









-
Lilies are perennial bulbous flowering plants in the Liliaceae family with fragrant and brightly colored blossoms highly desired by consumers. Lilies are widely used as cut flowers, and are planted in parks and courtyards. There are many varieties of lily each with unique characteristics. Asian hybrid lilies are rich in color but lacking fragrance. The flower of the Oriental hybrid lily is large and beautiful with an intense aroma, but the color of the flower is relatively simple. The main goal of lily breeding is to cultivate richly colored and fragrant lilies[1].
Brightly-colored plants tend to attract pollinators and resist abiotic and biological stresses[2−4]. Some cultivar lily petals are rich in anthocyanins, which are secondary metabolites in the biosynthesis pathway of flavonoids. Anthocyanins directly influence the color of flowers as a result of the coordinated expression of structural genes, such as CHS, CHI, F3H, F3 'H, DFR, ANS, 3GT, etc., which encode enzymes in the flavonoid biosynthesis pathway[5]. These structural genes (TFs) can be regulated by the MBW complex, which is composed of the transcription factors MYB, bHLH, and WD[6]. The flavonoid biosynthesis pathway related to anthocyanin synthesis has been extensively studied. Numerous structural and regulatory genes involved in the accumulation of anthocyanins have been found and isolated in model plants[7,8] and fruit cultivars[9,10], and the specific process of the anthocyanin biosynthesis pathway has been defined. The genes that influence lily flower color are currently being explored. The MBW complex can regulate the coloration in lily petals as LhMYB12, LhMYB12-lat, LrMYB15, and LhMYB18 affect anthocyanin pigmentation in the petals, ovary, bud, and spots, respectively[11−13]. However, SPL[14,15], MADS-box[16,17], NAC[18−20], and ARF[21] can also affect anthocyanins synthesis in many plants by regulating the expression of the MBW complex. For example, in blueberry and Litchi chinensis, the miR156-SPL module coordinates the accumulation of anthocyanins during fruit ripening[14, 15], and a MADS-box gene is involved in the regulation of anthocyanin accumulation in bilberry fruits and sweet potatoes[16, 17], a novel NAC transcription factor regulates anthocyanin accumulation in red-fleshed apples by interacting with MYB[18, 19], NAC can regulate anthocyanin accumulation in Arabidopsis[20], and ARF regulates anthocyanin biosynthesis in apples[21]. Therefore, the regulatory network of anthocyanins in lilies remains to be fully elucidated.
miRNA, which is common in eukaryotes, is a type of endogenous non-coding small RNA with a length of 18−24 nt. miRNAs cleave target mRNAs or prevent gene translation at the post-transcriptional level to influence gene expression[7,22]. miRNAs play a role in a variety of biological processes, including growth and development, stress responses, auxin signaling, and secondary metabolism[23,24], in which it regulates the synthesis and accumulation of secondary metabolites in plants, such as the metabolism of anthocyanin[23,25,26]. For example, miR156[15,24,27], miR396[28], miR828[29−31], and miR858[32] are involved in the biosynthesis of anthocyanins in plants[33].
The miRNA-regulated anthocyanin synthesis pathway has been thoroughly studied in model plants. As positive regulatory factors, miRNA can promote anthocyanin accumulation by inhibiting the expression of negative regulatory factors. MIR858a in Arabidopsis thaliana positively regulates anthocyanin synthesis by inhibiting MyBL2 in seedlings[34]. Overexpression of miR408, a positive regulatory factor, can increase anthocyanin accumulation in Arabidopsis seedlings[35]. Conversely, miRNA can act as a negative regulator to inhibit the accumulation of anthocyanins by inhibiting the expression of positive regulators. Overexpression of miR828 inhibits MYB expression in Arabidopsis thaliana, reducing anthocyanin accumulation[36]. In summary, miRNAs in plants indirectly affect anthocyanin accumulation by targeting and regulating genes in different ways.
Research on microRNAs therefore focuses on model plants and is rarely seen in flowering plants. The identification of the regulatory network of miRNA in lilies is currently incomplete. In the current study, we examined the regulation mechanism of miRNAs on lily coloration using lily petals in different stages as experimental material to perform miRNA sequencing. We then analyzed the expression of miRNA and its target genes in the development process of the lily flower based on existing transcriptome data[37]. We discovered a variety of miRNAs related to anthocyanin biosynthesis in lily petals, providing a foundation for future research.
-
Plants of the lily cultivar Vivian were cultivated in the Shenyang Agricultural University (Shenyang, China) greenhouse. Plant materials included bud stage petals (S1), coloring bud petals (S2) and blooming stage petals (S3) with three biological replicates, and were stored at −80 °C for further study.
Measurement of total anthocyanin content
-
Total anthocyanin content were measured using the method provided by Shin et al.[38]. In brief, the sample powder was first incubated in an acidic methanol solution overnight, then chloroform was added to remove the chlorophyll. The extract's absorbance (530 and 657 nm) was measured, and the formula A530 − 0.33 × A657 was used to analyze the total anthocyanin content.
Identification of anthocyanin metabolites
-
All samples were measured in a random order using the UPLC-MS/MS system. Metabolites were identified and quantified as described by Yin et al.[37].
MicroRNA-seq and analysis
-
The same lily petal samples were used for miRAN-seq and mRNA-seq, including S1, S2, and S3 with three biological replicates. The Illumina Hiseq2000/2500 system was used to perform the miRNA sequencing. To identify known and novel miRNAs, the miRBase 22.0 and RNAfold software were used to analyze unique sequences. The DEMs were screened according to the difference in expression |fold change| > 2). We also analyzed the miRNA target gene in the RNA-seq data using psRNATarget. We used BLASTN to predict target genes. We then analyzed the data using the Gene Ontology (GO) database and KEGG.
