








HTML
-
The diversity for flora of seed plants in Yunnan Province, P.R. China is observably high, and the endemic species of woody plants are rich, in which both supply good substrates for wood-decaying fungi. Wood-decaying fungi are kind of large basidiomycetes that grow on various kinds of wood, such as the living trees, dead standing trees, fallen trunk, fallen branch and stump (Dai 2012a), which can be used for industrial value, medicinal value, edible value and economic value (Russell & Paterson 2006, Dai et al. 2015, Vinay et al. 2015, M'Barek et al. 2020, Wu et al. 2020, Runnel et al. 2021).
Haikou Forestry Farm is located in Haikou Town, Kunming, belonging to the Jinsha River system and the geographical location is between 102°28'-102°38' E and 24°4'-24°56' N, with the altitude of 1800-2400 m (Xu et al. 2015, Zhang et al. 2017). It has a subtropical monsoon climate and the native tree species with mostly subtropical evergreen broadleaf trees (Xiong & Zhou 2019). The main vegetation includes warm coniferous forest (Pinus yunnanensis Franch., P. armandii Franch. and Keteleeria evelyniana Mast.), deciduous broad-leaved forest (Alnus nepalensis D. Don), and semi-humid evergreen broad-leaved forest (Lithocarpus dealbatus (J. D. Hooker et Thomson ex Miquel) Rehder and Castanopsis delavayi Franch.).
However, the previously documented wood-decaying fungi are mostly in northwest Yunnan Province, China, and few polypore and corticioid fungi have been reported in here so far. According to the modern taxonomy (Dai 2012a), wood-decaying fungi mainly belong to ten orders of Agaricomycetes, viz., Agaricales, Auriculariales, Cantharellales, Corticiales, Gloeophyllales, Hymenochaetales, Polyporales, Russulales, Thelephorales and Trechisporales. Therefore, the current wood-decaying fungi catalogues include poroid and corticioid hymenophore. In the present study, nine field trips were carried out in different areas of Haikou Forestry Farm, and about 681 specimens were collected, in which 52 species belonging to 37 genera, 16 families, 6 orders, which were identified from these materials. This paper is going to summarize the distribution of wood-decaying fungi and enrich the fungal diversity in this area.
-
The studied specimens are deposited at the herbarium of Southwest Forestry University (SWFC), Kunming, Yunnan Province, P.R. China. Macromorphological descriptions were based on field notes. Colour terms are from Petersen (1996). Micromorphological data were obtained from the dried specimens, and observed under a light microscope following Dai (2012a). The following abbreviations were used for the micro characteristics description: KOH = 5% potassium hydroxide, CB = Cotton Blue, CB– = acyanophilous, CB+ = cyanophilous, IKI = Melzer's reagent, IKI– = both inamyloid and indextrinoid, L = mean spore length (arithmetic average of all spores), W = mean spore width (arithmetic average of all spores), Q = variation in the L/W ratios between the specimens studied, n (a/b) = number of spores (a) measured from given number (b) of specimens.
Molecular procedures and phylogenetic analysis
-
CTAB rapid plant genome extraction kit-DN14 (Aidlab Biotechnologies Co., Ltd, Beijing) was used to obtain genomic DNA from dried specimens, according to the manufacturer's instructions that a small piece of dried fungal specimen (about 30 mg) was ground to powder with liquid nitrogen. The powder was transferred to a 1.5 mL centrifuge tube, suspended in 0.4 mL of lysis buffer, and incubated in a 65℃ water bath for 60 min. After that, 0.4 mL phenol-chloroform (24:1) was added to each tube and the suspension was shaken vigorously. After centrifugation at 13, 000 rpm for 5 min, 0.3 mL supernatant was transferred to a new tube and mixed with 0.45 mL binding buffer. The mixture was then transferred to an adsorbing column (AC) for centrifugation at 13, 000 rpm for 0.5 min. Then, 0.5 mL inhibitor removal fluid was added in AC for a centrifugation at 12, 000 rpm for 0.5 min. After washing twice with 0.5 mL washing buffer, the AC was transferred to a clean centrifuge tube, and 100 mL elution buffer was added to the middle of adsorbed film to elute the genome DNA. ITS region was amplified with primer pair ITS5 and ITS4 (White et al. 1990). The PCR procedure for ITS was as follows: initial denaturation at 95℃ for 3 min, followed by 35 cycles at 94℃ for 40 s, 58℃ for 45 s and 72℃ for 1 min, and a final extension of 72℃ for 10 min. The PCR products were purified and directly sequenced at Kunming Tsingke Biological Technology Limited Company, Kunming Yunnan Province, P.R. China. All newly generated sequences were deposited at GenBank (Table 1).
