








-
The genus Cyathus (Nidulariaceae, Nidulariales) was first introduced by Haller[1] and later was adopted by Persoon[2], typified by C. striatus (Huds.) Willd. Cyathus together with Crucibulum Tul. & C. Tul., Mycocalia J.T. Palmer, Nidula V.S. White, and Nidularia Fr., are commonly known as bird's nest fungi due to their cup-like basidiomata resembling bird nest and lenticular periodioles resembling eggs[3−5]. It is characterized by having deeper or cuped, inverted bell-like basidiomata covered with shaggy or tomentose hairs on the outside; peridium composed of three layers of tissues, inside peridium filled with a number of dark-colored, small, hard lentil-shaped peridioles attached with funicular cords; colorless, thin-walled or thick-walled, smooth basidiospores[3,6−11]. The species of Cyathus are saprobic, usually growing in decaying wood, on manure or directly on soil are a cosmopolitan group and have a rich diversity related to the high diversity of plants growing in boreal, temperate, subtropical, and tropical regions[3,5,12−14]. Both MycoBank database (
www.MycoBank.org ; 23 June 2022) and Index Fungorum (www.indexfungorum.org ; 23 June 2022) register 204 specific and infraspecific names in the genus Cyathus, but the actual number of species are about 60[15], including 35 species from China[5,16].Molecular systematic studies of the genus Cyathus have been carried out previously[14,17,18]. An overview of the phylogeny of the Agaricales presented based on a multilocus analysis of a six-gene region supermatrix revealed that the family Nidulariaceae was sister to Cystodermateae, in which Cyathus striatus and Crucibulum laeve grouped together within Nidulariaceae[17]. Phylogenetic relationships within the genus Cyathus (bird's nest fungi) were investigated with neighbor joining, maximum likelihood, weighted maximum parsimony and MrBayes analyses of the internal transcribed spacers (ITS) and large subunit (LSU) of ribosomal DNA sequences datasets, in which the morphological characters of the peridium plications and variations in peridium hair anatomy, peridiole structure and fruit-body color were not supported by the molecular data, while the ITS and LSU datasets supported the recognition of three infrageneric groups herein named the ollum, pallidum and striatum groups[18]. Phylogenetic analyses based on ITS and LSU ribosomal DNA sequences revealed that three taxa C. cheliensis, C. gansuensis, and C. megasporus were respectively accepted as synonyms of C. limbatus, C. pygmaeus, and C. poeppigii[5]. On the basis of the morphological and molecular data, Martin et al.[19] discussed affinities among Cyathus species, which showed that this group formed a monophyletic group with high support. Phylogenetic reconstruction of Cyathus species based on alignment of 641 nucleotides of the ITS region indicated that three new species as C. batistae and C. apiculatus, C. pedunculatus were proposed, and discussed relationships with other species of Cyathus[20]. Phylogenetic relationships of bird's nest fungi investigated with four commonly used loci (ITS, LSU, translation elongation factor (TEF), and RNA polymerase II second largest subunit (RPB2)) revealed that the family Nidulariaceae was resolved as a monophyletic group with Squamanitaceae as a potential sister taxon, and suggested that species concepts needed to be revisited and refined throughout Nidulariaceae and several bird's nest fungi species had global geographical distributions, whereas others may have more limited ranges, and the basic morphological characters of bird's nest fungi had likely been lost or gained multiple times[21]. The phylogenetic study using five loci (ITS, LSU, SSU, translation elongation factor 1-alpha (TEF1) and RPB2) revealed that a new genus Retiperidiolia to accommodate this phylogenetically and morphologically unique bird's nest fungus lineage, in which Cyathus formed a monophyletic lineage and then was sister to the genus Retiperidiolia[14].
-
The fresh fruiting bodies of the bird's nest fungi were collected from Wenshan (Yunnan Province, P. R. China). The fresh specimens were dried in an electric food dehydrator at 40 °C, then sealed and stored in an envelope bag and deposited in the herbarium of the Southwest Forestry University (SWFC, Kunming, Yunnan Province, P.R. China).
The macromorphological descriptions were based on field notes and photos captured in the field and lab. The macromorphological descriptions are based on Brodie[3]. The micromorphological data were obtained from the dried specimens and observed under Nikon Eclipse E100 light microscope following the methods of Zhao & Wu[22]. Color terms follow Kornerup & Wanscher[23]. Drawings were made with the aid of a fungus plotter. The measurements and drawings were made from slide preparations stained with Cotton Blue (0.1 mg aniline blue dissolved in 60 g pure lactic acid), Melzer's reagent (3 g potassium iodide, 1 g crystalline iodine, 44 g chloral hydrate, aq. dest. 40 ml) and 5% potassium hydroxide. In presenting spore size data, 5% of the measurements excluded from each end of the range are shown in parentheses. The following abbreviations are used: KOH = 5% potassium hydroxide; CB = cotton blue; CB– = acyanophilous; IKI = Melzer's reagent; IKI– = non-amyloid and non-dextrinoid; L = mean spore length (arithmetic average of all spores); W = mean spore width (arithmetic average of all spores); Q = L/W ratio; n = number of spores/measured from a given number of specimens.