The FPKM values of the genes were used for analysis. A heatmap was created after performing a row standardization. The heatmap and Sankey plot of the miRNA and the target genes were generated using the TBtools software[39]. The NCBI project PRJNA649743 hosts the transcriptome datasets created and examined for this investigation.
qRT-PCR and phylogenetic analysis
-
A qRT-PCR and statistical analysis were conducted following Yin et al.[37]. The supplemental table includes a list of the primers used in the study (Supplemental Table S1 & S2). SYBR Green Master Mix was used for qRT-PCR. The reference genes were GAPDH and Actin. The 2−ΔΔCᴛ method was used to calculate gene relative expression[40]. The SPSS 16.0 software package was used to perform statistical analysis. A phylogenetic analysis was conducted using MEGA 5.0 (ML method with 1,000 bootstrap replicates)[41].
Correlation analysis
-
Correlation analyses were performed to obtain the correlation between miRNA and the target genes. Using the 'correlate' function in SPSS, a Pearson correlation analysis was performed between miRNA and the target genes. Then, for visualization, we built a network in Cytoscape[42].
-
A determination of anthocyanin content was performed using three-period petals in S1 (bud stage), S2 (coloring stage), and S3 (blooming stage) to measure the pigmentation in lily petals during development. Each sample contained three replicates of different triennial plants. The color of the S1 petal samples was relatively light compared to the other two samples (Fig. 1a). This parallels the total anthocyanin content measured by anthocyanin content extracted, which was lower in the S1 sample compared to S3 (Fig. 1b). The anthocyanins content in the petals were lower in S1 and higher in S3. The content trend of each cyanidin derivative, including cyanidin, cyanidin 3-O-glucoside, and cyanidin 3-O-rutinoside, was the same as that of the total anthocyanins (Fig. 1c). Overall, the content of total anthocyanins and their components in lily petals gradually accumulated with the development of the petals.
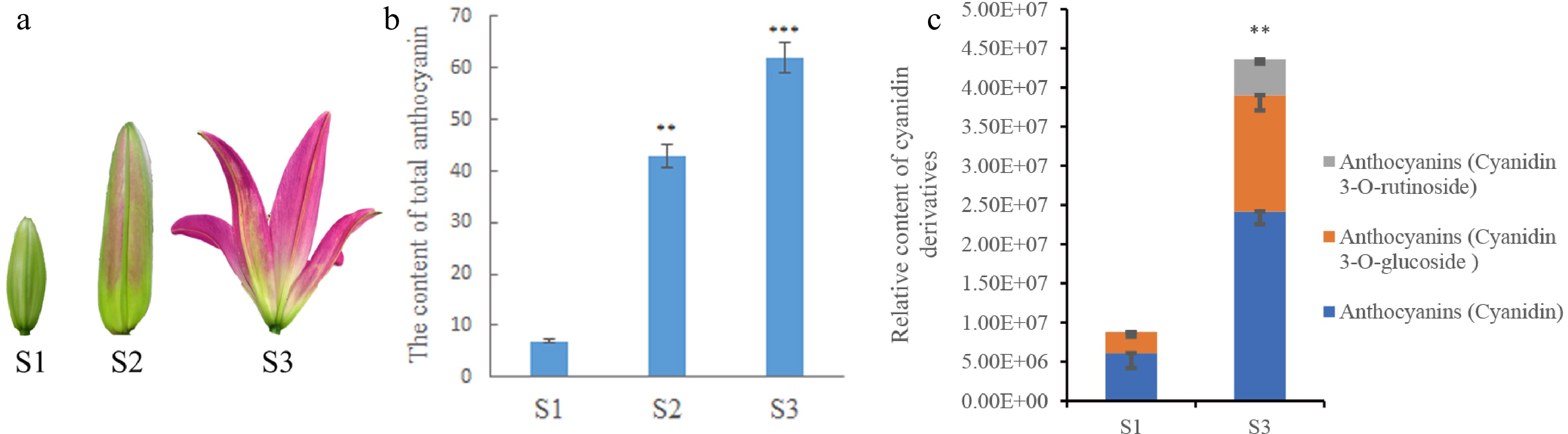
Figure 1.
Determination of anthocyanin content in lily petals. (a) Lily petals in different periods. (b) Anthocyanin content in lily petals at different periods of lily development. (c) The relative content of cyanidin and its compounds in petals determined in metabolomics analysis. * Denotes statistically significant differences between samples. * p-value ≤ 0.1; ** p-value ≤ 0.05; *** p-value ≤ 0.01.
miRNA sequencing and the target genes prediction
-
miRNA sequencing was carried out on lily flower petals to investigate the genes involved in color creation. We constructed a miRNA library and 10 M clean data generated from sequencing. The Rfam database was selected to annotate and sequence small RNA sequences. Subsequently, unique sequences were analyzed by miRBase 22.0 and RNAfold software to identify the known and novel miRNAs. Finally, it was mapped to the corresponding lily transcriptome using UniGene. A total of 326 miRNAs were obtained through miRNA sequencing from nine libraries, including 285 known miRNAs and 41 new miRNAs. The 285 known miRNAs belonged to 41 families (Fig. 2a). The miR396, miR166, miR156, etc. gene families had the most members.