Table 1. Names, sample numbers and corresponding GenBank accession numbers of ITS sequences used in this study
Species name Sample no. GenBank accession no. References Acanthofungus rimosus Wu 9601-1 MF043521 He et al. (2019) Acanthophysellum cerussatum He 2208 KX306874 He et al. (2019) Aleurobotrys botryosus He 2712 KX306877 He et al. (2019) Antrodia tanakae CLZhao 720 MG231457 This study Armillaria aotearoa NZFS 2425 NR151846 Hood & Ramsfield 2016 Auricularia angiospermarum BJFC 017274 NR151847 Wu et al. 2015 Auricularia villosula CLZhao 1296 MG231464 This study Bankera fuligineoalba REB-285 JN135196 He et al. (2019) Boletinellus merulioides 2630a KM248952 He et al. (2019) Bondarzewia berkeleyi Dai 12759 KJ583202 He et al. (2019) Boreostereum radiatum RLG-9717-Sp HM536085 Garcia-Sandoval et al. 2011 Brunneoporus malicola CLZhao 1530 MG231451 This study Candelabrochaete langloisii FP-110343 KY948793 He et al. (2019) Ceraceomyces serpens HHB-15692-Sp KP135031 He et al. (2019) Cinereomyces lindbladii CBS 290.71 MH860129 Vu et al. 2019 Cinereomyces lindbladii CLZhao 1523 MG231489 This study Coniolepiota spongodes ECV-2010a HM488756 He et al. (2019) Coprinellus curtus SZMC-NL-2339 FM878016 He et al. (2019) Coprinus comatus AFTOL-ID 626 AY854066 He et al. (2019) Coriolopsis polyzona Cui 11040 KR605824 He et al. (2019) Crepatura ellipsospora CLZhao 697 MK343695 Ma & Zhao 2019 Cyclomyces lamellatus Cui 7629 JQ279603 He et al. (2019) Cylindrobasidium laeve CLZhao 767 MG231497 This study Daedalea quercina FFUI-4 MN596945 Direct Submission Daedaleopsis confragosa CLZhao 1481 MG231506 This study Efibula americana FP-102165 KP135016 He et al. (2019) Eichleriella alliciens HHB 7194 KX262120 He et al. (2019) Exidiopsis effusa OM 19136 KX262145 He et al. (2019) Flammulina velutipes AFTOL-ID 558 AY854073 He et al. (2019) Fomitiporia mediterranea AFTOL-ID 688 AY854080 He et al. (2019) Funalia gallica CLZhao 1306 MG231491 This study Funalia trogii CLZhao 1557 MG231874 This study Gloeophyllum sepiarium CLZhao 732 MG231532 This study Gloeophyllum trabeum 1320 HM536094 He et al. (2019) Gloeoporus taxicola CLZhao 1441 MG231549 This study Grifola frondosa AFTOL-ID 701 AY854084 He et al. (2019) Gymnopus confluens ZRL 20151148 LT716054 He et al. (2019) Heliocybe sulcata IBUG 9930 HM536095 He et al. (2019) Heterobasidion annosum 06129/6 KJ583211 He et al. (2019) Heterobasidion insulare CLZhao 2899 MK268944 This study Heterobasidion orientale CLZhao 696 MG231561 This study Hymenochaetopsis yasudae CLZhao 1422 MG231607 This study Hymenopellis radicata AFTOL-ID 561 DQ241780 He et al. (2019) Hyphoderma macaronesicum TFC Mic 15939 NR119817 Schoch et al. 2014 Hyphoderma setigerum CBS 421.72 MH860512 Vu et al. 2019 Hyphoderma transiens CLZhao 1365 MK404378 This study Hyphoderma variolosum CBS 735.91 MH862321 Vu et al. 2019 Hyphodermella rosae FP-150552 KP134978 He et al. (2019) Inocutis dryophilus DLL 2012-001 KU139186 He et al. (2019) Irpex lacteus CLZhao 1258 MG231709 This study Junghuhnia crustacea X 262 JN710553 Miettinen et al. 2012 Lacrymaria lacrymabunda CBS 211.31 MH855192 He et al. (2019) Laurilia sulcata CBS 365.49 MH856552 Vu et al. 2019 Lenzitopsis daii