









HTML
-
The growth and productivity of temperate plants is commonly limited by heat stress. Over the next hundred years, the global temperature is projected to rise by 6.9 °C, which could diminish crop yield by 15%−35%[1,2]. Extensive efforts have been made to study the physiological and molecular factors regulating tolerance to heat stress in a multitude of plants (for reviews, see[3−8]). In the past decade, automated equipment designed for rapid testing has become prevalent, generating an appeal for conducting assays seeking to analyze how '–omic' technologies (associated with genes, mRNA, proteins, and metabolites) factor into the heat tolerance of plants.
Transcriptomic analysis is a powerful approach for the identification of genes associated with plant responses or resistance to abiotic stresses by RNA sequencing (RNA-seq), a technique previously utilized to study responses to heat stress in a variety of temperate grasses, including Kentucky bluegrass (Poa pratensis)[9], perennial ryegrass (Lolium perenne)[10], creeping bentgrass (Agrostis stolonifera)[11], and tall fescue (Festuca arundinacea Schreb.)[12]. Most of the previous studies on transcriptomic analysis in species of cool-season grasses focused on examining transcript changes in a single genotype responding to heat stress which reflected the heat responses of transcriptomes, and a majority of the heat-responsive genes in these studies was associated with primary metabolism of photosynthesis, respiration, proteins, and hormones, as well as transcriptional factors associated with response to heat stress, namely heat shock factors[9−10, 12]. Secondary metabolism for biosynthesis of compounds, such as phenolics, is well known to play roles in plant defense against biotic stresses, but limited information is available on key genes or networks involved in the metabolic pathways of secondary metabolites for plant tolerance to heat stress and are not well documented[3, 5]. Comparative analysis of transcriptomic changes under prolonged periods of heat stress for genotypes differing in tolerance to heat stress, particularly in stress-tolerant grass species, will allow for the classification of unique genes that correlate to genetic variations in heat tolerance, which could be useful for the utilization of genes regulating plant heat tolerance through genomic manipulation or molecular breeding.
Fine fescues, including hard fescue, are low-maintenance cool-season grass species in terms of fertility, irrigation, and mowing, and also exhibit superb heat tolerance with a broad variety of genetic variability among genotypes. Previous studies[13,14] that compared heat tolerance for 26 genotypes, including seven hard fescues, two slender creeping red fescues (Festuca rubra subsp. litoralis), two sheep fescues (Festuca ovina subsp. hirtula), seven strong creeping red fescues (Festuca rubra subsp. rubra), and eight chewings fescues (Festuca rubra subsp. commutata), demonstrated significant genetic variability in heat tolerance, with 'Reliant IV' hard fescue ranked highly as a heat-tolerant genotype and 'Predator' being heat-sensitive relative to 'Reliant IV', based on the evaluation of turf quality (TQ), photochemical efficiency (Fv/Fm) chlorophyll (Chl) content, relative water content (RWC), and electrolyte leakage (EL). Quantitative PCR analysis showed that there was a direct correlation between the abundance of transcripts for genes associated with energy production (ATP synthase), cellular respiration (Sucrose synthase), light-harvesting (Photosystem II CP47 reaction center protein, RuBisCO activase), oxidative defense (Catalase), cell structure and division (Actin), and stress defense (Heat shock protein 90) and heat tolerance, as manifested by the aforementioned physiological traits[14]. Therefore, we hypothesized that a more-detailed comparative analysis on transcriptomes of such two hard fescue cultivars can reveal a more comprehensive gene network and potential central hub genes that are responsible for their contrasting heat tolerances. For further understanding of the molecular factors and gene networks linked to heat tolerance in hard fescue, we used comparative transcriptomics by RNA-seq and conducted a weighted gene comparison network analysis for the heat-tolerant 'Reliant IV' and heat-sensitive 'Predator' cultivars to establish central hub and unique genes and underlying metabolic pathways that may relate to genetic differences in heat tolerance for hard fescue. Such information will be substantial in the efforts towards genetic augmentation of heat tolerance in cool-season grasses.
RESULTS -
Under non-stress temperature (0 d of heat stress) and at 7 d of heat stress, 'Predator' and 'Reliant IV' did not differ significantly in canopy density and color (Fig. 1a & b). 'Reliant IV' had more green leaves and a lower proportion of yellow leaves at 14 d of heat stress (Fig. 1c).

Figure 1. Plants of 'Predator' and 'Reliant IV' grown under (a) normal temperature (0 d of heat stress), (b) 7 d of heat stress, (c) 14 d of heat stress.
Leaf Chl content in 'Predator' and 'Reliant IV' decreased with heat stress at 14 and 21 d, and Chl content was significantly greater in 'Reliant IV' as compared with 'Predator' at 14 d of heat stress (Fig. 2a). Leaf Fv/Fm in 'Predator' and 'Reliant IV' declined during heat stress, but 'Reliant IV' maintained significantly higher Fv/Fm than 'Predator' from 7−21 d of heat stress (Fig. 2b). Heat stress caused increases in leaf EL in both 'Predator' and 'Reliant IV', but 'Reliant IV' had significantly lower EL than 'Predator' from 7−21 d of heat stress (Fig. 2c).