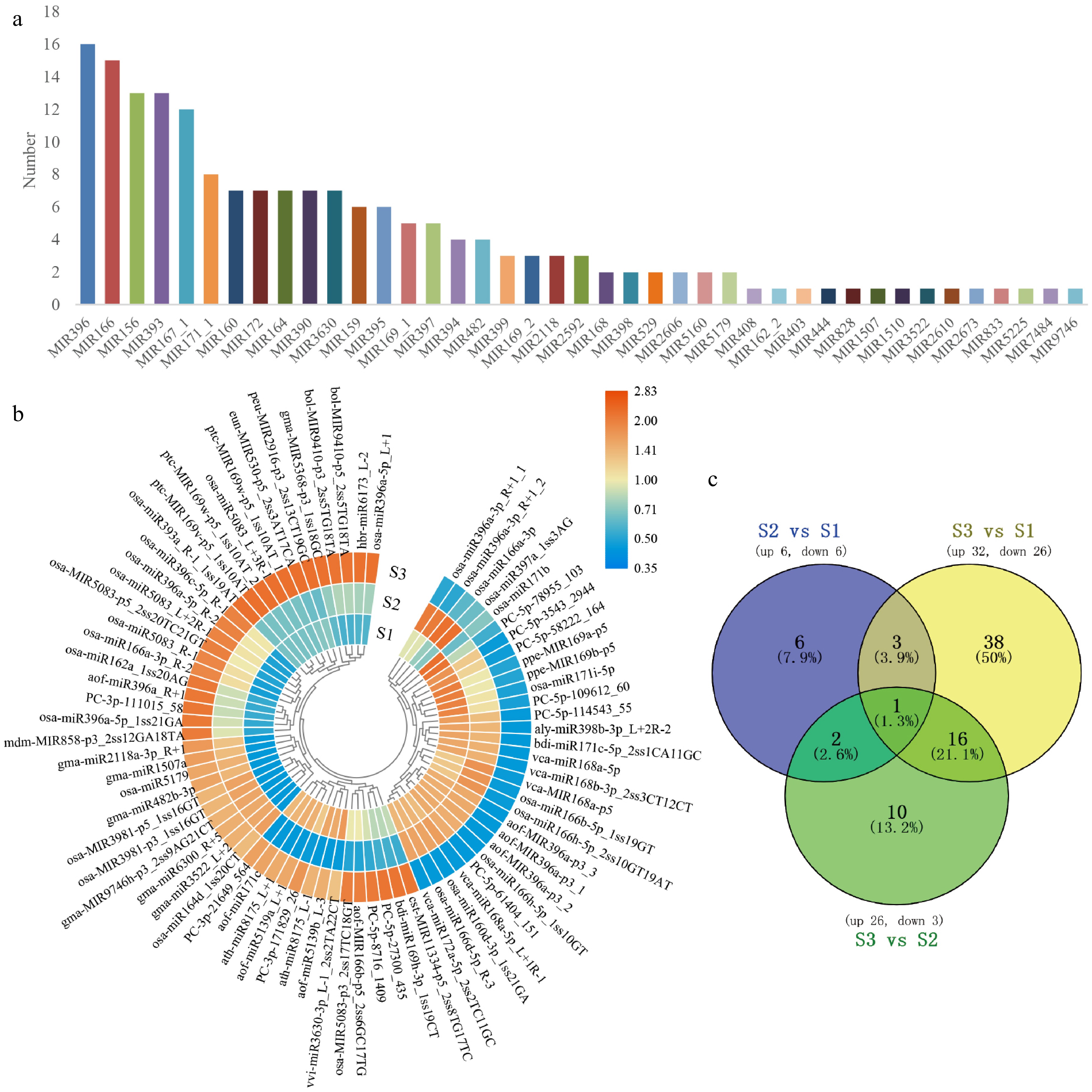
Figure 2.
The classification of miRNA and the target genes. (a) The selected 285 known miRNA belonged to 41 MiRNA families. (b) Heatmap of DEMs expression in the development of flower. After performing row clusters, each colored cell shows the average log2 (FPKM) value of each miRNA. (c) DEMs in floral development is depicted in a Venn diagram.
To further understand the role of miRNA in the process of flower color formation, we used psRNATarget to predict the miRNA target genes from the data of RNA-seq unigenes and found that 193 known miRNAs targeted 1,374 transcripts. There were 75 significant differentially expressed miRNAs (DEMs) selected by the threshold of a fold change > 2 with a p-value < 0.05 in all the compared groups (Fig. 2b). A total of 12 DEMs (six up, six down) in S2 vs S1; 58 DEMs (32 up, 26 down) in S3 vs S1; 29 DEMs (26 up, 3 down) in S2 vs S3 (Fig. 2c).
According to the psRNATarget prediction, a total of 898 potential target genes were identified in lily flower petals, which were targeted by the 75 DEMs. GO analysis and KEGG annotation were performed to evaluate the potential functions of these miRNA target genes. The GO analysis showed that these target genes were divided into three categories: cellular components, molecular functions, and biological processes (Supplemental Fig. S1a). In biological processes, the oxidation-reduction process was one of the biggest groups. In cellular components, the integral component of the membrane was the major group. Molecular functions concentrated on zinc ion binding, DNA binding, and ATP binding. KEGG analysis showed that the significantly enriched pathways of the target genes were in metabolism progress (Supplemental Fig. S1b), indicating that target genes regulated by miRNA would have a certain influence on plant metabolic activities. In addition, some target genes were enriched in flavone and flavonol biosynthesis, suggesting that miRNA may indirectly affect the flavonoid biosynthesis pathway.
Screening and verification of miRNA target genes related to petal color in lilies
-
We further analyzed the annotated target genes and found among them many genes encoding transcription factors. The expression levels were similar to the accumulation trend of total anthocyanins and cyanidin derivatives (Fig. 1b, c), and opposite to the expression levels of the corresponding miRNA (Fig. 3a, b), indicating that miRNA negatively regulates the expression of target genes. Therefore, we analyzed the correlation of expression trend between miRNA and the target genes. Gene pairs with p-value ≤ −0.5 were selected for the study data. A correlation network diagram was then created using the Cytoscape program (Fig. 3c).
