA adenosine deaminase, it was possible to obtain an enzyme that works on single-stranded DNA[104]. This gave rise to the ABE platforms and further expanded the capabilities of base editing. ABE platforms have an advantage over CBE platforms, as cellular inosine base excision repair activity is very low compared to uracil base excision repair[97,104]. This limits off-target or unintended base editing effects. However, as with CBE platforms, the by-stander effect of modifying unintended A that is within the activity window is still present in ABE platforms[105]. Similar to CBE platforms, protein engineering has enabled the increase of the efficiency of ABE platforms by combining Cas variants and adenosine deaminase variants[106].
Agronomic traits in crop plants are commonly controlled by single nucleotide variations or single nucleotide polymorphisms (SNPs), that generate allelic diversity. SNPs can alter a protein sequence, altering an amino acid critical for protein function, creating stop codons within a protein sequence, or altering cis-regulatory elements. In this regard, BE technology is a useful tool to induce point mutations to explore functional differences in alleles. This is particularly important in polyploid genomes, where alleles are highly homologous with variation occurring in a few nucleotides. Several groups have used BE technology to induce point mutations in polyploid crops such as wheat and cotton[107,108]. In wheat, Zhang et al. induced point mutations in the genes encoding the enzymes acetolactate synthase (ALS) and acetyl-coenzyme A carboxylase, thus creating herbicide-resistant lines[108]. Similarly, CBE technology was used in allotetraploid Brassica napus to induce C to T conversions in the ALS gene, altering a conserved proline at position 197 coded by the triplet CCT to TCT, and TTT, which alters the amino acid to serine and phenylalanine, respectively, leading to herbicide tolerance[109]. There are several other examples of base editing for different traits, such as flowering time, crop yield, and disease resistance[110]. In summary, base editing continues to develop as a powerful tool for plant breeding, as protein engineering has enabled adaptations for high efficiency and specificity[3].
-
Although Base Editing has provided a means to control alterations without generating DSBs, there are some drawbacks with this technology. For instance, there is the possibility of a bystander effect, when nucleotides close to the target are also unintentionally modified. Additionally, there is the constraint of a small activity window within the R-loop, which limits the creation of several modifications. Lastly, BE is limited to six conversion possibilities, out of the 12 combinations of possible conversions. These limitations have prompted the development of another, more robust technology – primer editing (PE)[111].
Similar to BE, PE promotes changes to a DNA sequence without the need of DSBs. The technology uses a nCas9 variant that can nick ssDNA at the opposite DNA strand of the target. The differences lie with the enzyme that is fused with the nCas9. Instead of base modifiers, PE uses a reverse transcriptase (RT) along with a prime editing guide (pegRNA)[112]. pegRNA are designed to contain the spacer sequence for Cas9 function, a sequence complementary to the target DNA, and on its 3' end, a primer binding site (PBS) along with the template containing the desired edit. The sequence of events of this mechanism is as follows: (1) the target is recognized by DNA pairing, nCas9 forms the R-loop structure and nicks the opposite strand; (2) PBS binds to the opposite strand and the template, carrying the desired edit, is used by the RT for DNA synthesis; (3) the result of RT activity is a 3' flap containing the edit and a 5' adjacent flap which is displaced by intracellular exonuclease activity[111]. The result of this sequence of events is a heteroduplex in which one of the DNA strands contains the desired edit. This heteroduplex is resolved by a DNA repair mechanism or DNA replication, which fixes the edit permanently[113]. The template can be designed to contain any kind of base change or small indels. The possibility of incorporating edits on a template is a specific feature of PE technology. Template design makes it possible to incorporate any base modification; it can be designed to have multiple modifications within one template sequence and can be designed to incorporate small indels. Therefore, template design is what makes this system versatile in comparison with BE technology or homologous repair technology, which requires DSBs and a DNA donor[111].
PE was initially described in human cell lines. In this initial report, several modifications were made to increase PE efficiency[114]. Mutations were incorporated in the Moloney murine leukaemia virus (MMLV) RT to increase stability, processivity, and binding to the primer-template sequence. This alteration increased efficiency when compared to wild-type MMLV RT and was termed PE2. One of the constraints of PE1 and PE2 is relying on cellular DNA repair mechanisms to incorporate the edit on the complementary strand. To address this, an additional sgRNA was added, which directs a nick to the non-edited strand. This modification further increased editing efficiency and was named PE3[114]. The PE3 system was further improved by directing the sgRNA to the edited strand after the edited DNA was incorporated and inducing a nick on the non-edited strand, which avoids simultaneous nicking of both strands that can lead to the generation of the DSB[114]. This alteration was denominated PE3b. Zhang et al. designed a homologous 3' extension mediated prime editor (HOPE) by using paired pegRNAs, in which the desired edit is included in both strands, which further improved PE efficiency in human cell lines[102].