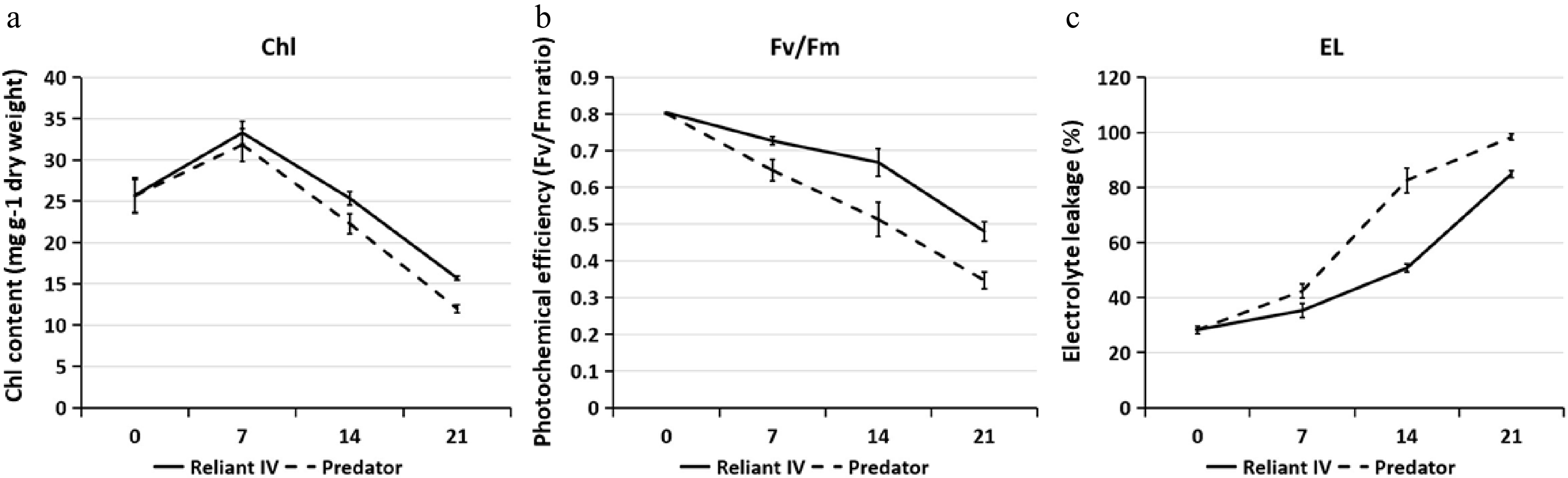
Figure 2. (a) Leaf chlorophyll (Chl) content, (b) photochemical efficiency (Fv/Fm), and (c) electrolyte leakage (EL) of 'Predator' and 'Reliant IV' in response to heat stress. The error bars represent standard error (SE) for three biological replicates (n = 3).
Differential transcriptomic responses of 'Predator' and 'Reliant IV' to heat stress
-
The RNA sequencing analysis yielded a total of more than 574 and 513 million reads in 'Predator' and 'Reliant IV', respectively, which was about 90% of the Q30 rate. Both cultivars had over a 97% alignment rate with a similar total contig number, N50 value, average contig length, total assembled bases, GC content (Supplemental Table S1), and gene annotations (Supplemental Table S2), indicating that the transcriptome assembly could mostly represent their transcript profile.
In 'Predator, a total of 2,184 and 4,324 genes were up-regulated at 7 d while 2,093 and 2,352 genes were down-regulated at 7 d and 14 d of heat stress, respectively (Fig. 3a & b; Supplemental Table S3). Among the differentially expressed genes (DEGs) in 'Predator', 1,792 were commonly up-regulated while 1,585 were commonly down-regulated in response to heat stress at both 7 and 14 d.
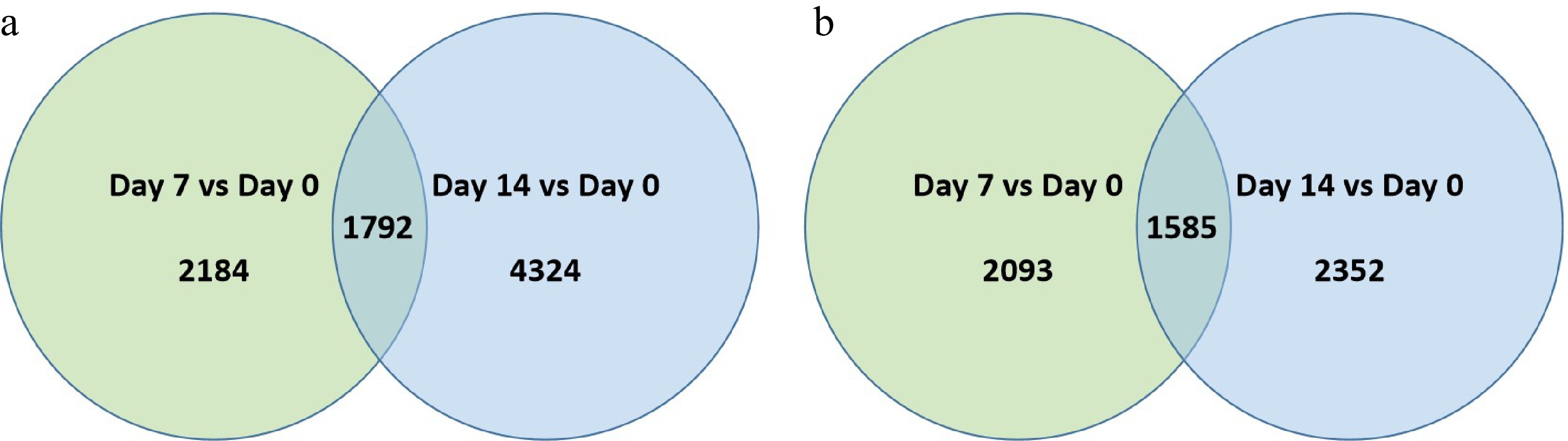
Figure 3. Venn diagram illustrating the number of differentially expressed genes (DEGs) that were (a) up-regulated or, (b) down-regulation at 7 d or 14 d of heat stress in comparison to that at 0 d of heat stress (Day 7 vs. Day 0 or Day 14 vs. Day 0) for 'Predator'. The numbers in each circle and overlapping area represent unique and common DEGs between days of heat stress, respectively.
At 7 and 14 d of heat stress, 2,351 and 1,998 genes were up-regulated and 2,227 and 853 were down-regulated, respectively, in 'Reliant IV' (Fig. 4a & b; Supplemental Table S4). Among the DEGs regulated in response to heat stress in 'Reliant IV', 779 were commonly up-regulated and 665 were commonly down-regulated at both 7 and 14 d of heat stress. While a similar number of DEGs were found in 'Reliant IV' and 'Predator' at 7 d of heat stress, fewer up- or down-regulated DEGs were found in 'Reliant IV' than 'Predator' in response to heat stress at 14 d.