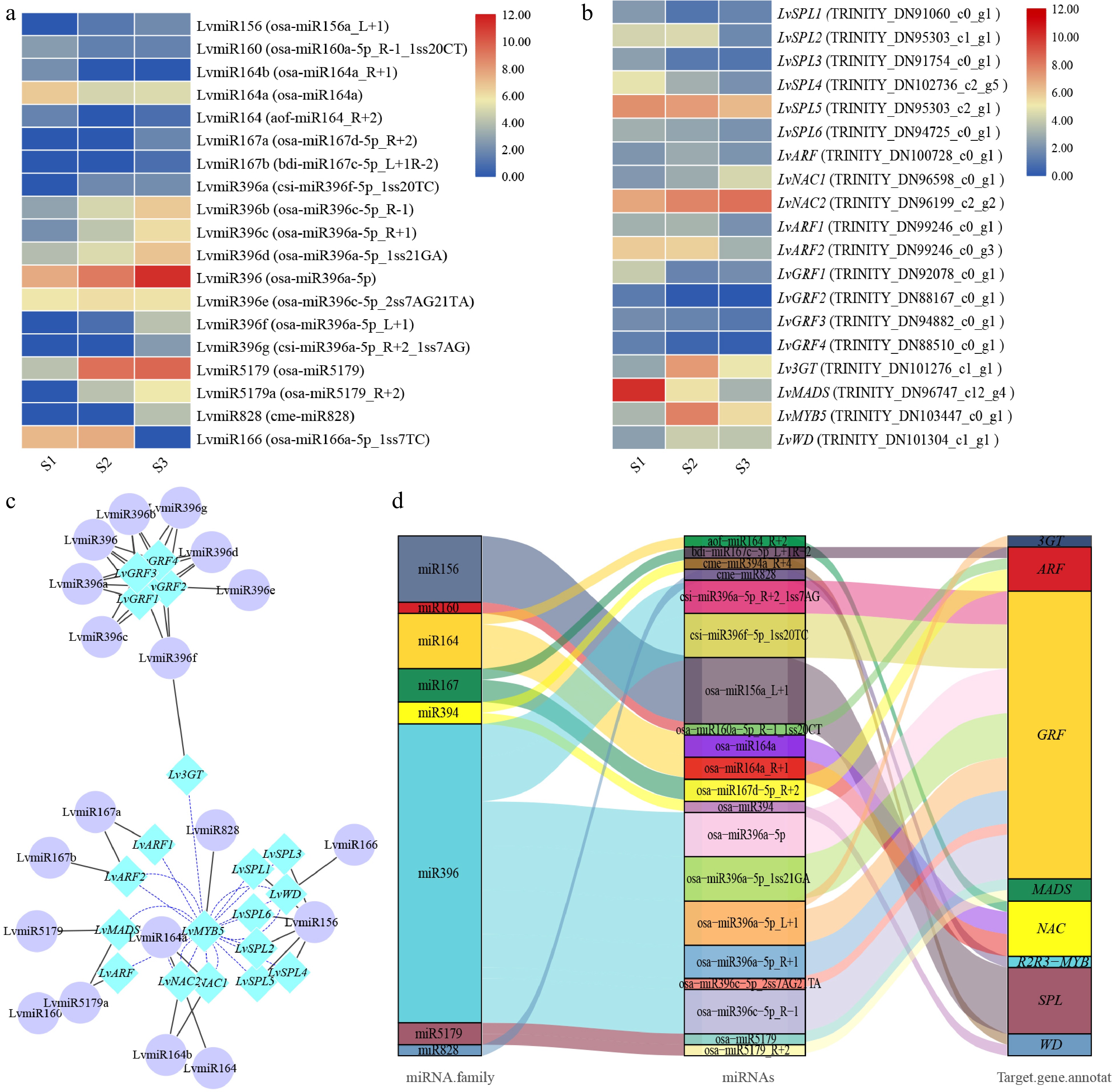
Figure 3.
The correlation between miRNA and target genes screened from lily petals related to anthocyanin synthesis. (a) Heatmap of miRNA expression. (b) MiRNA target gene expression heatmap. The average log2 (FPKM) value of each gene is shown by the color of each cell. (c) The network of target genes and miRNAs controls the progress of anthocyanins synthesis. TF genes are represented by the blue rhombic nodes, miRNA are represented by the purple oval nodes; black lines represent negative correlation; and blue lines represent positive correlation. (d) The Sankey plot of miRNA and target genes.
Finally, we screened the target genes including SPL (SQUAMOSA promoter-binding protein-like), MYB, WD, 3GT (anthocyanidin 3-O-glucosyltransferase), GRF (growth regulation factor), ARF (Auxin response factor), NAC,and MADS, which were targeted by miR156, miR828, miR166, miR396, miR160, miR167, miR164, and miR5179, respectively (Table 1). The regulation model of these miRNAs and target genes can be divided into two categories: one miRNA target regulates multiple gene transcripts, or several miRNAs target regulate the same gene transcript (Fig. 3d). We found that, miR159 can target multiple SPL, miR164 can target multiple NAC, miR396 can target 3GT and GRF, and both miR160 and miR167 can target ARF, miR828, miR166, miR5179 can target MYB, WD, MADS, respectively (Table 1). In future research, we will select these miRNAs and their target genes for further study.