Molecular procedures and phylogenetic analysis
-
The CTAB rapid plant genome extraction kit-DN14 (Aidlab Biotechnologies Co., Ltd, Beijing, China) was used to obtain genomic DNA from dried fungal specimens, according to the manufacturer's instructions. The ITS region was amplified with the primer pair ITS5 and ITS4[24]. The PCR cycling procedure for ITS was as follows: initial denaturation at 95 °C for 3 min, followed by 35 cycles at 94 °C for 40 s, 58 °C for 45 s and 72 °C for 1 min, and a final extension of 72 °C for 10 min. The PCR products were purified and directly sequenced at Kunming Tsingke Biological Technology Limited Company (Yunnan Province, P.R. China). All newly generated sequences were deposited in GenBank (Table 1).
Table 1. List of species, specimens, and GenBank accession numbers of ITS sequences used in this study.
Species name Sample no. GenBank
accession no.References Crucibulum laeve SWFC 21261 DQ463357 Zhao et al.[18] Cyathus africanus DAOM 200370[T] DQ463347 Zhao et al.[18] C. albinus UFRN-Fungos 2239 KY176371 Accioly et al.[25] C. amazonicus URM 80036[T] KY495280 Accioly et al.[25] C. amazonicus UFRN-Fungos 2798 KY176375 Accioly et al.[25] C. annulatus MichaelKuo-8200901 MT444076 Kraisitudomsook et al.[21] C. apiculatus UFRN:Fungos 1448 KT365516 da Silva et al.[20] C. aurantogriseocarpus UFRN:Fungos:2798 KX966026 da Cruz et al.[26] C. badius KH:JPN15-1321 KX906250 da Cruz et al.[27] C. batistae UFRN:Fungos 1449 KT365515 daSilva et al.[20] C. berkeleyanus SWFC 20789 DQ463355 Zhao et al.[18] C. bulleri DAOMC 195062 MK020156 Vats & Mishra[28] C. canna CBS 370.80 MH861275 Vu et al.[29] C. colensoi DAOM 200423 DQ463344 Zhao et al.[18] C. crassimurus DAOM 200372[T] DQ463350 Zhao et al.[18] C. discoideus AB 7831 KY652080 da Cruz[30] C. gansuensis SWFC 20880[T] DQ463348 Zhao et al.[18] C. gansuensis Strain 69 KC869661 da Cruz et al.[27] C. gracilis AB7873 KY652081 da Cruz[30] C. hookeri SWFC 20799 DQ463346 Zhao et al.[18] C. hortensis UFRN:Fungos:1819 KX906252 da Cruz et al.[27] C. ibericus AH:48138 KX858598 Crous et al.[31] C. ibericus AH:48137[T] KX858597 Crous et al.[31] C. intermedius UFRN:Fungos 1033 KT365519 da Silva et al.[20] C. jiayuguanensis SWFC 20846[T] DQ463341 Zhao et al.[18] C. lignilantanae MA Fungi 87327 NR_154827 da Cruz et al.[27] C. limbatus UFRN-Fungos 2238 KY176373 Accioly et al.[25] C. magnomuralis UFRN:Fungos:1817 KX906251 da Cruz et al.[27] C. minimus AB7868 KY652082 da Cruz[30] C. novae-zeelandiae PDD-76442 MT444096 Kraisitudomsook et al.[21] C. olla PDD-86833 MT444086 Kraisitudomsook et al.[21] C. olla BPI 727227 DQ463345 Zhao et al.[18] C. pallidus KKUITN2 KU202745 Sutthisa & Sanoamuang[32] C. pallidus KKUITN3 KU202751 Sutthisa & Sanoamuang[32] C. parvocinereus UFRN:Fungos:1814 KX906253 da Cruz et al.[27] C. pedunculatus UFRN:Fungos 403 KT365518 da Silva et al.[20] C. poeppigii cp-457 KT962176 da Silva et al.[20] C. pyristriatus MFLUCC:14-0770 KU865513 Richter et al.[33] C. renweii SWFC 201406[T] DQ463352 Zhao et al.[18] C. setosus DAOM 200815[T] DQ463349 Zhao et al.[18] C. stercoreus NK-08 MT444037 Kraisitudomsook et al.[21] C. stercoreus DM4 KY706156 Hay et al.[34] C. striatus NK-61 MT444056 Kraisitudomsook et al.[21] C. subglobisporus BBH-14815 MT444063 Kraisitudomsook et al.[21] C. subglobisporu BBH18348 EF613553 Zhao et al.[5] C. triplex SWFC 21077 DQ463353 Zhao et al.[18] C. uniperidiolus AMH:10196 MN398297 Boonmee et al.[35] C. wenshanensis CLZhao 20202[T] ON795104 This study Nidula niveotomentosa SWFC 3000 DQ463358 Zhao et al.[18] Sequencher 4.6 (GeneCodes, Ann Arbor, MI, USA) was used to assemble and edit the generated sequence reads. Sequences were aligned in MAFFT 7 (
https://mafft.cbrc.jp/alignment/server/ ) using the 'G-INS-I' strategy and manually adjusted in BioEdit[36]. Crucibulum laeve (Huds.) Kambly and Nidula niveotomentosa (Henn.) Lloyd were selected as an outgroup for the phylogenetic analysis of the ITS phylogenetic tree[25].Maximum parsimony (MP), Maximum Likelihood (ML) and Bayesian Inference (BI) analyses were applied to the ITS dataset sequences. Approaches to phylogenetic analyses followed[22]. MP analysis was performed in PAUP* version 4.0b10[37]. All of the characters were equally weighted and gaps were treated as missing data. Trees were inferred using the heuristic search option with TBR branch swapping and 1000 random sequence additions. Maxtrees were set to 5000, branches of zero length were collapsed and all most-parsimonious trees were saved. Clade robustness was assessed using bootstrap (BT) analysis with 1,000 replicates[38]. Descriptive tree statistics tree length (TL), the consistency index (CI), the retention index (RI), the rescaled consistency index (RC) and the homoplasy index (HI) were calculated for each most-parsimonious tree generated. ML was inferred using RAxML-HPC2 through the Cipres Science Gateway (
www.phylo.org )[39]. Branch support (BS) for ML analysis was determined by 1000 bootstrap replicates and evaluated under the gamma model.MrModeltest 2.3[40] was used to determine the best-fit evolution model for the dataset for Bayesian Inference (BI). Bayesian Inference was performed with MrBayes 3.1.2 with a general time reversible (GTR+I+G) model of DNA substitution and a gamma distribution rate variation across sites[41]. Four Markov chains were used in each of two runs from random starting trees for 1.5 million generations (Fig. 1), with trees and parameters sampled every 100 generations. The first quarter of the generations were discarded as burn-in. A majority rule consensus tree of all remaining trees and posterior probabilities were calculated. Branches were considered significantly supported if they received maximum likelihood bootstrap value (BS) of > 70%, a maximum parsimony bootstrap value (BT) of > 70%, or Bayesian posterior probabilities (BPP) of > 0.95.