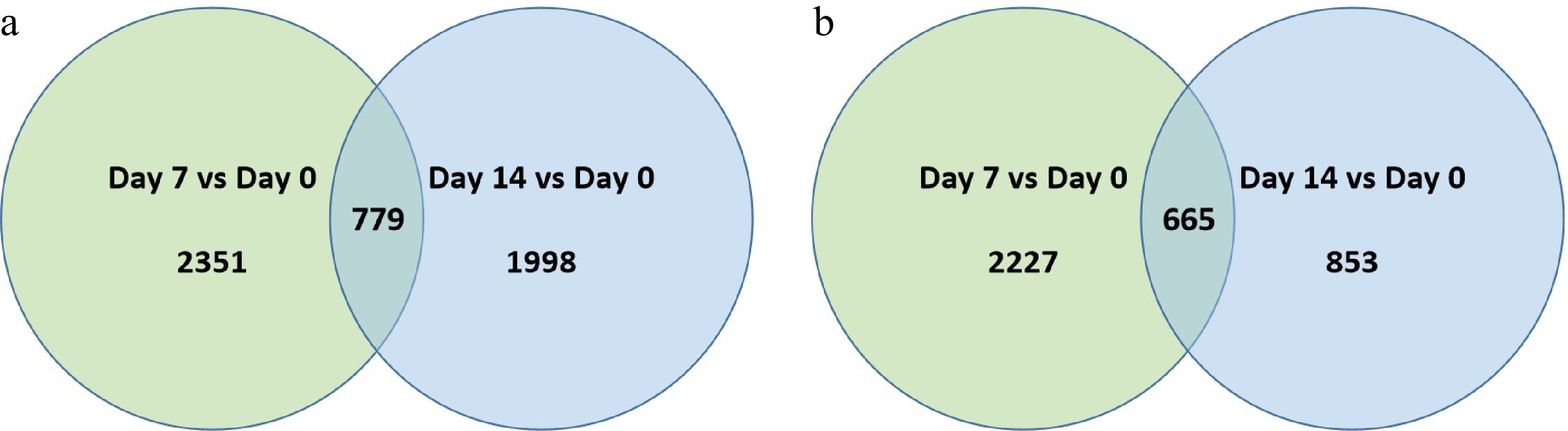
Figure 4. Venn diagram illustrating the number of differentially expressed genes (DEGs) that were (a) up-regulated or, (b) down-regulation at 7 d or 14 d of heat stress in comparison to that at 0 d of heat stress (Day 7 vs. Day 0 or Day 14 vs. Day 0) for 'Reliant IV'. The numbers in each circle and overlapping area represent unique and common DEGs between days of heat stress, respectively.
In order to identify major DEGs and their associated biological pathways in relation to genotypic variations in heat tolerance, all up- and down-regulated DEGs at 7 and 14 d of heat stress in 'Predator' and 'Reliant IV' were analyzed using a GO term enrichment program. Among the DEGs up-regulated at 7 d of heat stress, the GO biological process term 'response to oxygen-containing compounds' and GO molecular function term 'oxidoreductase activity' were uniquely enriched in 'Reliant IV', while most enriched GO terms were commonly found in both cultivars, including 'unfolded protein folding', 'heat shock protein folding', 'response to temperature stimulus', 'response to heat', and 'extracellular region' (Fig. 5a; Supplemental Table S5). At 14 d of heat stress, 'Reliant IV' had several uniquely up-regulated and enriched DEGs in 12 GO cellular components, biological processes and molecular terms that were not present in 'Predator', including 'response to oxygen-containing compounds', 'oxidation-reduction process', 'oxylipin biosynthetic process', 'oxylipin metabolic process', 'response to chemical', 'extracellular region', 'intracellular', 'vacuolar lumen', 'ammonia-lyase activity', 'nucleic acid binding transcription factor activity', 'transcriptional factor activity sequence-specific DNA binding', and 'oxidoreductase activity' (Fig. 5b; Supplemental Table S5).
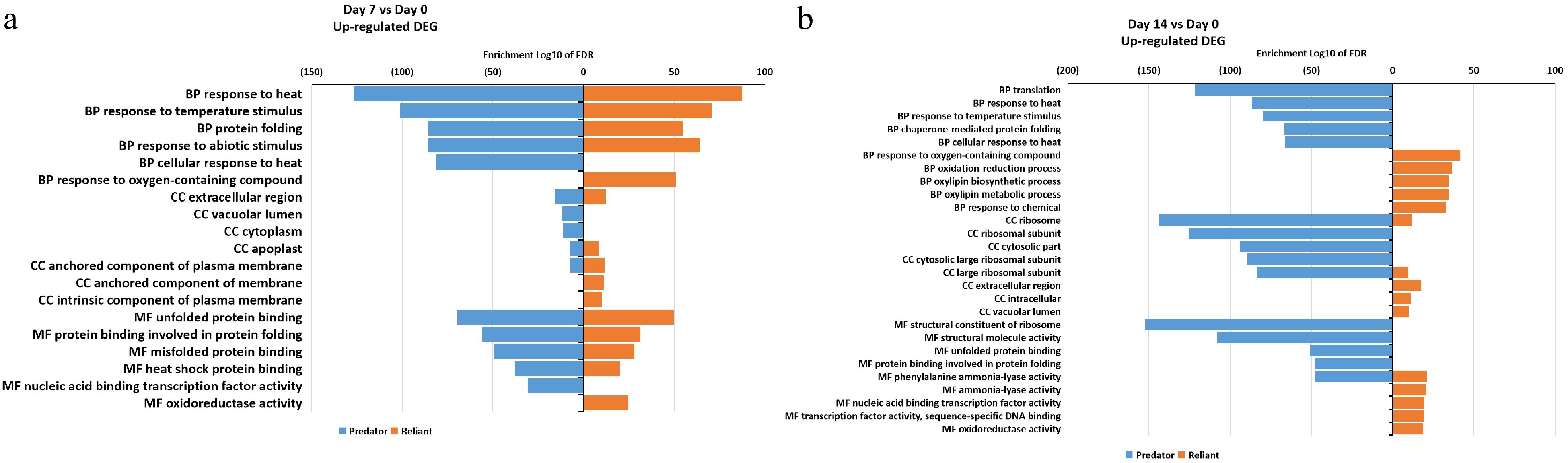
Figure 5. Gene ontology (GO) enrichment analysis of DEGs that were up-regulated at (a) 7 d of heat stress and (b) 14 d of heat stress in 'Predator' and 'Reliant IV'.