Yuan 2959 JN169799 He et al. (2019) Lycoperdon ericaeum ZRL 20151498 LT716030 He et al. (2019) Macrolepiota dolichaula xml 2013058 LT716021 He et al. (2019) Megalocystidium luridum CBS 106.71 MH860024 Vu et al. 2019 Megasporoporiella CLZhao 1438 MG231737 This study subcavernulosa Melanogaster rivularis S 190 HQ714731 He et al. (2019) Meripilus giganteus FP-135344 KP135307 He et al. (2019) Meruliopsis albostramineus HHB-10729 KP135051 He et al. (2019) Microporus xanthopus CLZhao 1285 MG231749 This study Nigroporus vinosus KA17-0261 MN294801 Direct Submission Oligoporus farinosus CIEFAP 91 JX090117 Pildain & Rajchenberg 2013 Omphalotus olearius AFTOL-ID 1718 DQ494681 He et al. (2019) Perenniporia hainaniana Cui 6364 JQ861743 He et al. (2019) Phaeophlebiopsis caribbeana HHB-6990 KP135415 He et al. (2019) Phanerochaete concrescens CLZhao 1430 MG231768 This study Phanerochaete sordida CLZhao 1459 MG231774 This study Phellinus ellipsoideus Cui 4270 JQ837948 He et al. (2019) Phlebiopsis crassa CLZhao 786 MG231791 This study Phylloporus pelletieri Pp 1 DQ534566 He et al. (2019) Pisolithus tinctorius AWW 219 EU718114 He et al. (2019) Polyozellus multiplex AFTOL-ID 677 DQ411528 He et al. (2019) Polyporus arcularius CLZhao 1338 MG231798 This study Porphyrellus porphyrosporus MB 97-023 DQ534563 He et al. (2019) Postia lactea Cui 9319 KX900894 Direct Submission Psathyrella candolleana ZRL 20151400 LT716063 He et al. (2019) Pseudochaete subrigidula He 1157 JQ716403 He et al. (2019) Pycnoporus sanguineus ZRL 2015009 LT716078 He et al. (2019) Rhizochaete americana FP-102188 KP135409 He et al. (2019) Rhizomarasmius oreinus AQUI 6763 NR132910 Moreau et al. 2015 Rhodocollybia maculata AFTOL-ID 540 DQ404383 He et al. (2019) Rhodotus asperior HKAS 56754 KC179737 He et al. (2019) Rigidoporus undatus Miettinen 13591 KY948731 He et al. (2019) Sarcodon joeides REB-270 KC571772 He et al. (2019) Schizophyllum commune CLZhao 1528 MG231811 This study Schizophyllum leprieurii ROBLEDO 1313 KM098065 Direct Submission Scleroderma areolatum AWW 211 EU718115 He et al. (2019) Sparsitubus nelumbiformis Cui 8497 KX880631 He et al. (2019) Steccherinum bourdotii CLZhao 1347 MG231820 This study Steccherinum ochraceum CLZhao 2897 MK269280 This study Stereum hirsutum CLZhao 1411 MG231830 This study Stereum rugosum CLZhao 1310 MG231836 This study Stereum sanguinolentum CLZhao 668 MG231838 This study Trametes hirsuta CLZhao 1544 MG231868 This study Trametopsis cervina TJV 93 216T JN165020 He et al. (2019) Tremella flava CBS 8471 KY105681 He et al. (2019) Tremella mesenterica CBS 6973 NR155937 He et al. (2019) Trulla dentipora AS 2288 KY970064 Direct Submission Veluticeps fimbriata L-10628-Sp HM536100 He et al. (2019) Xylobolus frustulatus He 2231 KU881905 He et al. (2019) Sequencher 4.6 (GeneCodes, Ann Arbor, MI, USA) was used to edit the DNA sequence. Sequences were aligned in MAFFT 7 (http://mafft.cbrc.jp/alignment/server/) using the "G-INS-i" strategy and manually adjusted in BioEdit (Hall 1999). Sequences of Tremella flava Chee J. Chen and T. mesenterica Retz. obtained from GenBank was used as an outgroup to root tree following He et al. (2019) in ITS analysis (Fig. 1).