Table 1. Target genes of anthocyanin biosynthesis related miRNAs in lily petals.
miRNA family miRNAs Target genes Target gene annotation miR156 osa-miR156a_L+1 TRINITY_DN91060_c0_g1, TRINITY_DN95303_c1_g1, TRINITY_DN91754_c0_g1, TRINITY_DN95303_c2_g1, TRINITY_DN94725_c0_g1, TRINITY_DN102736_c2_g5 SPL miR828 cme-miR828 TRINITY_DN103447_c0_g1 R2R3-MYB miR166 osa-miR166a-5p_1ss7TC TRINITY_DN101304_c1_g1 WD miR396 osa-miR396a-5p_L+1 TRINITY_DN101276_c1_g1 3GT csi-miR396f-5p_1ss20TC, osa-miR396c-5p_R-1, osa-miR396a-5p_R+1, osa-miR396a-5p_1ss21GA, osa-miR396a-5p TRINITY_DN92078_c0_g1 GRF csi-miR396f-5p_1ss20TC, osa-miR396c-5p_2ss7AG21TA, osa-miR396c-5p_R-1, osa-miR396a-5p_R+1, osa-miR396a-5p_1ss21GA, osa-miR396a-5p, osa-miR396a-5p_L+1, csi-miR396a-5p_R+2_1ss7AG TRINITY_DN88167_c0_g1 GRF osa-miR396c-5p_R-1, osa-miR396a-5p_R+1, osa-miR396a-5p_1ss21GA, osa-miR396a-5p, csi-miR396f-5p_1ss20TC, osa-miR396a-5p_L+1,
csi-miR396a-5p_R+2_1ss7AGTRINITY_DN94882_c0_g1 GRF csi-miR396f-5p_1ss20TC, osa-miR396c-5p_R-1, osa-miR396a-5p_1ss21GA, osa-miR396a-5p, osa-miR396a-5p_L+1, csi-miR396a-5p_R+2_1ss7AG TRINITY_DN88510_c0_g1 GRF miR5179 osa-miR5179, osa-miR5179_R+2 TRINITY_DN96747_c12_g4 MADS miR164 osa-miR164a, osa-miR164a_R+1 TRINITY_DN96598_c0_g1 NAC osa-miR164a_R+1, osa-miR164a, aof-miR164_R+2 TRINITY_DN96199_c2_g2 NAC miR160 osa-miR160a-5p_R-1_1ss20CT TRINITY_DN100728_c0_g1 ARF miR167 osa-miR167d-5p_R+2 TRINITY_DN99246_c0_g1 ARF bdi-miR167c-5p_L+1R-2, osa-miR167d-5p_R+2 TRINITY_DN99246_c0_g3 ARF To reveal the expression patterns of anthocyanin biosynthesis-related miRNAs in lily petals, nine miRNAs and 11 unigenes were selected for RT-qPCR validation (Fig. 4). Generally, the expression patterns of miRNA and unigenes were consistent with the results of the high throughput sequencing, indicating that the sequencing data were reliable. In the process of anthocyanin synthesis in lily petals, LvmiR164 (osa-miR164a_R+1) and LvmiR166 (osa-miR166a-5p_1ss7TC) showed down-regulated pattern, and LvmiR160 (osa-miR160a-5p_R-1_1ss20CT), LvmiR167c (bdi-miR167c-5p_L+1R-2), LvmiR5179 (osa-miR5179_R+2), LvmiR828 (cme-miR828), LvmiR156 (osa-miR156a_L+1) and LvmiR396 (osa-miR396a-5p_L+1) showed up-regulated pattern (Fig. 4). RT-qPCR was also used to confirm the target genes' corresponding expression patterns, including LvNAC1 (TRINITY_DN96598_c0_g1) and LvNAC2 (TRINITY_DN96199_c2_g2) for LvmiR164, LvWD (TRINITY_DN101304_c1_g1) for LvmiR166, LvARF1 (TRINITY_DN100728_c0_g1) and LvARF2 (TRINITY_DN99246_c0_g3) for LvmiR160 and LvmiR167c, LvMADS (TRINITY_DN96747_c12_g4) for LvmiR5179, LvMYB5 (TRINITY_DN103447_c0_g1) for LvmiR828, Lv3GT (TRINITY_DN101276_c1_g1) for LvmiR396, and LvSPL1 (TRINITY_DN91754_c0_g1), LvSPL2 (TRINITY_DN102736_c2_g5), and LvSPL3 (TRINITY_DN95303_c2_g1) for LvmiR156. The expression levels increased in LvNAC1/2 and LvWD and decreased in LvARF1/2, LvMADS, and LvSPL1/2/3. LvMYB5 and, Lv3GT increased in S2 and then decreased slightly in the S3 stage (Fig. 4). In general, the expression trend of the target genes was opposite to that of the miRNA.