In plants, the PE system was tested initially in rice and wheat[115]. Modifications of the system were made such as optimized codon usage for plants. Other RT were tested, the Cauliflower Mosaic virus (CaMV) RT and a retrotransposon RT. No significant increase in PE activity was observed, although CaMV RT showed similar efficiency to MMLV RT[115]. Several conclusions were drawn from extensive testing of PE efficiency in plants. PE3 and PE3b were not more efficient than PE2 in plants, contrary to mammalian cell lines. All 12 base conversions and small deletions and insertions were possible with the optimized plant PE system (PPE). PPE was less efficient than BE and increasing insertion or deletion length proportionately decreases PPE efficiency. There was also variability of efficiency depending on the target gene and increased temperature also favored PPE efficiency. Together, these results show that PPE efficiency can vary and optimization is required on a case-by-case basis[115].
Further optimization in PPE efficiency was described by Zong et al.[111], by modifying MMLV RT to remove its RNase H domain and also including a viral RNA chaperone in the system. RNase H activity degrades DNA-RNA duplex, which is undesirable for editing. RNA chaperones decrease secondary and tertiary RNA structures that can inhibit RT activity[111]. With these modifications, base changes were made to produce a herbicide-resistant rice plant[111]. Multiplex modifications in several gene targets were obtained using a surrogate PE system in rice[116]. This system is based on the recovery of a non-functional hygromicin resistant gene or by modifying endogenous ALS gene for herbicide resistance or both. Modifications were introduced in up to eight gene targets with increased efficiency compared to the normal PE3 system[116]. The double-surrogate system, with hptII and modified ALS, was more efficient than individual systems, suggesting a synergistic effect[116]. Yet another optimized system was developed that enabled multiplexing in hexaploid wheat[117]. Optimization of protein architecture and nCas9 activity increased efficiency, allowing for multiplexing of up to ten genes[117]. In summary, although modifications and improvements have substantially increased PPE efficiency, there is still an open and active field of optimization of PPE systems in the near future. Presently, there is already a suite of PPE systems available for editing several crop plants[118]. This system will greatly improve the precision of editing and provide a robust tool for breeding programs to design more efficient and productive crops.
-
Extensive editing of the plant genome simultaneously at multiple sites can be achieved through multiplexed genome editing. Multiplexing is the expression and delivery of multiple sgRNAs by the CRISPR/Cas system, capable of offering multiple guides for Cas to access different editing sites with a high degree of flexibility[74]. The delivery of multiple sgRNAs can be accomplished basically by two methods: via particle bombardment or through ribonucleoproteins (RNPs)[53].
In this context, the CRISPR/Cas RNPs system is extremely interesting, since it can be introduced free of plasmids in protoplasts of different plants, such as Arabidopsis, wild tobacco (Nicotiana attenuata), lettuce (Lactuca sativa), rice[53,119], apple (Malus pumila), grape (Vitis vinifera)[49], Petunia hybrida[120], and soybean[53]. RNP introduction into the plant genome is achieved through polyethylene glycol (PEG)-mediated transformation into the plant callus by particle bombardment and into plant zygotes produced by in vitro fertilization[53]. The use of RNPs for gene delivery at the expense of exogenous DNA vectors presents advantages in the context of multiplexing, since it minimizes off-targets due to the shorter exposure of the editing sites in the genomic DNA to the components of the CRISPR/Cas system[121].
Despite recent technical advances in sgRNA delivery methods, there is still an efficiency gap for plant transformation aiming at multiplexing, since the traditional method of transforming most crops is by transferring the transfer DNA (T-DNA) via Agrobacterium[122].
The most conventional way of performing multiplexing is through costly and laborious achievement of large constructs with multiple coding sgRNA repetitive elements, under the control of a single promoter and a terminator, which leads to the stacking of multiple sgRNA expression units. In addition to being complex, this type of construction can also decrease the transformation efficiency, given the large extension of the expression cassette[123].
Four alternatives have already been described in the literature to increase the viability and ease of engineering constructs for multiplexing[14,57]. The first approach is to use the CRISPR/Cas12a system carrying a crRNA under the control of a strong promoter and the tracrRNA under the control of another independent promoter, or tandem sgRNAs under the control of the same promoter[57]. The second, third, and fourth subsequent approaches use the CRISPR/9 system for multiplexing. The second strategy is to flank the sgRNAs with hammerhead (HH) ribozymes and hepatitis delta virus (HDV) ribozyme sequences, producing tandem repetitions of the ribozyme/sgRNA ribozyme (RGR) system[39]. The cluster with several RGRs allows the expression of a long transcript carried from the sgRNA units, which can be processed and separated from each other. This strategy has already been successfully used in rice[39].