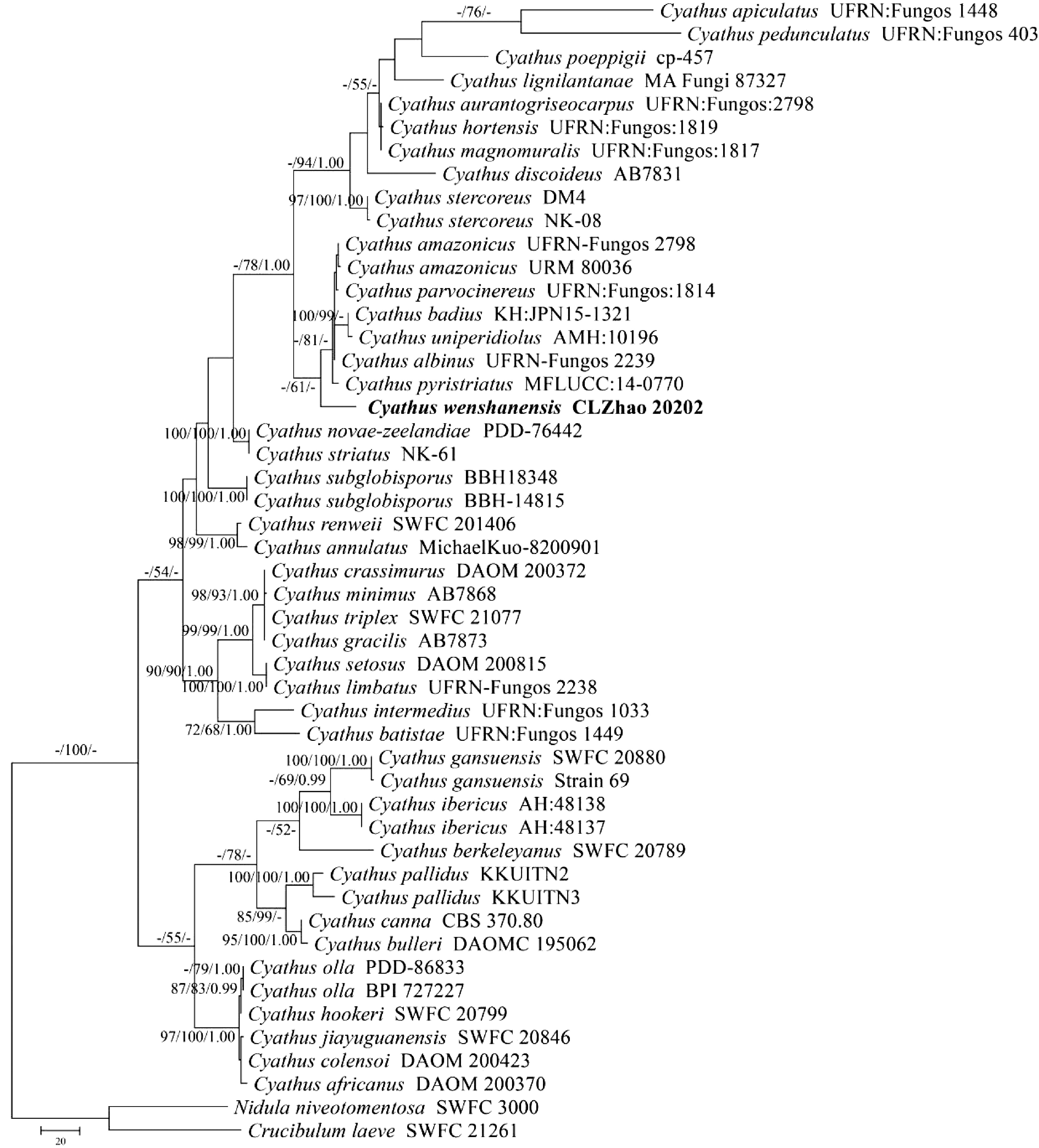
Figure 1.
Maximum parsimony strict consensus tree illustrating the phylogeny of the new species and related species in genus Cyathus based on ITS sequences. Branches are labelled with maximum likelihood bootstrap value > 70%, parsimony bootstrap value > 50% and Bayesian posterior probabilities > 0.95, respectively. The present species are in bold.