The down-regulated DEGs at 7 d of heat stress mostly overlapped between two cultivars, including those categorized in photosynthesis, energy, photosystems, and chlorophyll binding, while DEGs in the biological process 'generation of precursor metabolites and energy', cellular component 'chloroplast part' and 'plastid part', as well as molecular function 'oxidoreductase activity' and 'carbon-carbon lyase activity' categories were down-regulated only in 'Predator' (Fig. 6a; Supplemental Table S5). At 14 d of heat stress, DEGs in four molecular function and biological process categories, including 'photosynthesis', 'ribulose-bisphosphate carboxylase activity', and 'lyase activity', were uniquely down-regulated in 'Predator', while most of the other processes were shared by both cultivars (Fig. 6b, Supplemental Table S5).
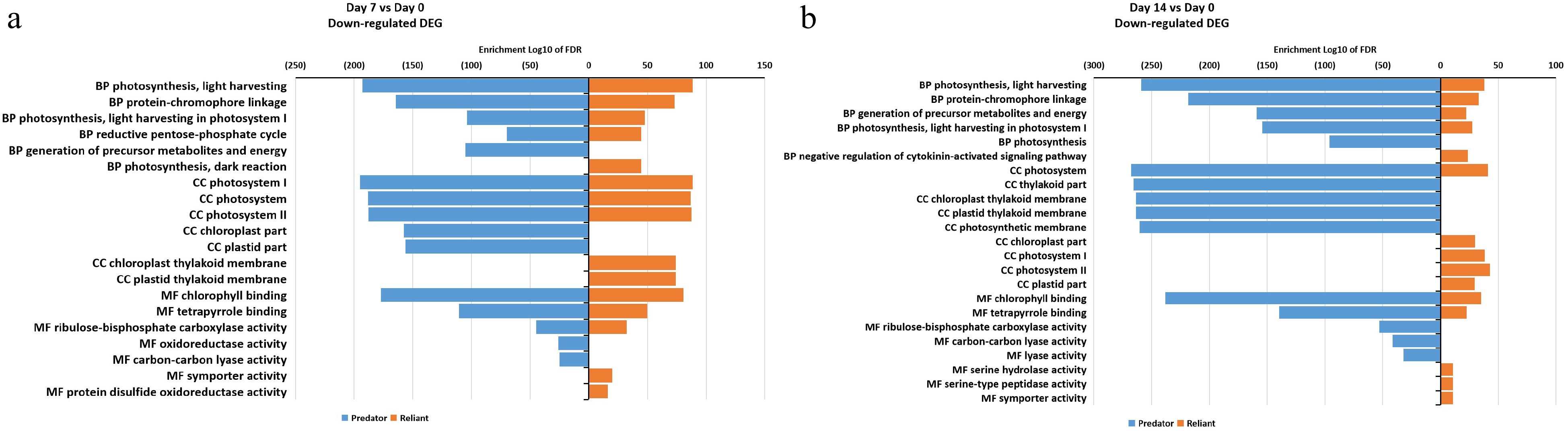
Figure 6. Gene ontology (GO) enrichment analysis of DEGs that were down-regulated at (a) 7 d of heat stress and (b) 14 d of heat stress in 'Predator' and 'Reliant IV'.
The KEGG enrichment analysis demonstrated that most of the enriched KEGG terms were identical between 'Predator' and 'Reliant IV' (Fig. 7a & b), whereas the main difference in gene functional enrichment between two cultivars was the up-regulated DEGs at 7 d of heat stress, with unique enrichment in 'Reliant IV' for genes involved in primary and secondary metabolism.