The third approach involves using a polycistronic tRNA/sgRNA system, in which each sgRNA is fused with an individual tRNA. After the production of a single transcript containing all the elements simultaneously, the formation of mature sgRNA is obtained after processing mediated by endogenous RNaseZ and RNaseP, as observed in cereals (rice[124], wheat[125], and maize[126]).
The last strategy is to flank sgRNAs or crRNAs by Csy4 excision sites, which are processed by the Csy4 endonuclease. All of these multiplexing strategies allow for different types of genomic editing, such as gene replacement, gene insertion, gene knockout, and gene deletion[127].
-
The diversification of the CRISPR/Cas toolbox and the editing alternatives by other technologies have increased the possibilities for large-scale manipulation of plant genes[128]. Reports of applications of multiple editing approaches for the production of phenotypic changes in plants, based on new genetic circuits and their metabolic developments in host organisms, have pointed to a range of new applications for genome editing[3,129].
The applications are staggering. CRISPR/Cas9 has made it possible to alter the expression of genes by generating precise point mutations in the genome, thus allowing greater speed in studies of gene function. It can even be used in multiplex systems, or individually in multicopy genes. This precision is thanks to a meticulously designed sgRNA sequence that can direct the Cas enzyme to generate a cut in a very specific and previously defined homologous region, determined by computer programs, avoiding off-target action as much as possible[130].
Despite technical advances, genome editing tools in plants still present challenges and bottlenecks. Table 5 summarizes several interesting applications of the editing of plant genomes in the context of synthetic biology, pointing out the main limitations and challenges of achieving each one.
Table 5. Recent applications of CRISPR/Cas for genomic manipulation in plants and their main challenges.
Application Main features Purposes Limitations Edited plants Ref. Thermal sensitivity of CRISPR/Cas9 and Cas12a Optimal editing efficiency under higher temperatures Increased efficiency of CRISPR/
Cas9 and Cas12a by thermal
sensitivityFew plants tested Arabidopsis (29 °C) and maize (28 °C) [131] Generation of transgene-free edited plants Elimination of CRISPR transgenes
by segregation associated with screening with selection marker
genes/reporter genes
Editing of plant genomes without
the integration of exogenous DNA,
by the transient expression of the CRISPR/Cas machineryMinimize regulatory issues,
enhance public acceptance
and improve biosafetyNot feasible for vegetatively propagated plants, trees, polyploids and self-incompatible plants Arabidopsis, wild tobacco lettuce. rice, apple, grape, Petunia, hybrid, and soybean [120] Editing of polyploid
plantsMultiallelic genome editing in polyploid plants Introduction of valuable
traits in polyploid plantsLow efficiency, labor
and time-consumingAutopolyploid lines of Arabidopsis, Tragopogon (Asteraceae), potato (Solanum tuberosum) and Rape (Brassica napus) [132] Generation of germline-edited plants Genome editing using CRISPR delivered by Agrobacterium before embryogenic cell division To improve downstream genetic
and trait analysisLow editing efficiency
in germlinesArabidopsis [133] Off-target minimization Use wild-type Cas9 and Cas12a,
nCas9s and high-fidelity versions of SpCas9 design of low-mismatch sgRNAs to limit the exposure of the genome to CRISPR componentsTechnical improvement of
editing systems
Minimize the major concern of
CRISPR technologyMany high-fidelity SpCas9s show intrinsically lower nuclease activities
in plantsrice [134,135] Improve plant breeding To express plant growth-stimulating genes use of haploid induction-edit and haploid-inducer mediated genome editing (IMGE) To enhance genome editing by
CRISPR in recalcitrant plant
species and varieties
To increase stable inheritable of desirable traits through many generationsInevitable segregation
in seed productionSorghum (Sorghum bicolor), sugarcane, and indica rice [136,137] -
The plant breeding landscape has changed significantly over the past four decades. It now includes genome-wide association mapping, transgenic crops, crops with stacked traits, and, more recently, genome editing techniques. Transgenics initially provided a quick way to introduce new traits, even cross-species traits, to plants. However, the technology proved to be labor-intensive, with the transgene copy number, and genome location of the transgene presenting major drawbacks. This eventually led to only a few major biotech companies with the capability to commercially release cultivars with traits aimed mainly at insect and herbicide tolerance. The discovery and use of site-specific nucleases made it possible to alter specific regions of the genome.