-
The ITS dataset (Fig. 1) included sequences from 49 fungal specimens representing 42 species. The dataset had an aligned length of 805 characters, of which 270 characters were constant, 232 were variable and parsimony-uninformative, and 303 parsimony-informative. The MP analysis yielded one equally parsimonious trees (TL = 1211, CI = 0.6474, HI = 0.3526, RI = 0.7855, RC = 0.5086). Best model for the ITS dataset estimated and applied in the Bayesian analysis: GTR+I+G, lset nst = 6, rates = invgamma; prset statefreqpr = dirichlet (1,1,1,1). The bayesian and ML analyses resulted in a similar topology as MP analysis, with an average standard deviation of split frequencies = 0.009975 (BI), and the effective sample size (ESS) across the two runs was double the average ESS (avg ESS) = 248. The phylogenetic tree (Fig. 1) inferred from ITS sequences revealed that C. wenshanensis nested within the genus Cyathus, in which it formed a monophyletic lineage and grouped with C. albinus, C. amazonicus, C. badius, C. parvocinereus, C. pyristriatus and C. uniperidiolus.
Taxonomy
-
Cyathus wenshanensis Z.Y. Duan & C.L. Zhao, sp. nov. Figs 2−5.

Figure 2.
Basidiomata of Cyathus wenshanensis. (a) Basidiomata, (b) outer corving of peridium. Scale bars: (a) = 1 cm, (b) = 1 mm.
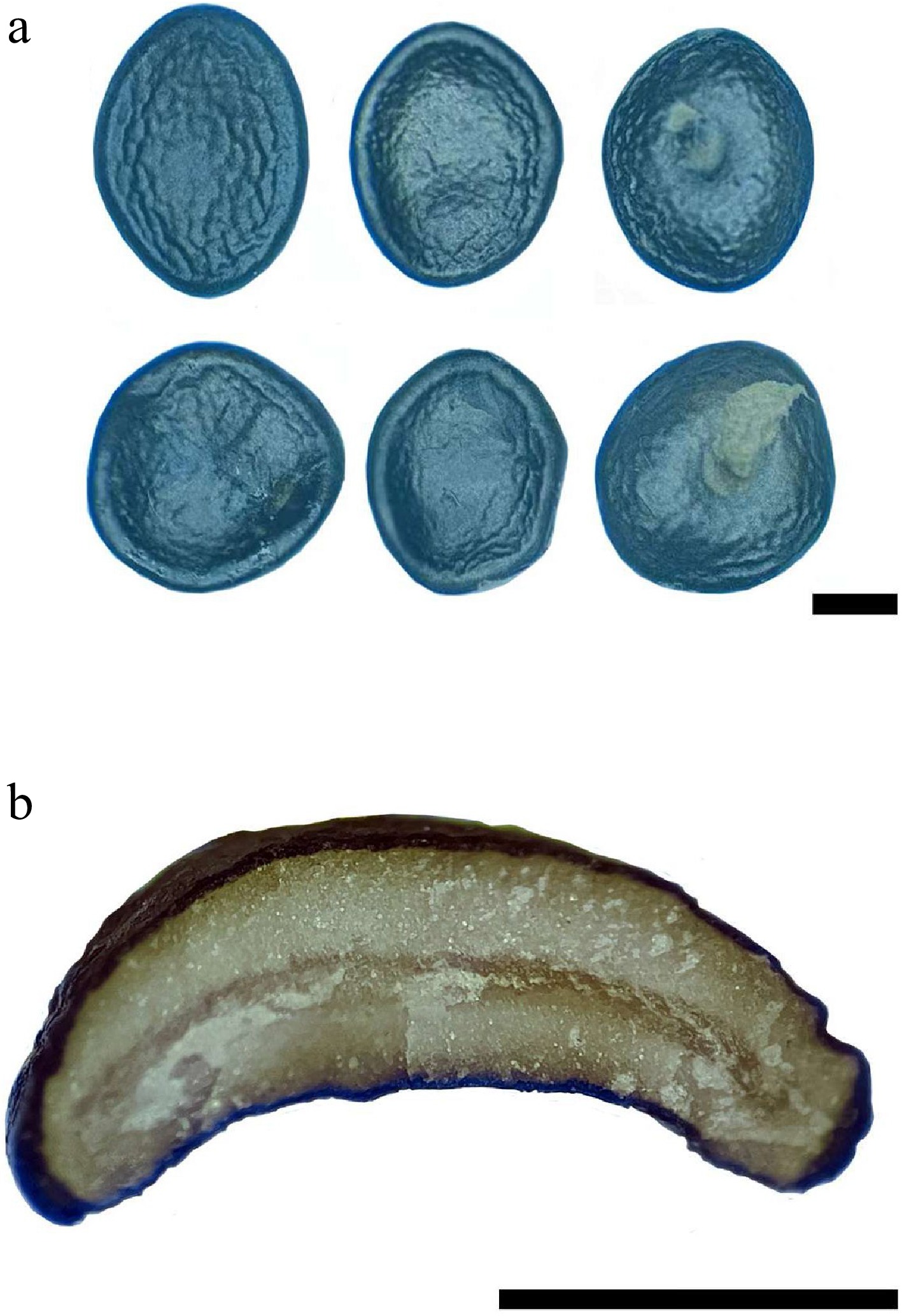
Figure 3.
Peridiole of Cyathus wenshanensis. (a) Peridioles with funicular cord, (b) transversal section of peridiole showing single-layered cortex. Scale bars: (a) = 1 mm, (b) = 1 mm.