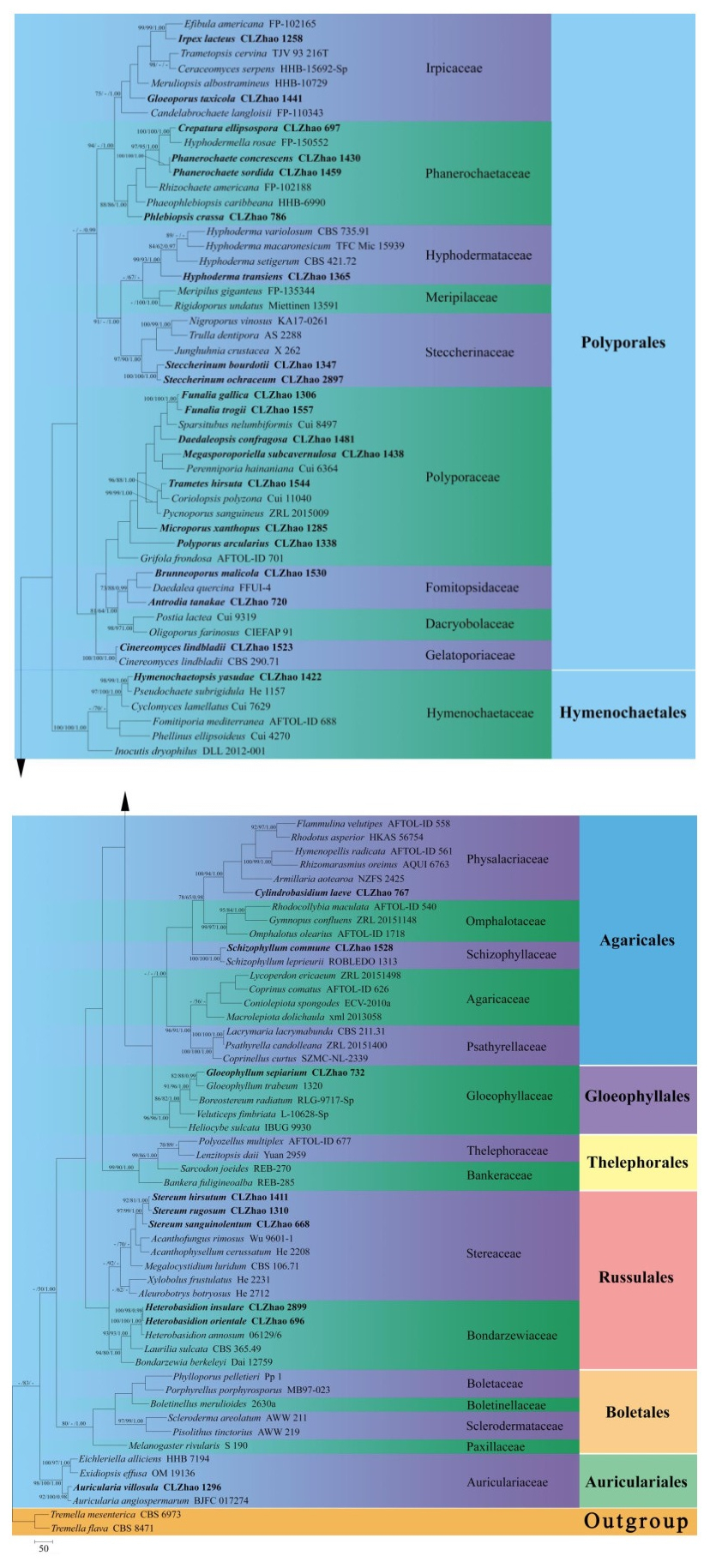
Figure 1. Maximum parsimony strict consensus tree illustrating the phylogeny of 52 species with related taxa in Agaricomycetes based on ITS sequences. Branches are labelled with maximum likelihood bootstrap equal to or greater than 70%, parsimony bootstrap equal to or greater than 50% and Bayesian posterior probabilities equal to or greater than 0.97, respectively. The taxa from the present study are indicated in black bold.
Maximum parsimony analysis was applied to the ITS dataset sequences. Approaches to phylogenetic analysis followed Zhao & Wu (2017) and the tree construction procedure was performed in PAUP* version 4.0b10 (Swofford 2002). All characters were equally weighted and gaps were treated as missing data. Trees were inferred using the heuristic search option with TBR branch swapping and 1000 random sequence additions. Max-trees were set to 5000, branches of zero length were collapsed and all parsimonious trees were saved. Clade robustness was assessed using a bootstrap (BT) analysis with 1, 000 replicates (Felsenstein 1985). Descriptive tree statistics tree length (TL), consistency index (CI), retention index (RI), rescaled consistency index (RC), and homoplasy index (HI) were calculated for each Maximum Parsimonious Tree (MPT) generated. Sequences were also analyzed using Maximum Likelihood (ML) with RAxML-HPC2 through the Cipres Science Gateway (www.phylo.org; Miller et al. 2009). Branch support (BS) for ML analysis was determined by 1000 bootstrap replicates.