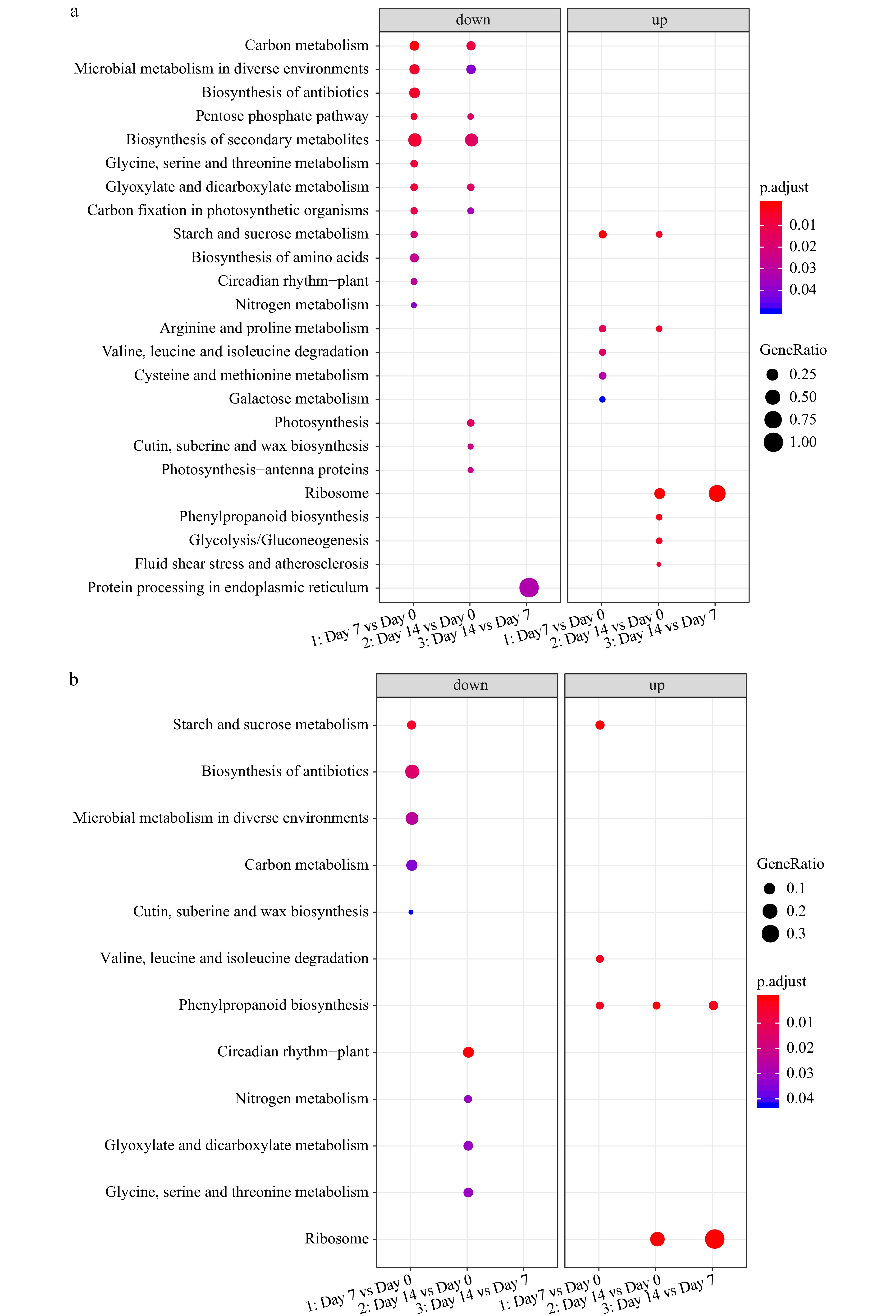
Figure 7. KEGG enrichment analysis of DEGs for (a) 'Predator' and (b) 'Reliant IV' at 7 and 14 d of heat stress in comparison to 0 d of heat stress.
To further explore gene networks and central hub genes related to heat stress tolerance in hard fescue, weighted gene co-expression network analysis (WGCNA) was performed on all heat-responsive DEGs in 'Predator' and 'Reliant IV' exposed to 7 or 14 d of heat stress. The WGCNA grouped DEGs having similar expression arrays under heat stress for 'Predator' and 'Reliant IV' into 15 and 13 module eigengenes, respectively (Supplemental Fig. S1). The gene networks and central hub genes linked to the DEGs up-regulated in only 'Reliant IV' or to DEGs regulated more in 'Reliant IV' with respect to 'Predator' at 7 and 14 d of heat stress, which were enriched in the GO-term functional categories (Fig. 5) for 'response to oxygen-containing compounds', 'oxidation-reduction process', 'oxidoreductase activity', 'oxylipin biosynthetic process', 'oxylipin metabolic process', 'extracellular region', and 'ammonia-lyase activity', were found mainly in two WGCNA modules, MElghtcyan and MEdarkmagenta.
The central hub genes and connecting genes in the MElightcyan and MEdarkmagenta modules had a log2 fold change in gene expression levels ranging from 1.59 to 7.2 in 'Reliant IV' subjected to heat stress for 7 and 14 d in comparison to that at 0 d of heat stress. The central hub genes in MElightcyan and MEdarkmagenta were BBE22 (Berberine bridge enzyme-like 22 protein) and ALPL (transposase-derived ALP1-like protein), respectively (Fig. 8a & b; Supplemental Table S6 & S7). The unique DEGs in heat-stressed 'Reliant IV' in MElightcyan included LOX1 (lipoxygenase 1) and LOX3 (lipoxygenase 3) in the pathways of oxylipin metabolism, and PAL2 (phenylalanine ammonia-lyase 2) in the processes involving ammonia-lyase activity. The unique DEGs found in MEdarkmagenta included two genes associated with the oxidation-reduction process or oxidoreductase activity for dhurrin biosynthesis, C71E1 (4-hydroxyphenylacetaldehyde oxime monooxygenase) and C79A1 (tyrosine N-monooxygenase), respectively.