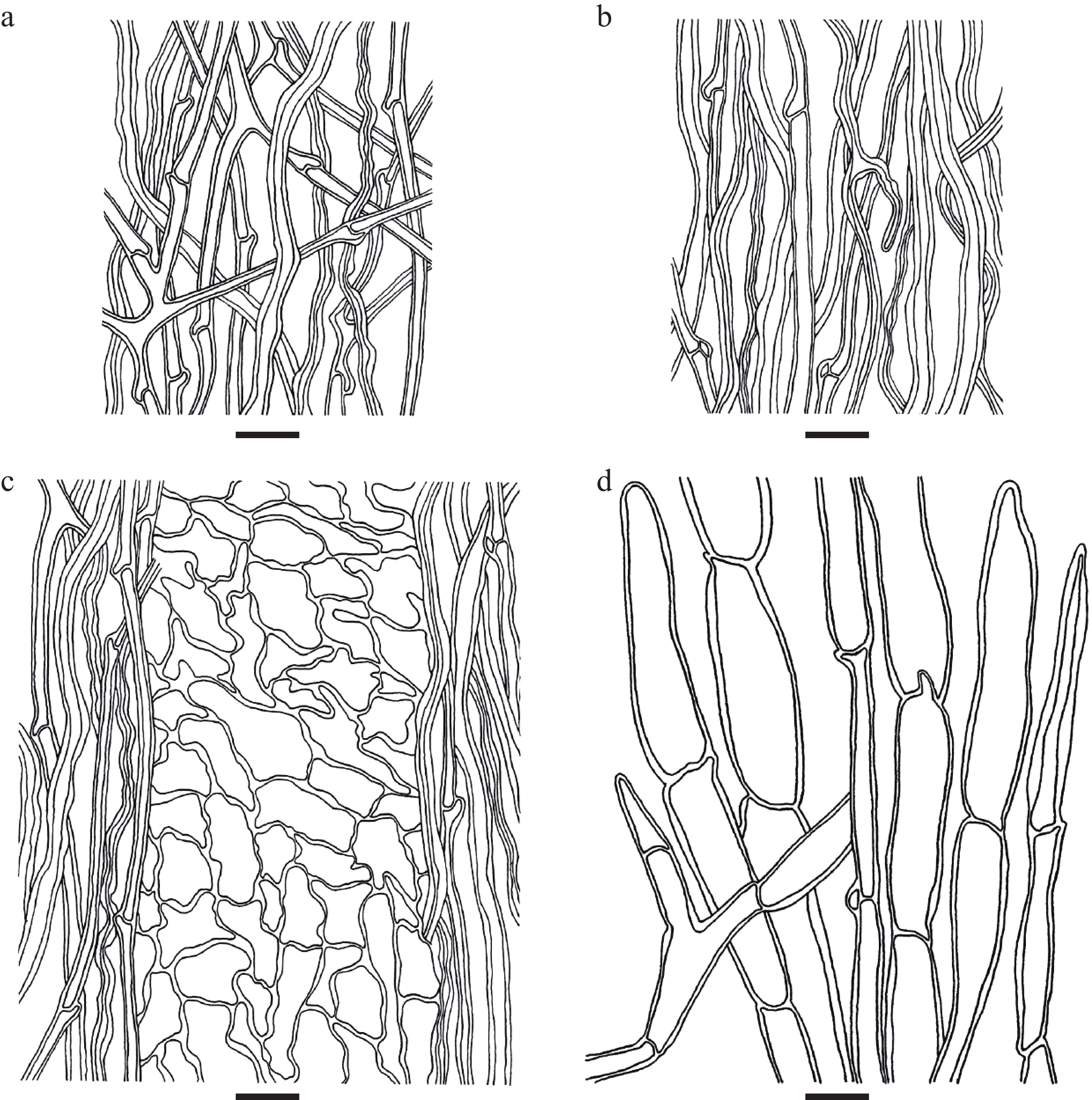
Figure 4.
Microscopic structures of Cyathus wenshanensis. (a) Outer wall of peridium, (b) inner wall of peridium, (c) three-layered peridium, (d) the structure of the hair. Scale bars: (a)–(d) = 10 μm.

Figure 5.
Microscopic structures of Cyathus wenshanensis. (a) Basidiospores. (b) Funicular cord. (c) The internal structure of peridiole. Scale bars: (a)–(c) = 10 μm.
Index Fungorum number: IF844702; Facesoffungi number: FoF12564
Etymology – wenshanensis (Lat.): referring to the provenance (Wenshan) of the type specimens.
Basidiomata obconical to cupulate, 5–15 mm high, 5–10 mm wide at the mouth, without expanding at the top or tapering abruptly at the base; the base usually attached to the substrate by a slightly conspicuous emplacement, brown (5E6) to beige (4C3); exoperidium brown (5E6), hirsute, external wall striate near the mouth, 0.4–0.7 mm between folds, covered with brown (5E6) to dark brown (7F6), irregular and flexible tufts of hair; hair hyphae with clamp connections, colorless, thick-walled (wall up to 0.5–2 μm thick), 3.5–13.5 µm in diameter; endoperidium greyish brown (8F3) to black brown (7F4), conspicuously striate with 0.4–0.8 mm between the groves; mouth finely fimbriate; peridium walls consist of three different layers: (1) outer wall layer, hyphal system trimitic, CB–, IKI–, tissues unchanged in KOH; generative hyphae with clamp connections, colorless to pale brown, slight thick-walled, frequently branched, 1.5–4 µm in diameter; skeletal hyphae colorless to pale brown, thick-walled, unbranched, 2–4 µm in diameter; binding hyphae colorless to pale brown, thick-walled, unbranched, 1.5–2.5 µm in diameter; (2) inner wall layer, hyphal system trimitic, CB–, IKI–, tissues unchanged in KOH; generative hyphae with clamp connections, colorless to pale brown, slight thick-walled, rarely branched, 2–4 µm in diameter; skeletal hyphae colorless to pale brown, thick-walled, unbranched, 2.5–4.5 µm in diameter; binding hyphae colorless to pale brown, thick-walled, rarely branched, 1.5–3 µm in diameter; (3) pseudopare-chymatous layer.