MrModeltest 2.3 (Nylander 2004) was used to determine the best-fit evolution model for each data set for Bayesian inference (BI). Bayesian inference was calculated with MrBayes3.1.2 with a general time reversible (GTR+I+G) model of DNA substitution and a gamma distribution rate variation across sites (Ronquist & Huelsenbeck 2003). Four Markov chains were run for 2 runs from random starting trees for 1500 thousand generations (Fig. 1), and trees were sampled every 100 generations. The first one-fourth generations were discarded as burn-in. A majority rule consensus tree of all remaining trees was calculated. A majority rule consensus tree of all remaining trees was calculated. Branches were considered as significantly supported if they received maximum likelihood bootstrap (BS) > 70%, maximum parsimony bootstrap (BT) > 50%, or Bayesian posterior probabilities (BPP) > 0.95.
Morphological studies
-
The ITS dataset (Fig. 1) included sequences from 102 fungal specimens representing 101 species. The dataset had an aligned length of 1389 characters, of which 308 characters were constant, 283 parsimony-uninformative, and 798 parsimony-informative. Maximum parsimony analysis yielded 1 equally parsimonious tree (TL = 8644, CI = 0.2678, HI = 0.7322, RI = 0.4535, RC = 0.1215). The best-fit model for ITS alignment estimated and applied in the Bayesian was GTR+I+G, lset nst = 6, rates = invgamma; prset statefreqpr = dirichlet (1, 1, 1, 1). Bayesian resulted in a similar topology with an average standard deviation of split frequencies = 0.029534 (BI), and the effective sample size (ESS) across the two runs is the double of the average ESS (avg ESS) = 354.
The phylogeny (Fig. 1) inferred from ITS sequences demonstrated that fifty-two species nested in sixteen families, Auriculariaceae, Bondarzewiaceae, Dacryobolaceae, Fomitopsidaceae, Gelatoporiaceae, Gloeophyllaceae, Hymenochaetaceae, Hyphodermataceae, Irpicaceae, Phanerochaetaceae, Physalacriaceae, Polyporaceae, Schizophyllaceae, Schizoporaceae, Steccherinaceae and Stereaceae, belonging to six orders Agaricales, Auriculariales, Gloeophyllales, Hymenochaetales, Polyporales, Russulales in Agaricomycetes.
Checklist
-
An alphabetical list (according to genus name) of wood-decaying fungi identified in these investigations is given below. The authors of scientific names are according to the second edition of Authors of Fungal Names (http://www.indexfungorum.org/AuthorsOfFungalNames.html). Substrate and collecting data are provided after the name of each species. The hosts are listed alphabetically, and within the same host tree, they are arranged by the order: living tree, dead standing tree, trunk, fallen branch and stump. The collectors and collection numbers are listed alphabetically, too (Dai 2011, 2012a).
1. Antrodia tanakae (Murrill) Spirin & Miettinen, on the fallen branch of Acacia dealbata Link, CLZhao 720; on the stump of Acacia dealbata, CLZhao 1536
2. Auricularia villosula Malysheva, on the fallen branch of Alnus nepalensis, CLZhao 1428; on the trunk of Juglans regia L., CLZhao 743; on the trunk of Quercus, CLZhao 1296; on the fallen branch of Quercus, CLZhao 1340
3. Basidioradulum crustosum (Pers.) Zmitr., Malysheva & Spirin, on the fallen angiosperm branch, CLZhao 3028
4. Bjerkandera adusta (Willd.) P. Karst, on the stump of Pinus yunnanensis, CLZhao 1555; on the dead tree of Quercus, CLZhao 1275
5. Brunneoporus malicola (Berk. & M.A. Curtis) Audet, on the stump of Acacia dealbata, CLZhao 1524; on the trunk of Quercus acutissima Carr., CLZhao 1530