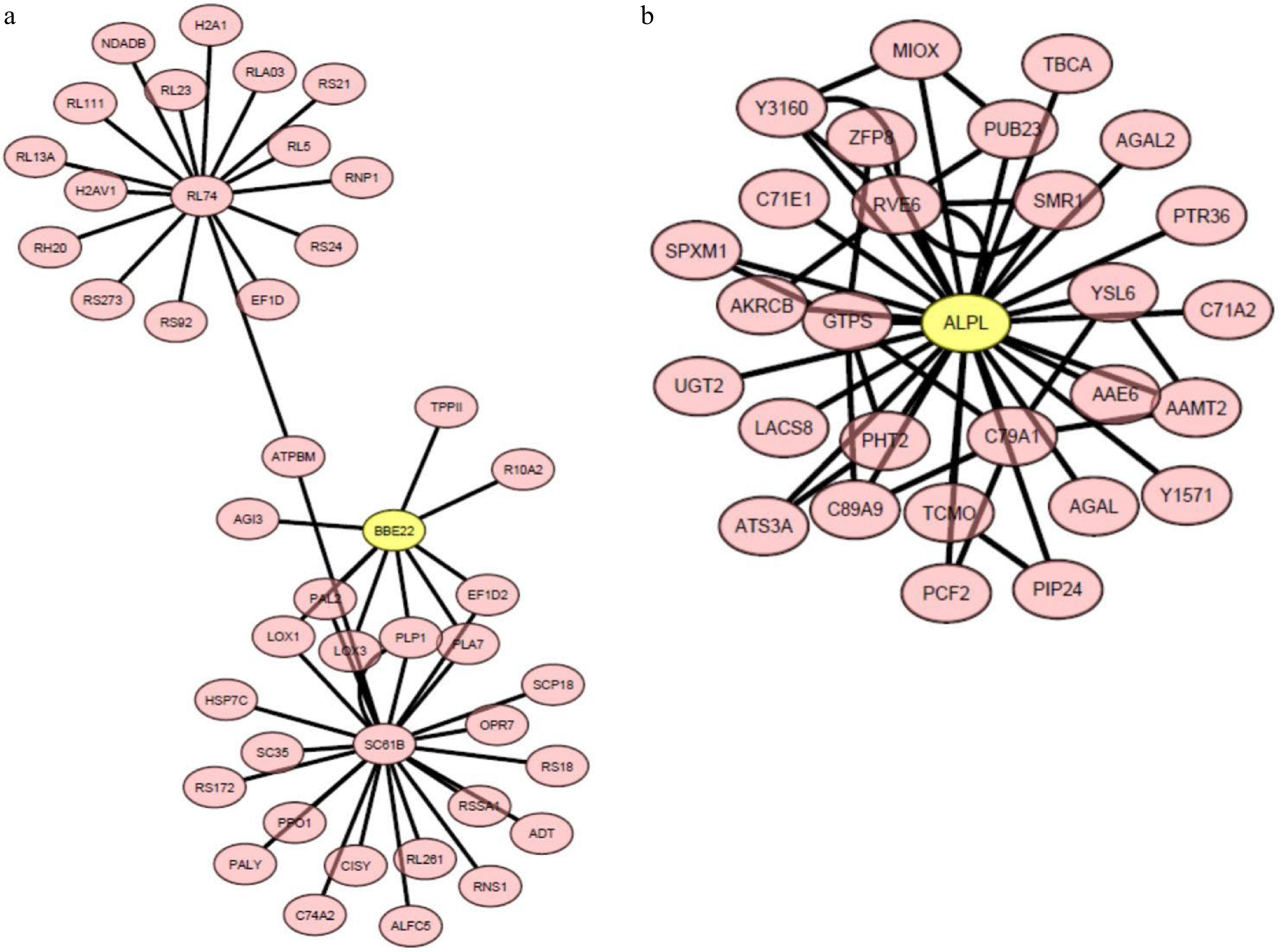
Figure 8. The heat-upregulated central hub genes and the connecting genes in module (a) MElightcyan and (b) MEdarkmagenta in 'Reliant IV' exposed to 7 and 14 d of heat stress. The genes in the yellow circles represent central hub genes.
Differential physiological responses of 'Predator' and 'Reliant IV' to heat stress
DISCUSSION -
The correlative analysis of transcriptional alterations as a result of heat stress for two hard fescue cultivars differing in heat tolerance allowed for the identification of several unique heat-responsive DEGs in heat-tolerant 'Reliant IV', while a majority of the heat-responsive DEGs were common in both cultivars, including those with putative functions in primary metabolism, such as photosynthesis and respiration, hormone metabolism, and transcription factors relative to stress that were previously reported in transcriptomic profiling of heat-responsive genes in other plant species[9−12,14]. The WGCNA and GO-term enrichment analysis revealed three unique central hub genes (BBE22 and ALPL) involved in secondary metabolism for stress protection, only up-regulated in 'Reliant IV' under heat stress in this study; however, the manners in which those genes are related to heat tolerance have not previously been examined. Therefore, the discussion below focuses only on those unique heat-responsive DEGs with functions categorized in secondary metabolism, as found in the GO enrichment analysis and the central hubs (BBE22 and ALPL) which may have critical functions in conferring heat tolerance to the heat-tolerant cultivar of hard fescue in order to further understand novel mechanisms of heat tolerance in temperate grasses.
A central hub gene, BBE22, and three genes, LOX1, LOX3, and PAL, linked with BBE2, were found to be uniquely enriched and up-regulated in the heat-tolerant cultivar, 'Reliant IV', but not in the heat-sensitive 'Predator', under heat stress. The four genes have been associated with secondary metabolism for stress defenses, particularly biotic stress, while their functions for heat stress tolerance are not well-known. LOXs are a family of genes encoding enzymes responsible for catalyzing the hydroperoxidation of polyunsaturated fatty acids, acting on linolenic and linoleic acid in secondary metabolic pathways to synthesize oxylipins, such as jasmonic acid (JA) and green leaf volatiles (GLVs)[15,16]. LOXs play important roles in secondary metabolism, response to naturally-occurring and stress-induced senescence, and defense responses to pathogen and insect attack[17]. LOX1 is inducible by abscisic acid, pathogens, JA[18], and cadmium exposure in Arabidopsis (Arabidopsis thaliana)[19]. Arabidopsis lox1 mutants were shown to have enhanced oxidative damage induced by cadmium[19]. LOX3 encodes an enzyme participating in the synthesis of JA[20] and has previously been exhibited to play a role in JA-mediated salt stress response, as lox3 mutants of Arabidopsis with lower JA content were hypersensitive to salt stress and had slower seed germination rate and lower seedling survival rate, whereas JA treatment rescued the salt-sensitive phenotypes in lox3 mutants[21]. Some studies have also reported that LOX genes (LOX1, LOX3, and LOX5) regulate organ development (i.e. seed germination and lateral rooting) and the levels of LOX activity have been positively associated with cell elongation in rapidly growing tissues of plants[17,22−24]. At the start of the phenylpropanoid pathway, phenylalanine ammonia-lyase (PAL) catalyzes the removal of an amino group from phenylalanine to produce precursors for many secondary metabolites having critical functions in plant defense against biotic and abiotic stresses[25,26]. The PAL2 gene mediates the biosynthesis of lignin and phenolics and was previously reported to be up-regulated in Achillea pachycephala[27] subjected to drought stress and in Arabidopsis[28] exposed to salt stress. The berberine bridge enzyme (BBE) has been associated with plant immunity and nitric oxide responses[29−32]. In a recent study of Arabidopsis response to spider mite, BBE22 was found to be involved in H2O2 homeostasis[33]. lox1 mutants of Arabidopsis exhibited down-regulation of transcription levels of H2O2-scavenging enzymes and significant increases in H2O2 accumulation in leaves and roots exposed to cadmium[19]. The up-regulation of PAL transcripts was associated with increases in the enzymatic activity of peroxidase in barley (Hordeum vulgare L.) exposed to salt stress[34]. The unique up-regulation of the central hub gene of BBE22 linked with LOX1, LOX3, and PAL in the heat-tolerant cultivar of hard fescue suggested that stress defenses involving secondary metabolism could be activated through the central control of BBE22, involving H2O2 scavenging and homeostasis; however, this notation deserves further investigation.