Peridioles depressed, shiny, angular to irregular, suborbicular, broadly ellipsoid to ovoid, dark grey (8F1) to black (6F3), surface smooth to wrinkled, tunica present, often inconspicuous, cortex single-layered, 2.5–3.5 × 2–3 mm; funicular cord present, funiculus hyphae with clamp connections, thick-walled, unbranched, pale yellowish, 1–3.5 µm in diameter; hyphal system of peridiole middle dimitic, generative hyphae with clamp connections, colorless, thin-walled, frequently branched, with oil drops inside, 1–3 µm in diameter, CB–, IKI–, tissues unchanged in KOH; skeletal hyphae colorless, slight thick-walled, unbranched, with oil drops inside, 1.5–4 µm in diameter, CB–, IKI–, tissues unchanged in KOH.
Basidiospores subglobose, elliptical to ellipsoid-elongate, colorless, smooth, thick-walled (wall up to 1–5 μm thick), CB–, IKI–, with inclusions or oil-like globule, without apiculus, (10–)11–21(–22) × 9–14(–15) µm, L = 16.34 µm, W = 11.51 µm, Q = 1.4 (n = 60/1). Basidia not observed.
Known distribution – Thus far known only from China.
Material examined – China. Yunnan Province, Wenshan, Pingba Town, Huguangqing Village, 23.26°N, 104.06°E, on the fallen branch of angiosperm, 12 August 2020, collected by C.L. Zhao. Specimen voucher number: CLZhao 20202 (SWFC 020202).
-
In the present study, C. wenshanensis sp. nov. is described based on the phylogenetic analyses and morphological characteristics.
Phylogenetically, the molecular systematics and taxonomic overview of the bird's nest fungi revealed that the family Nidulariaceae was resolved as a monophyletic group with Squamanitaceae as a potential sister taxon, in which Cyathus and Crucibulum each formed its own independent and well-supported clade, and Nidula and Nidularia formed a clade together, but each genus is polyphyletic[21]. In the present study, C. wenshanensis nests within the genus Cyathus located in the family Nidulariaceae, in which it forms a monophyletic lineage and then groups with taxa C. albinus, C. amazonicus, C. badius, C. parvocinereus, C. pyristriatus and C. uniperidiolus. However, morphologically C. albinus differs from C. wenshanensis by having the golden blond to dark blond exoperidium, brownish gray peridioles with double-layered cortex, and basidiospores with conspicuous apiculous[25]. C. amazonicus differs in having very dark brown to grayish dark brown exoperidium and and gray, shiny endoperidium[42]. C. badius differs in having the smooth exoperidium, light brown to orange endoperidium, ovoid basidiospores[43]. C. parvocinereus differs in having the campanulate, smaller basidiomata (4–7 × 3.5–5 mm), pearl grey to brightness silvery endoperidium and greyish brown to grey peridioles with double-layered cortex[44]. C. pyristriatus differs in its clavate basidiomata with yellowish-brown or buff exoperidium, grey to dark grey endoperidium, greyish-brown peridioles, and ovoid basidiospores[45]. C. uniperidiolus distinct from C. wenshanensis in having the globose to sub-globose basidiomata with serrate margin at mouth, smooth peridium walls, and globose, smooth peridioles[35] (see Table 2).
Table 2. The comparison among Cyathus wenshanensis and phylogenetically related species.