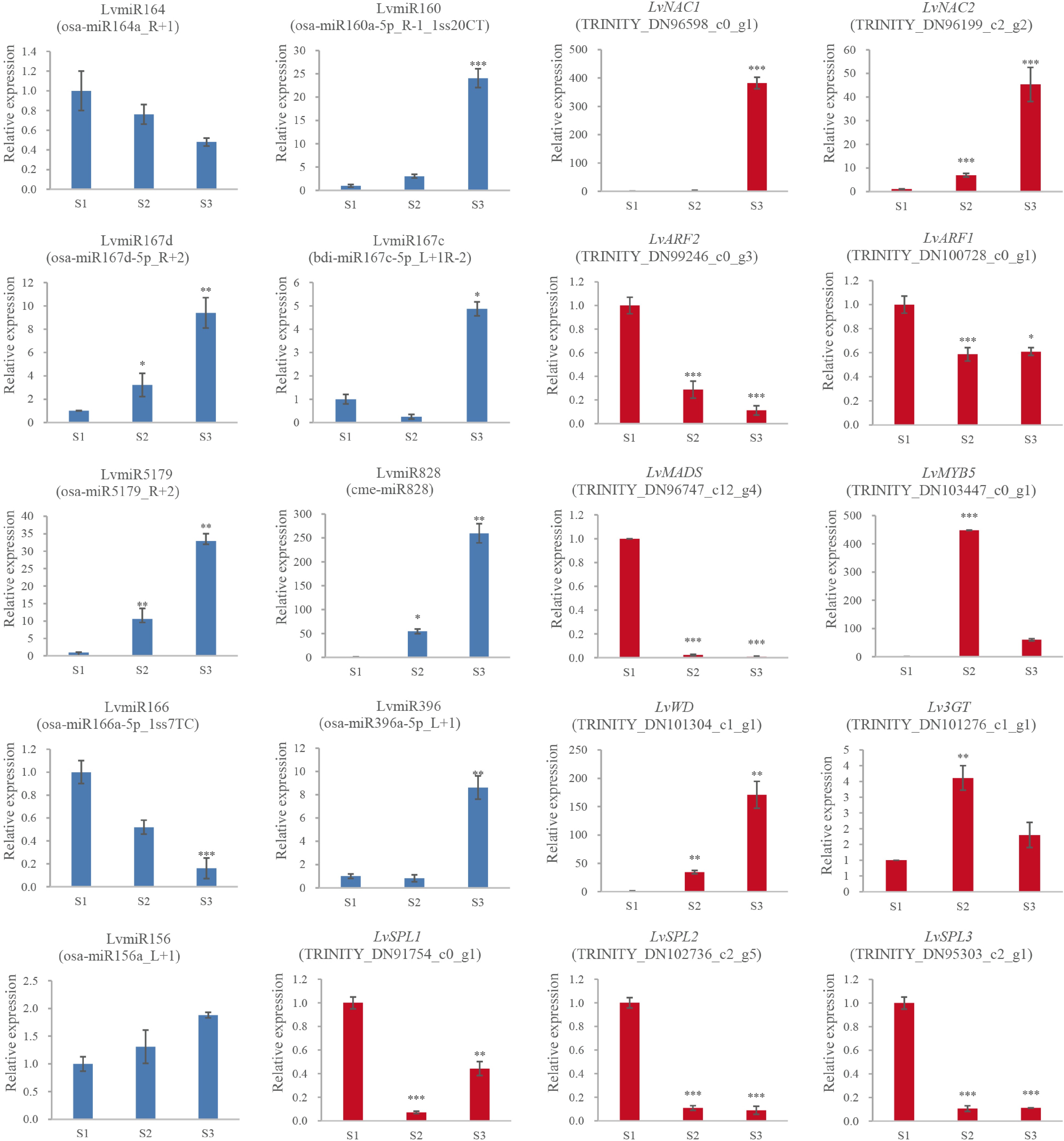
Figure 4.
The expression and the regulation of miRNA and the target genes. 2−ΔΔCᴛ method was used to measure genes' relative expression in S1, S2, S3 stage of lily cultivar Vivian. * Denotes statistically significant differences between samples.
Consistent with previous experiments, the content of the anthocyanin substances was lowest in the S1 stage and gradually increased in the S2 and S3 stages. This parallels the expression levels of Lv3GT and LvMYB5, which were perhaps the positive regulation genes related to anthocyanin biosynthesis in the lily petals[37,38]. In contrast, for flower coloring, significant inverse relationships between 3GT and LvMYB5 expression levels and LvmiR396 and LvmiR828 expression levels were found in the S2 and S3 stages. These findings suggest that the expression levels of these target genes and miRNAs are related. miRNAs in the pigmentation of lily flowers negatively regulated their target genes.
Analysis of the targeted regulation of LvMYB5 by miR828
-
Lv-miR828 targeted LvMYB5 as analyzed by psRNATarget (Table 1), which is directly related to the anthocyanin synthesis pathway[37,43]. This regulatory pathway is therefore likely to affect lily pigmentation, and so we conducted further analysis on these genes.
From the multi-sequence alignment analysis of mature LvmiR828, we found it shares a high identity with miR828 in other plants, where it functions in anthocyanin biosynthesis. There was only one nucleotide difference between the mature miR828 (Fig. 5a), indicating that LvmiR828 might play a similar role here as in other species. The cleavage site was predicted using psRNATarget to be in the CDS sequence of LvMYB5 and the motif site of LvMYB5, coding for helix 3 of specific R3 domain, was targeted by LvmiR828 (Fig. 5b). We further compared the amino acid sequences of the target gene MYB in Arabidopsis thaliana and grapes with LvMYB5 in lilies, and found that the target sites were located at the end of R3 region with high similarity (Fig. 5c). We therefore speculate that miR828 targets LvMYB5 transcriptional translation through the cleavage site and inhibits anthocyanin synthesis.

Figure 5.
A preliminary regulation analysis of LvmiR828 target LvMYB5. (a) Multiple sequence alignment of the mature miR828 in plant. (b) Predicted conserved binding site of LvMYB5 targeted by miR828. (c) The binding sites of MYB in plant targeted by miR828.
-
We found that there was no anthocyanin synthesis at the S1 stage. And the expression of Lv3GT and LvMYB5 was at a lower level. So, there wasn't regulation of post-transcriptional genes. However, transcriptional level and post-transcriptional level regulation appeared since the gene expression was in the stage of lily color formation. Therefore, miRNAs target Lv3GT and LvMYB5 in the stage where the petals start to color (S2 and S3 stage). So, some miRNAs, act as negative regulators and inhibit the expression of MYB activators to limit anthocyanin synthesis at flower developmental stages[44].