The ALPL (transposase-derived ALP1-like protein) was another central hub gene upregulated in heat-tolerant 'Reliant IV' exposed to heat stress, which was linked with two DEGs, CYP71E1 (4-hydroxyphenylacetaldehyde oxime monooxygenase) and CYP79A1 (tyrosine N-monooxygenase) in the pathway of dhurrin biosynthesis. Little is known about the biological function of ALPL, except that ALPL1 was identified as a stress-responsive transcription factor related to herbicide resistance[35,36]. The two DEGs linked to ALPL, CYP79A1 and CYP71E1, are localized in the membrane and activate the conversion of tyrosine to dhurrin, which can account for 30% of the dry mass in sorghum (Sorghum bicolor) seedlings[37−39]. Dhurrin can also serve as a stored form of nitrogen that can be remobilized in order to avoid hydrogen cyanide formation and provide a vital source of reduced nitrogen[40,41]. Dhurrin is an insect-defense compound and can be hydrolyzed by β-glucosidase to release toxic hydrogen cyanide gas upon tissue disruption caused by feeding insect larvae[42]. It was previously shown that an accumulation of dhurrin level in leaves is directly correlated to better tolerance to drought in post-flowering sorghum lines[43,44]. The unique upregulation of ALPL, CYP79A1, and CYP71E1 in heat-tolerant cultivars of hard fescue suggests their possible involvement in the heat tolerance of plants; however, the direct link between ALPL, CYP79A1, and CYP71E1 and heat stress tolerance deserves further investigation.
In summary, unique heat-responsive DEGs found in 'Reliant IV' by functional enrichment and WGCNA analysis include central hub genes (BBE22 and ALPL) and their connecting genes involved in secondary metabolism for biosynthesis of oxylipins (LOX1 and LOX3), phenolic compounds (PAL2), and dhurrin (C79A1 and C71E1), which could be involved in mediating defense against heat stress in plants. The direct causal relationship of those unique heat-responsive genes to heat tolerance warrants further investigation, as they could be useful for the determination of molecular markers associated with superior heat tolerance in hard fescue and other cool-season turfgrass species.
MATERIALS AND METHODS -
Plants of two cultivars of hard fescue, 'Predator' and 'Reliant IV', were transported from a Rutgers University turfgrass research farm (North Brunswick, NJ) to a greenhouse maintained at a daily temperature of 23/20 °C (day/night), with natural and artificial sodium-vapor lighting of 750 μmol m−2 s−1 photosynthetically active radiation (PAR). Experimental plants were transplanted into 15 cm in diameter × 20 cm deep plastic pots filled with an even soil and peat moss mixture. During the 6-week establishment period, plants were irrigated daily, trimmed biweekly to a canopy height of 7 cm, and fertilized biweekly with an even volume of of half-strength Hoagland's nutrient solution[45]. Prior to induction of heat stress treatment, mature plants were transported to controlled-environment growth chambers (Environmental Growth Chambers, Chagrin Falls, OH) set to a 14-h photoperiod at 700 μmol m−2 s−1 PAR, 50% relative humidity, and 22/18 °C (day/night) for plant establishment.
Temperature treatments and experimental design
-
Plants subjected to heat stress were maintained at a temperature of 38/33 °C (day/night) for 21 d, and non-stress control plants were kept at an optimal temperature of 22/18 °C (day/night). Both temperature regiments were replicated across three growth chambers, and for each cultivar, there were three individual replicates randomly dispersed among the three chambers for each temperature treatment. Plants were irrigated each day and half-strength Hoagland's nutrient solution was applied as a fertilizer weekly. This experiment was arranged as a split-plot randomized design, having temperature conditions defined as the main plots and cultivars defined as the sub-plots.
Physiological analyses
-
To measure the chlorophyll (Chl) content of leaf tissue, the methods of Hiscox and Israelstam[46] were followed with minor changes made to the procedure. Leaf tissue (100 mg) was excised from plants and submerged in 10 mL dimethyl sulfoxide for 5 d until Chl was completely extracted. After measuring the absorption of the Chl extract at wavelengths of 663 and 645 nm via spectrophotometer (Spectronoic Instruments, Rochester, NY), the leaf tissue was oven-dried for 3 d at 80 °C, and leaf Chl content was determined using the equations provided by Arnon[47].
A chlorophyll fluorescence meter (FIM 1500, ADC BioScientific Ltd., Herts, UK) was utilized to measure the variable fluorescence (Fv) and maximal fluorescence (Fm) of plants after leaves were dark-adapted for 30 min, and photochemical efficiency (Fv/Fm) was expressed as the ratio of Fv to Fm.
Leaf electrolyte leakage (EL) was quantified as a measure of membrane stability using the procedure detailed by Blum and Ebercon[48] with certain modifications. Leaf tissue (100 mg) was excised from each plant, submerged in 30 mL of pure, deionized water, and kept on an orbital platform shaker for 16 h. A conductance meter (Model 32, YSI Inc., Yellow Springs, OH) equipped with a probe was used to measure initial conductance (Ci) of each solution. After killing leaf tissue in an autoclave set to a cycle of 20 min at 120 °C, tissue solutions were shaken for 16 h and maximal conductance (Cmax) was measured. EL was calculated by dividing Ci by Cmax and multiplying by 100 to express values as a percentage.