C. wenshanensis C. albinus C. amazonicus C. badius C. parvocinereus C. pyristriatus C. uniperidiolus Basidiomata Size (high
× wide)5–15 × 5–10 mm 6–8.5 × 5–6.52 mm 9–11 × 5–7 mm 8–10 × 5–8 mm 4–7 × 3.5–5 mm 5.5–7 × 4–6 mm 2–12 × 2–3.5 mm Shape Obconical to
cupulateInfundibuliform Obconical Infundibuliform Campanulate Clavate to broadly obconic Globose to sub-globose Exoperidium Colour Brown Golden blond to dark blond Very dark brown to grayish dark brown Brown Reddish brown Yellowish-brown or buff Dark brown Surface Strigose Tufts; striate, 0.4–0.7 mm Strigose tufts; striate; 0.3–0.5 mm Strigose tufts; striate Shaggy, wooly tufts; smooth to striate;
0.3 mmStrigose tufts; striate; 0.4–0.5 mm shaggy or fluffy hairs Smooth to velvety Mouth Fimbriate Fimbriate Fimbriate Fimbriate Fimbriate Serrate Endoperidium Colour Greyish brown to black brown Grayish brown Gray to brownish gray Light brown to orange Platinum Grey to dark grey Dark brown Surface Striate, 0.4–0.8 mm Striate, 0.3–0.6 mm Striate Smooth to minutely striate; 0.5 mm Striate; 0.5 mm Striate Smooth Peridioles Shape Lentil-shaped Lentil-shaped Lentil-shaped Lentil-shaped Lentil-shaped Lentil-shaped Globose Size 2–3.5 mm 1.8–2.6 mm 1.7–3 mm 2–2.5 mm 1–2 mm 3–3.5 mm 2–2.5 mm Color Dark grey to black Brownish gray Dark gray Light grey to black Greyish brown to grey Greyish-brown to dark grey Black Cortex Single layered Double layered Single layered Single layered Double layered Double layered Basidiospores Shape Subglobose, elliptical to ellipsoid-elongate; apiculum absent Ovoid to ellipsoid; apiculus present Subglobose to broadly ellipsoid Subglobose, ovoid to elliptical elliptical, globose; apiculum absent Ovoid, subglobose, ellipsoid to broadly ellipsoid Oval, sub-globose, broadly ellipsoid to ellipsoid-elongate Size 11–21 × 9–14 µm 14.8–20 × 10.4–14.3 µm 14–19 × 12–16 µm 13–19 × 9–11 µm 11.43–17.78 × 9–15.24 µm 14–17 × 8–10 µm 14.2–28.7 × 11.7–23.7 µm Walls 1–5 μm thick 0.8–1.3 μm thick thick-walled 1.9–3.2 μm thick 2–3.5 μm thick 1.5–3 μm thick thick-walled Distribution China Brazil Amazon rainforest Japan, Brazil Brazil Thailand India Reference Present study Accioly et al.[25] Trierveiler-Pereira et al.[42] da Cruz et al.[43] da Cruz & Baseia[44] Hyde et al.[45] Boonmee et al.[35] Morphologically, six taxa of Cyathus as C. apiculatus, C. hortensis, C. limbatus, C. lignilantanae, C. pedunculatus, and C. poeppigii are similar to C. wenshanensis on the basis of the character by having the obvious stripes on the inner and outer walls of peridium. However, C. apiculatus differs from C. wenshanensis by the basidiomata being expanded at the mouth and abruptly tapering to the base, silvery endoperidium, smaller peridioles (1–1.5 × 1.5–2 mm), and longer basidiospores (22–37 × 10–22 μm)[20]; C. hortensis is distinguished from C. wenshanensis by its basidiomata constricting abruptly at the base and forming a slender stipe, cinnamon exoperidium, smaller peridioles (1.2–2 × 1–1.5 mm) with double-layered cortex, and ovoid, wider basidiospores (17–34 × 13–20 μm)[44]; C. limbatus differs from C. wenshanensis by its double-layered peridioles, and basidiospores with apiculus[46]; C. lignilantanae is different from C. wenshanensis by having a reddish brown exoperidium, brownish grey to greyish brown, smaller peridioles (2.1–2.3 × 1.8–2 mm) with double-layered cortex[19]; C. pedunculatus is separated from C. wenshanensis by having the basidiomata abruptly tapering in the base forming a conspicuous pedicel, pale yellow to dark blond exoperidium, double-layered cortex, brownish grey, smaller peridioles (1.5–2 × 1–1.5 mm), and larger basidiospores (25–34 × 22–29 μm)[20]; C. poeppigii is distinguished from C. wenshanensis by having the narrowly obconical basidiomata with incurved mouths and a slender stipe at the base, and dark brown, smaller peridioles (1.5–2 mm) with double cortex, and larger basidiospores (30–45 × 18–30 μm)[47].
Several taxa, Cyathus batistae, C. discoideus, C. gracilis, C. hookeri, C. magnomuralis, C. renweii and C. triplex are similar to C. wenshanensis based on the character having the fimbriate of basidiomata mouth. However, C. batistae differs from C. wenshanensis by its expanded mouth of basidiomata, with the stipe, smooth exoperidium wall, double-layered cortex peridioles, and smaller basidiospores (9–13 × 5–8 μm) with apiculus[20]; C. discoideus differs from C. wenshanensis by having grey brown, smaller peridioles (1.56–2.16 × 1.41–1.74 mm)[30]; C. gracilis is distinguished from C. wenshanensis by the basidiomata with slender base, umber to rusty outer surface of peridium, double-layered cortex peridioles, and basidiospores with apical notch[48]; C. hookeri differs from C. wenshanensis by its smooth peridium walls, and smaller basidiospores (9–13 × 5–8 μm)[49]; C. magnomuralis is distinguished from C. wenshanensis by having the dark blond exoperidium, smaller peridioles (1–1.5 × 1–1.5 mm) with double-layered cortex, and ovoid, larger basidiospores (27–49 × 23–41 μm) with small apiculus[44]; C. renweii differs from C. wenshanensis by its greyish peridioles with the brown tunica, and longer basidiospores (21–31 × 10.5–13.5 μm)[50]; C. triplex is separated from C. wenshanensis by its smaller basidiomata (5–8 × 4.5–5 mm) with the slender base orbicular, flattened peridioles with double-layered cortex, and basidiospores with the apical notch[25].