With the recent advent of genome editing techniques, promising possibilities for plant improvement have emerged, allowing the generation of varieties with desired agronomic characteristics. This evolution meets the growing global demand for food that is healthier and fits into a more efficient and sustainable production system, which has been achieved in recent years.
Among the various methods for gene editing known today as MNs, ZFNs, and TALENs, the CRISPR system stands out for its greater efficiency and simplicity of execution. Although ZFNs and TALENs were first discovered for gene editing, their complexities and limitations have made them less practical for widespread use in plants. In contrast, the CRISPR/Cas system has emerged as a more accessible and versatile tool, driving significant advances in crop improvement[24]. This system has been used extensively in animal products, products for human health, and in agriculture. The feasibility, reduced cost, and relative easiness of the system provides for a promising future for agricultural products. Modifications in delivery systems, vector design, and Cas engineering have enhanced the application, scope, and efficiency of the system.
Furthermore, this technology allows products to be obtained free of foreign DNA since it is possible in species propagated by seeds, especially, to get rid of the gene complex from the editing system and separate only the events containing the induced mutation without insertions or unwanted and off-target deletions. However, the potential application of this precision gene editing technology to obtain commercial products depends on the establishment of a regulatory system that is clear and internationally recognized for crops edited by CRISPR/Cas[24], mainly when it comes to commodities.
The system is quickly becoming an essential tool for metabolic engineering and synthetic biology. As the possibility of altering several genes at once is further improved, entire metabolic pathways can be modified for a desired outcome. Similarly, gene replacement and gene introduction using this system can open new frontiers to explore novel, synthetic pathways. As our understanding of system biology continues to advance, gene editing methods will need to be modified and/or adjusted. New tools will be developed and novel, tailored solutions will become available. They will play a major role in the future of food production, sustainability, and food security.
-
The authors confirm their contribution to the paper as follows: literature collection: da Cunha NB, Leite ML; tables and figures design: da Cunha NB, Costa FF; manuscript layout and format: da Cunha NB, Vianna GR, Rodrigues JCM; draft manuscript preparation: da Cunha NB, Vianna GR, Rodrigues JCM; manuscript modification and review: Leite ML, Carrijo J, Dias SC, Rech EL. All authors reviewed the results and approved the final version of the manuscript.
-
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
-
This work was supported by fellowships from Universidade de Brasília (UnB), Universidade Católica de Brasília (UCB), Empresa Brasileira de Pesquisa Agropecuária (EMBRAPA), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Fundação Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) and Fundação de Apoio à Pesquisa do Distrito Federal (FAPDF). Elíbio Leopoldo Rech is supported by Embrapa Genetic Resources and Biotechnology/National Institute of Science and Technology in Synthetic Biology, National Council for Scientific and Technological Development (465603/2014-9), Research Support Foundation of the Federal District (0193.001.262/2017), and Coordination for the Improvement of Higher Education Personnel.
-
The authors declare that they have no conflict of interest.
- Copyright: © 2025 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
da Cunha NB, Leite ML, Rodrigues JCM, Carrijo J, Costa FF, et al. 2025. Molecular tools for genome editing in plants: a synthetic overview. Vegetable Research 5: e010 doi: 10.48130/vegres-0025-0005 

Molecular tools for genome editing in plants: a synthetic overview
- Received: 24 August 2024
- Revised: 21 January 2025
- Accepted: 23 January 2025
- Published online: 02 April 2025
Abstract: The modification of plant genomes is an essential step towards the development of new crops to increase food production. Initially, genome modification relied on conventional plant breeding and the introduction of genetic traits using crossing techniques to generate new varieties. In the 1980s, trans and cis modification allowed the incorporation of specific traits into plant genomes, largely due to the development of DNA delivery systems such as Agrobacterium and biolistics. This technological breakthrough has boosted a second generation of genetically modified crops that have had a major impact on agriculture. However, the stochastic nature of the DNA delivery systems, with little control over where the genome modification occurred, required regulatory measures to ensure environmental, human, and animal safety, which has hindered the ability and speed to generate new and more adapted varieties. New technological advances have made it possible to increase the precision of genome modifications leading to a third generation of agricultural products. These technological advances rely on enzymes targeting specific genomic regions, making it possible to introduce mutations and new traits into the plant genome. Meganucleases, zinc-finger nucleases, transcription activator-like effector nucleases, and CRISPR/Cas9 are valuable tools that allow for specific genomic modifications. These tools have made it possible to develop new, safer varieties in a shorter timeframe. In this review, we explore the functional mechanisms of these new breeding tools, their advantages and drawbacks, and their potential to explore metabolic engineering and synthetic biology in plants.
-
Key words:
- Crop improvement /
- Genome editing /
- Plant genetic engineering /
- New breeding tools