In this study, we found LvmiR828 was negatively correlated with its target gene LvMYB5 during the flower coloration. The prediction results of the targeted regulation of LvMYB5 by LvmiR828 showed that the binding site was at R3 domain 3’ end of LvMYB5, which probably affected the DNA binding activity of LvMYB5 and directly mediated the cleavage of MYB in mRNA level, thus affecting the synthesis of anthocyanins. Similar results have been obtained in Arabidopsis thaliana and potato, where miR828 targets MYB TFs to affect anthocyanin synthesis[29,36]. miR828 targeting AtMYB75, AtMYB113 regulates DFR and LDOX in Arabidopsis[30]. As a negative regulator, miR828 inhibited the biosynthesis of anthocyanin in tomatoes at different developmental stages, and the content of anthocyanin in transgenic plants overexpressing AtmiR828 was also significantly reduced[36]. Similarly, miR828 and miR858 have also been shown to regulate MYB114 resulting in the accumulation of anthocyanins in grapes[44]. Our results are consistent with these findings and the miR828 is conserved in plants. So, LvmiR828 participates in anthocyanin biosynthesis through the targeted regulation LvMYB5. We also found that the abundance of LvmiR828 was also lower than that of other miRNAs such as LvmiR156, and LvmiR159 detected by high throughput sequencing. Additionally, we've seen similar performants in other plants like Malus × domestical[23], Arabidopsis[36], and Vitis[44] among others. The expression of miRNA in plants is the result of many factors. The final expression level of miRNA and its target genes is likely related to a variety of regulatory factors such as space and time[45]. Moreover, miRNA can be expressed under different environmental stimuli[46]. Similarly, other miRNAs that regulate genes related to anthocyanin biosynthesis have been found in lily petal coloring. LvmiR396 and LvmiR166 target Lv3GT and LvWD, respectively, which may affect anthocyanin synthesis. WD can also form complexes with MYB and bHLH to regulate the biosynthesis of anthocyanins[6]. 3GT (Anthocyanidin 3-O-glucosyltransferase) is one of the key enzymes of anthocyanin biosynthesis and plays an important role in anthocyanin accumulation[47]. Therefore, the regulatory network of lily flower color synthesis is very complex, and the synthesis of anthocyanidin may be generated under the combined action of multiple regulatory factors.
miRNAs and target participate in various secondary metabolisms to regulate coloration indirectly
-
Flower blooming and petal pigmentation are the important developmental stages of ornamental plants, along with the transitional period from vegetative growth to reproductive growth[48]. A variety of internal and external signal regulation mechanisms ensure blooming and pigmentation in plants, including light, temperature, sugar and plant hormones, and other regulations[49]. A large amount of evidence indicates that MBW complexes related to anthocyanin synthesis are regulated by both exogenous environmental and endogenous developmental signals[50,51]. Several transcription factors (TFs) have been found to influence anthocyanin biosynthesis by regulating MBW complexes, such as NAC[18,19], MADS[16,17], ARF[20,21] and SPL[14,15] among others. These genes form a complex regulatory network that regulates anthocyanin biosynthesis.
SPL targeted by miR156 negatively regulates the expression of anthocyanin biosynthesis genes by disrupting the stability of the MBW complex[24]. For example, miR156 suppressed the expression of SPL13 and increased the expression of WD40-1, then enhanced anthocyanin biosynthesis in alfalfa[27]. In blueberries, VcMIR156a-VcSPL12 interacts with VCMYBPA1 to regulate the coloration of the fruit[14]. Similar results were obtained in Arabidopsis thaliana, lychee, and peony[15,24,52].
In the present study, we found that Lv-mir5179 targets the MADS-box gene. The MADS-box TF family may play an important function in regulating floral organogenesis and flowering time in plants[53,54]. The MADS-box protein is involved in anthocyanin biosynthesis in sweet potatoes (Ipomoea batatas (L.) Lam)[17,55]. And in O. italica, miR5179 inhibited OitaDEF2 (MADS-box) expression and was involved in the development of perianth organs of O. italica[56]. As lilies and O. italica belong to the monocotyledons, they were likely to have similar functions. As a result, the LvmiR5179-mediated MADS-box regulation found in this study may play an important role in pigmentation.
ARFs may regulate the expression of the MBW complex by binding to auxin response elements in the promoters of MYB[57,58]. Growing evidence suggests that auxin-suppressed anthocyanin biosynthesis in Arabidopsis[58] and Malus[21]. MiR160 and miR167 were found to target ARFs (Table 1)in the process of lily coloration, suggesting that they may indirectly affect the coloration.
NAC TFs can regulate the pathway of disease resistance, plant development, abiotic stress response, and the phenylpropanoid pathway[59−61]. In Arabidopsis thaliana, ANAC032 inhibits anthocyanin accumulation. while ANAC078 promotes coloration. In peaches, PpNAC1 is an activator in pigmentation[61−63]. IbNAC56 regulates pigmentation in purple-fleshed sweet potatoes by interacting with the complexes of IbMYB340 and IbbHLH2[64]. Additionally, the miR164-NAC regulatory pathway controls ripening in Rosaceae fruit, grapes, and citrus[65]. We found that LvmiR164 targeted NAC TFs, which may also indirectly regulate anthocyanin biosynthesis via interaction with MYB.
This study examined miRNA during the coloration progress of lily flowers. A total of 75 miRNAs with different expressions were identified in the process of anthocyanin accumulation. A variety of DEMs may play a regulatory role in the anthocyanin biosynthesis. In this study, miR828 and miR396 targeted LvMYB5 and Lv3GT, respectively, the expression of which could function on the anthocyanin biosynthesis pathway, and impacted the pigment of the petals. In addition, we predict that some miRNA target genes may be indirectly involved in the biosynthesis of anthocyanin in lily petals (Table 1). Such as, LvmiR156 targets LvSPL, LvmiR164 targets LvNAC and LvmiR5179 targets LvMADS, which may regulate MYB TFs to impact the pigment of petals indirectly. In addition, LvmiR167 and LvmiR160 target LvARF, which may activate the auxin signal pathway and indirectly affect the expression of MYB TFs and structural genes involved in the anthocyanin biosynthesis pathway to regulate the formation of flower color. These miRNAs and their target genes may also be involved in regulating other secondary metabolic pathways, forming a complex regulatory network of anthocyanin biosynthesis under the impact of various metabolic pathways.