6. Byssomerulius corium (Pers.) Parmasto, on the fallen angiosperm branch, CLZhao 1560; on the fallen branch of Pinus yunnanensis, CLZhao 734, CLZhao 781; on the fallen branch of Quercus, CLZhao 693, CLZhao 1266, CLZhao 1274, CLZhao 1313; on the stump of Quercus, CLZhao 1341
7. Cinereomyces lindbladii (Berk.) Jülich, on the trunk of Pinus yunnanensis, CLZhao 1523
8. Crepatura ellipsospora C.L. Zhao, on the trunk of Alnus, CLZhao 697; on the fallen branch of Quercus, CLZhao 868, CLZhao 1260, CLZhao 1265
9. Cylindrobasidium laeve (Pers.) Chamuris, on the dead bamboo, CLZhao 756, CLZhao 767
10. Daedaleopsis confragosa (Bolton) J. Schröt, on the trunk of Alnus nepalensis, CLZhao 1481
11. Funalia gallica (Fr.) Bondartsev & Singer, on the trunk of Alnus nepalensis, CLZhao 1309; on the fallen branch of Alnus nepalensis, CLZhao 1306
12. Funalia trogii Berk., on the trunk of Alnus nepalensis, CLZhao 1540; on the angiosperm trunk, CLZhao 741, CLZhao 3009, CLZhao 1557; on the fallen angiosperm branch CLZhao 1552
13. Fuscoporia torulosa (Pers.) T. Wagner & M. Fisch, on the fallen branch of Quercus, CLZhao 1305
14. Gloeophyllum sepiarium (Wulfen) P. Karst, on the trunk of Pinus yunnanensis, CLZhao 732, CLZhao 764; on the fallen branch of Pinus yunnanensis, CLZhao 774, CLZhao 904; on the stump of Pinus yunnanensis, CLZhao 731, CLZhao 784
15. Gloeoporus dichrous (Fr.) Bres., on the fallen branch of Alnus nepalensis, CLZhao 1471
16. Gloeoporus taxicola (Pers.) Gilb. & Ryvarden, on the trunk of Pinus armandii, CLZhao 1441
17. Heterobasidion insulare (Murrill) Ryvarden, on the stump of Pinus yunnanensis, CLZhao 2899
18. Heterobasidion orientale Tokuda, T. Hatt. & Y.C. Dai, trunk of Pinus yunnanensis, CLZhao 696
19. Hymenochaete adusta (Lév.) Har. & Pat., J. Bot, on the trunk of Alnus, CLZhao 700
20. Hymenochaete rheicolor (Mont.) Lév., on the trunk of Alnus, CLZhao 672, CLZhao 679; on the fallen branch of Quercus, CLZhao 666, CLZhao 671, CLZhao 682
21. Hymenochaete villosa (Lév.) Bres., on the trunk of Quercus acutissima, CLZhao 1533
22. Hymenochaetopsis corrugata (Fr.) S.H. He & Jiao Yang, on the fallen angiosperm branch, CLZhao 2893
23. Hymenochaetopsis yasudae (Imazeki) S.H. He & Jiao Yang, on the fallen branch of Alnus nepalensis, CLZhao 1422, CLZhao 1445; on the living tree of Pinus armandii, CLZhao 1475; on the fallen branch of Pinus armandii, CLZhao 1486, CLZhao 1495, CLZhao 1549
24. Hyphoderma transiens (Bres.) Parmasto, on the fallen angiosperm branch, CLZhao 1493; on the trunk of Populus yunnanensis Dode, CLZhao 1365
25. Hyphodontia tropica Sheng H. Wu, on the fallen angiosperm branch, CLZhao 2898, CLZhao 2901
26. Irpex lacteus (Fr.) Fr., on the trunk of Acacia dealbata, CLZhao 1258
27. Lopharia cinerascens (Schwein.) G. Cunn., on the fallen branch of Alnus nepalensis, CLZhao 1499
28. Megasporoporiella subcavernulosa (Y.C. Dai & Sheng H. Wu) B.K. Cui, on the fallen branch of Alnus nepalensis, CLZhao 1438, CLZhao 1466; on the stump of Alnus nepalensis, CLZhao 1412; on the fallen angiosperm branch, CLZhao 2966, CLZhao 2984, CLZhao 3016; on the stump of Cupressus funebris Endl., CLZhao 1444, CLZhao 1491
29. Microporus xanthopus (Fr.) Kuntze, on the fallen branch of Alnus nepalensis, CLZhao 1503; on the fallen angiosperm branch, CLZhao 3012; on the trunk of Quercus, CLZhao 1253, CLZhao 1285; on the fallen branch of Quercus, CLZhao 1268, CLZhao 1304; on the stump of Quercus, CLZhao 1343
30. Phanerochaete concrescens Spirin & Volobuev, on the fallen branch of Alnus nepalensis, CLZhao 1541, CLZhao 1545; on the fallen angiosperm branch, CLZhao 2929, CLZhao 2931, CLZhao 2939, CLZhao 2940, CLZhao 2945, CLZhao 2946, CLZhao 2949; on the trunk of Pinus yunnanensis, CLZhao 2916