RNA extraction, library preparation, and RNA sequencing
-
Leaf tissue (0.2 g) was excised from plants at 0, 7, and 14 d of heat stress and total RNA was extracted and pooled into a library, and prepared for RNA sequencing using the methods previously described by Xu and Huang with no modifications[49]. RNA was extracted, and the Low Sample (LS) procedure from the Illumina TruSeq RNA Library Prep Kit v2 (Illumina, San Diego, CA) was utilized to construct pooled libraries before sequencing RNA with a MiSeq Reagent Kit v3 cartridge (Illumina, San Diego, CA).
Read alignment, counting, and identification of differentially expressed genes (DEGs)
-
Raw sequence reads from MiSeq sequencing were analyzed using the direct methods and program settings provided by Xu and Huang[49], by assembling and trimming reads and analyzing predicted transcripts using the 956 plantae Benchmarking Universal Single-Copy Orthologs (BUSCO) profiles[50]. Transcripts were analyzed for differential expression according to the methods specified in Xu and Huang[49], and edgeR was used for estimating genewise dispersion[51]. To obtain differentially expressed genes (DEGs), the ratios of transcript abundances for 7 d of heat stress to control conditions, 14 d of heat stress to control conditions, and 14 d to 7 d of heat stress conditions for each cultivar were filtered by applying a log2 fold-change threshold of > 2 or < −2 and a false discovery rate (FDR) of < 0.001, and coding regions of assembled transcripts were defined according to the methods specified previously in Xu and Huang[49].
Gene functional enrichment analysis
-
To determine the gene Ontology (GO) enrichment of DEGs, the GOseq package in R was utilized in order to amend gene length bias with transcriptome data[52]. In order for GO terms to be labeled as 'signifcantly enriched in DEGs', a false discovery rate (FDR) of < 0.05 was required.
The Kyoto Encyclopedia of Genes and Genomes (KEGG) is a database which includes a hierarchical structure of different levels of biological processes, molecular functions, and cellular components collected from all living organisms. For KEGG enrichment analysis, we used the ClusterProfile R package[53]. Since our species are not available for direct KEGG enrichment, the enricher function in the package was called to use customized KEGG terms in both DEGs and annotations and to calculate the over-represented KEGG terms using default parameters. For visualization of the final results, we used the dotplot function in the package and modified the figure with the ggplot2 function to compare cultivars side-by-side.
Weighted gene co-expression network analysis (WGCNA)
-
To construct a co-expression network of genes, the WGCNA package in R was used[54]. First, DEGs expressed in at least one pair-wise comparison of heat stress with control condition were included for co-expression network construction. Gene expression levels from all time points were initially clustered into multiple groups to calculate the sample height. A soft threshold of 35 was used to generate adjacency matrix as the scale-free topology criterion, and DEGs were further clustered in the above hierarchical structure using topological overlap method[55]. The module in the gene dendrogram was detected by the dynamic tree cut method, using the parameter of mergeCutHeight = 0.1 and minModuleSize = 30. To further create weighted gene co-expression network, gene connectivity was calculated from the edge weight in the topology overlap matrix, which represents the strength of interactions between two genes. Finally, the weights of all edges of a node (gene) in the network were added together to be the level of connectivity, and the nodes with the highest connectivity in each module, calculated using chooseTopHubInEachModule function in the package, were considered as central hub genes for each module.
Statistical analysis and data accessions
-
-way analysis of variance was implemented to separate the differences between temperature treatments, cultivars, and their interactive effects on different parameters using SAS (SAS Int. Cary, NC). Differences among treatments and cultivar variations were defined using Student's t-test at a probability level of p = 0.05.
Transcriptome shotgun assembly for 'Predator' and 'Reliant IV' were installed onto the GenBank Transcriptome Shotgun Assembly (TSA) database, using the accession number PRJNA632630. The reads from all replicates of 'Predator' and 'Reliant IV' are stored in GenBank accession SAMN14916095 - SAMN14916112.
Plant materials and growth conditions
- The authors wish to thank the National Institute of Food and Agriculture, USDA, Specialty Crop Research Initiative for funding (award number 2017-51181-27222).
- The authors declare that they have no conflict of interest.
- Supplemental Table S1 RNA-seq library and assembly statistics.
- Supplemental Table S2 Summary of RNA-seq annotations found in seven databases.
- Supplemental Table S3 Lists of up- and down-regulated DEGs found in ‘Predator’ at 7 and 14 d of heat stress.
- Supplemental Table S4 Lists of up- and down-regulated DEGs found in ‘Reliant IV’ at 7 and 14 d of heat stress.
- Supplemental Table S5 GO term enrichment of DEGs in both ‘Predator’ and ‘Reliant IV’ at 7 and 14 d of heat stress.
- Supplemental Table S6 WGCNA analysis of DEGs showed two modules (MElightcyan and MEdarkmagenta) and pair-wise gene connection weight in each of them.
- Supplemental Table S7 Gene expression pattern of the central hub genes and connecting genes in MElightcyan and MEdarkmagenta modules, showing as log of fold change(logFC) ± standard error(SE, n = 3). N.S. means ‘not significant’.
- Supplemental Fig. S1 Cluster dendrograms of WGCNA analysis for DEGs in ‘Predator’ (a) and ‘Reliant IV’ (b) exposed to 7 d and 14 d of heat stress. The resulting original and merged module eigengenes after dynamic tree cut method were also shown at the bottom.
- Copyright: © 2021 by the author(s). Exclusive Licensee Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
Acknowledgments Conflict of interest Supplementary information Rights and permissions (8) References(55) - About this article
Cite this articleXu Y, Rossi S, Huang B. 2021. Comparative transcriptomics and gene network analysis revealed secondary metabolism as preeminent metabolic pathways for heat tolerance in hard fescue. Grass Research 1: 12 doi: 10.48130/GR-2021-0012

-
{kind=link}