Eight species of the genus Cyathus as C. africanus, C. colensoi, C. gansuensis, C. ibericus, C. jiayuguanensis, C. novae-zeelandiae, C. olla, and C. pallidus are similar to C. wenshanensis in light of the characteristics of having single-layered cortex peridioles. However, C. africanus differs from C. wenshanensis by its peridium walls with woolly hairs, silvery peridioles, and broadly ovate, smaller basidiospores (8.5–12 × 6.5–8.5 μm) with apiculus[51]; C. colensoi differs from C. wenshanensis by its smooth peridium walls, and ovoid, smaller basidiospores (8.5–11.5 × 7–8.5 μm)[30,49]; C. gansuensis differs from C. wenshanensis by its narrow base basidiomata with grayish to dark smoke-gray interior, grayish, smaller peridioles (1.5–2 × 0.8–1.5 mm), and ovoid basidiospores[52]; C. ibericus differs in its whitish to pale brownish grey external peridium with woolly hairs, smaller peridioles (0.8–1.2 mm diam), and ovoid, smaller basidiospores (7–9 × 5–6 μm)[30]; C. jiayuguanensis differs from C. wenshanensis by its basidiomata with the short stipe, smoke-gray peridioles, and ovoid, smaller basidiospores (8–11.5 × 7–8.5 μm)[52]; C. novae-zeelandiae differs in C. wenshanensis by its basidiomata abruptly constricted into a stipe, peridioles with white tunica, and smaller basidiospores (11–13 × 5–6 μm)[4,53]; C. olla differs from C. wenshanensis by its peridium with tomentose outside, silver, smooth inside, pure silver peridioles, and the smaller basidiospores (9.8–11.2 × 6.4–8 μm)[54]; C. pallidus differs from C. wenshanensis by its smaller basidiospores (6.8–14.5 × 6–8.1 μm)[32].
Cyathus annulatus, C. aurantogriseocarpus, C. minimus, and C. stercoreus are similar to C. wenshanensis inferred from the characteristics of having thick-walled basidiospores without apiculus. However, C. annulatus is separated from C. wenshanensis by its expanded peridium at the top, ochraceous-tawny exoperidium, pale buff inner surface, subtriangular peridioles, and the striking dark-brown ring at the mouth[55]; C. aurantogriseocarpus differs from C. wenshanensis by the orange-grey exoperidium with long tomentum, brownish grey, smaller peridioles (1.5–1.75 × 1.2–1.5 mm) with double-layered cortex, and larger basidiospores (32.5–47 × 22.5–28.5 μm)[26]; C. minimus differs from C. wenshanensis by the clay brown exoperidium, yellowish brown endoperidium, and reddish brown, coffee or brown tobacco, smaller peridioles (1.3–1.37 × 1.13–1.23 mm)[30]; C. stercoreus differs in its smooth peridium walls, double-layered cortex peridioles and larger basidiospores (30–41 × 25–31 μm)[4].
The family Nidulariaceae is a characteristic group of Agaricomycetes (Basidiomycota), which has a number of macrofungi based on a result of the morphological, phylogenetic and cytological studies in China[56,57], but the species diversity of macrofungi are still not well known, especially in subtropical and tropical areas of the country[58−61]. The new species, Cyathus wenshanensis is from the subtropics. Therefore, the present paper enriches the fungal diversity in the Chinese ecosystem, and it is likely that more new taxa will be found after further fieldwork and molecular analyses.
In addition, the results of BLAST queries in NCBI based on ITS separately showed the sequences producing significant alignments descriptions: in ITS blast results, the top ten taxa are C. pyristriatus (Max score 1116; Total score 1116; Query cover 92%; E value 0.0; Ident 95.47%), C. parvocinereus (Maximum record descriptions: Max score 1081; Total score 1081; Query cover 89%; E value 0.0; Ident 95.71%), C. amazonicus (Maximum record descriptions: Max score 1077; Total score 1077; Query cover 87%; E value 0.0; Ident 96.11%), C. uniperidiolus (Maximum record descriptions: Max score 1075; Total score 1075; Query cover 91%; E value 1e-78; Ident 94.84%), C, albinus (Max score 1053; Total score 1053; Query cover 85%; E value 0.0; Ident 96.01%), and C. badius (Maximum record descriptions: Max score 1000; Total score 1000; Query cover 83%; E value 0.0; Ident 95.15%).
The research was supported by the National Natural Science Foundation of China (Project No. 32170004), Yunnan Fundamental Research Project (Grant No. 202001AS070043) and received support from Yunnan Academy of Biodiversity, Southwest Forestry University.
-
The authors declare that they have no conflict of interest.
- Copyright: © 2022 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Duan ZY, Yu J, Zhao CL. 2022. Molecular phylogeny and morphology reveal a new wood-rotting fungal species, Cyathus wenshanensis sp. nov. from the Yunnan-Guizhou Plateau. Studies in Fungi 7:8 doi: 10.48130/SIF-2022-0008 

Molecular phylogeny and morphology reveal a new wood-rotting fungal species, Cyathus wenshanensis sp. nov. from the Yunnan-Guizhou Plateau
- Received: 05 July 2022
- Accepted: 09 September 2022
- Published online: 10 October 2022
Abstract: A new species of bird's nest fungus, Cyathus wenshanensis is proposed based on a combination of the morphological and molecular evidence. It is characterised by the obconical to cupulate basidiomata covered with hirsute hairs, striations on the outer and inner surface of the peridium, funicular peridioles, a trimitic hyphal system of peridium with generative hyphae having clamp connections, a dimitic hyphal system of peridiole middle, and subglobose, elliptical to ellipsoid-elongate, thick-walled basidiospores. Sequence of the internal transcribed spacers (ITS) gene region was generated, and the phylogenetic analysis was performed with maximum likelihood, maximum parsimony and Bayesian inference methods. The phylogenetic analyses inferred from ITS dataset indicated that C. wenshanensis nested within the genus Cyathus, in which it formed a monophyletic lineage and grouped with C. albinus, C. amazonicus, C. badius, C. parvocinereus, C. pyristriatus and C. uniperidiolus.
-
Key words:
- Bird's nest fungi /
- Nidulariaceae /
- molecular phylogeny /
- taxonomy /
- Yunnan Province