31. Phanerochaete sordida (P. Karst.) J. Erikss. & Ryvarden, on the trunk of Alnus, CLZhao 698; on the fallen branch of Alnus nepalensis, CLZhao 1459, CLZhao 1541, CLZhao 1545; on the angiosperm trunk, CLZhao 1461; on the fallen angiosperm branch, CLZhao 2929, CLZhao 2931, CLZhao 2939, CLZhao 2940, CLZhao 2945, CLZhao 2946, CLZhao 2949, CLZhao 4738, CLZhao 4746, CLZhao 4754; on the fallen branch of Pinus armandii, CLZhao 1515; on the trunk of Pinus yunnanensis, CLZhao 2916
32. Phellinus gilvus (Schwein.) Pat., on the fallen branch of Alnus nepalensis, CLZhao 1334
33. Phlebiopsis crassa (Lév.) Floudas & Hibbett, on the angiosperm trunk, CLZhao 786; on the fallen branch of Coriaria nepalensis Wall., CLZhao 1295, CLZhao 1308; on the trunk of Pinus yunnanensis, CLZhao 724; on the trunk of Quercus, CLZhao 1269, CLZhao 1314; on the fallen branch of Quercus acutissima, CLZhao 1532
34. Polyporus arcularius (Batsch) Fr. on the stump of Coriaria nepalensis, CLZhao 1338; on the fallen branch of Quercus, CLZhao 1316
35. Postia hibernica (Berk. Broome) Jülich, on the fallen angiosperm branch, CLZhao 2903; on the fallen branch of Pinus yunnanensis, CLZhao 2909
36. Pulcherricium coeruleum (Lam.) Parmasto, on the fallen branch of Alnus nepalensis, CLZhao 1434; on the fallen angiosperm branch, CLZhao 3003, CLZhao 3020
37. Schizophyllum commune Fr., on the trunk of Acacia dealbata, CLZhao 1527; on the fallen branch of Acacia dealbata, CLZhao 1528, CLZhao 1529; on the trunk of Alnus nepalensis, CLZhao 1537; on the stump of Alnus nepalensis, CLZhao 1562; on the stump of Eucalyptus robusta, CLZhao 1561
38. Steccherinum bourdotii Saliba & A. David, on the fallen branch of Alnus nepalensis, CLZhao 1472; on the fallen branch of Pinus armandii, CLZhao 1347
39. Steccherinum ochraceum (Pers. ex J.F. Gmel.) Gray, on the fallen angiosperm branch, CLZhao 2897, CLZhao 2968
40. Stereum gausapatum (Fr.) Fr., on the trunk of Alder, 11 January 2017, CLZhao 668, CLZhao 669, CLZhao 673, CLZhao 677, CLZhao 683, CLZhao 694, CLZhao 707; on the dead tree of Quercus, 22 April 2017, CLZhao 1259, CLZhao 1270, CLZhao 1290, CLZhao 1300, CLZhao 1318, CLZhao 1320
41. Stereum hirsutum (Willd.) Pers., on the fallen branch of Alnus nepalensis, CLZhao 1404, CLZhao 1405, CLZhao 1427, CLZhao 1455, CLZhao 1457, CLZhao 1465, CLZhao 1469, CLZhao 1470, CLZhao 1479, CLZhao 1482, CLZhao 1489, CLZhao 1498; on the fallen angiosperm branch, CLZhao 740; on fallen branch of Coriaria nepalensis, CLZhao 1559; on fallen branch of Pinus yunnanensis, CLZhao 2906; on the fallen branch of Quercus, CLZhao 1291
42. Stereum rugosum Pers., on the trunk of Quercus, CLZhao 1310
43. Stereum sanguinolentum (Alb. & Schwein.) Fr., on the trunk of Alnus, CLZhao 673; on the stump of Pinus yunnanensis, CLZhao 669
44. Trametes hirsuta (Wulfen) Lloyd, on the fallen branch of Alnus nepalensis, CLZhao 1544; on the angiosperm trunk, CLZhao 1344; on the trunk of Pinus yunnanensis, CLZhao 739
45. Trametes strumosa (Fr.) Zmitr., on the angiosperm trunk, CLZhao 718
46. Trametes versicolor (L.) Lloyd, on the trunk of Alnus nepalensis, CLZhao 1539, CLZhao 1546; on the fallen branch of Alnus nepalensis, CLZhao 1431; on the stump of Alnus nepalensis, CLZhao 1510; on the angiosperm trunk, CLZhao 748, CLZhao 1293, CLZhao 1477; on the trunk of Cupressus funebris, CLZhao 1509; on the trunk of Quercus, CLZhao 1330; on the fallen branch of Quercus, CLZhao 686, CLZhao 714, CLZhao 1302, CLZhao 1307
47. Trametopsis cervina (Schwein.) Tomšovský, on the trunk of Alnus nepalensis, CLZhao 1246; on the fallen branch of Quercus, CLZhao 1315
48. Trichaptum abietinum (Dicks.) Ryvarden, on the angiosperm stump, CLZhao 719, CLZhao 723, CLZhao 736, CLZhao 777, CLZhao 3004; on the trunk of Pinus yunnanensis, CLZhao 730
49. Tubulicrinis xantha C.L. Zhao, on the fallen branch of Pinus yunnanensis, CLZhao 2868, CLZhao 2869, CLZhao 2883
50. Xylodon kunmingensis L. W. Zhou & C.L. Zhao, on the fallen angiosperm branch, CLZhao 3010, CLZhao 3019; on the stump of angiosperm, CLZhao 755; on the fallen branch of Pinus yunnanensis, CLZhao 752
51. Xylodon nespori (Bres.) Hjortstam & Ryvarden, on the trunk of Cupressus funebris, CLZhao 1492
52. Xylodon rimosissimus (Peck) Hjortstam & Ryvarden, on the fallen branch of Pinus armandii, CLZhao 1487
Molecular phylogeny
-
In the kingdom Fungi, the phyla Ascomycota and Basidiomycota cover around 97% of all fungal species (Willis 2018). According to the latest version of Ainsworth & Bisby's Dictionary of the Fungi (Kirk et al. 2008), there are 1589 genera and more than 30 thousand species of Basidiomycota, which comprise nearly 32% of all described fungal taxa (Dai et al. 2015), and more and more taxa were recorded worldwide every year, which observably increase the fungal diversity (Sarma & Hyde 2018, Yafetto 2018, Freitas-Neto et al. 2019, Hipol et al. 2019, Ayesha et al. 2020, De Leon AM et al. 2020, Huang et al. 2020, Himani & Krishnappa 2020, Blanco-Dios 2021, Kumar et al. 2021, Wang & Zhao 2021, Zong et al. 2021).
Although some notable explorations of wood-decaying fungi have been made from Yunnan Province (Cui & Dai 2012, Wu et al. 2017, Zhao & Wu 2017, Shen et al. 2018, Wu et al. 2018, Liu et al. 2019, Luo et al. 2019, Xu et al. 2019, Chen & Zhao 2020, Huang et al. 2020, Wang et al. 2020, Wu et al. 2021, Zong et al. 2021), only three new species were found from Haikou Forestry Farm (Ma & Zhao 2019, Shi et al. 2019, He et al. 2020). Species in the present list are mostly new to the studied area. The 681 specimens belong to 52 species, which are distributed in 37 genera, 16 families, 6 orders, in which 11 are pathogenic (Dai 2012b), and 28 are medicinal (Dai & Yang 2008), and a few are edible (Dai et al. 2010). Among them, 28 species belong to order Polyporales and 14 species belong to order Hymenochaetales and 6 species belong to order Russulales, and 2 species belong to order Agaricales and 1 species belong to order Auriculariales and 1 species belong to order Gloeophyllales.
The molecular phylogenetic analyses with combined nLSU, SSU, 5.8s, rpb1, rpb2, and tef1 datasets for the subphyla Agaricomycotina, Pucciniomycotina and Ustilaginomycotina revealed that 1928 currently used genera names are distributed in 241 families, 68 orders, 18 classes and four subphyla (He et al. 2019). In the present study, 52 species nested in 37 genera, 16 families, 6 orders based on ITS dataset, which is similar to the topology of the phylogenetic analyses in He et al. (2019).
This work will comprehensively improve the understanding of the diversity of wood-decaying fungi in this area, which is conducive to the rational utilization and effective protection of fungal resources, and provides scientific basis for the prevention and control of forest diseases in this farm.
- The researches were supported by the Yunnan Fundamental Research Project (Grant No. 202001AS070043) and the Science Foundation of Southwest Forestry University (Project No. 111715).
- Copyright: © 2021 by the author(s). This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
X He, JZ Chen, CL Zhao. 2021. Diversity of wood – decaying fungi in Haikou Forestry Farm, Yunnan Province, P.R. China. Studies in Fungi 6(1):365−377 doi: 10.5943/sif/6/1/27


|