It has been recently established that miRNA can affect anthocyanin synthesis, for example, miR164 inhibits anthocyanidin accumulation in grape berry cells[66], microRNA858 inhibits anthocyanin biosynthesis in kiwifruit by suppressing AaMYBC1 expression[67], and blackberry miRNAs play an important role in flavonoid and anthocyanin synthesis[68]. Based on past reports and the data analysis above, we hypothesized a possible miRNA-target module linked to the synthesis of anthocyanin (Fig. 6). These findings contribute to our understanding of the functional characterization of miRNAs and their targets in regulating anthocyanin biosynthesis in plants.
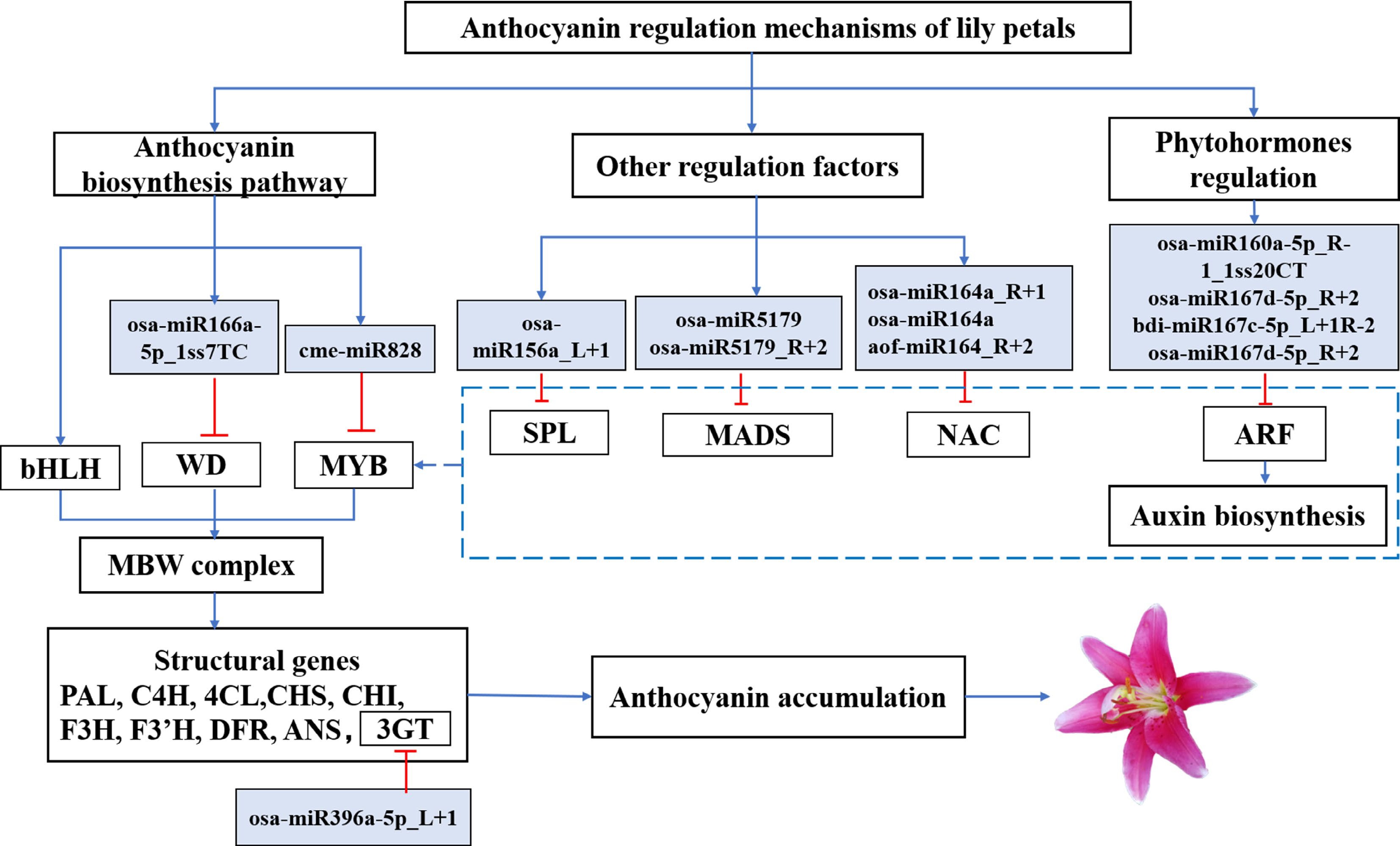
Figure 6.
An interaction model between the target genes for miRNAs involved in the progress of flower coloration in lily petals has been presented. Solid arrowhead lines: the proven regulatory roles. Dashed lines: potential roles.
-
MiRNAs are important post-transcriptional regulators, some of which participate in regulating anthocyanin biosynthesis and other pathways, including plant stress, growth and development, and internal and external hormones[23,24], however, the study of miRNAs involved in lily pigmentation is incomplete. In this study, we comprehensively performed the RNA-seq and miRNA sequencing of lily petals at three stages of flower coloring. A total of 326 miRNAs were identified from nine libraries, including 285 known and 41 new miRNAs, using the Illumina Next Seq 500 platform. The differential miRNAs were screened, and the functions of the target genes were analyzed. The expression patterns of candidate miRNAs and target genes were detected by qPCR to obtain the genes that may function in flower coloration in lily petals. Identification and functional analysis of miRNAs and target genes related to flower coloring will help reveal the coloration regulation mechanism in ornamental plants in complex and variable environments.
This work was financially supported by Discipline Construction of Professional Degree (Grant No. 880220039) China, Doctoral Research Foundation of Liaoning Provincial Natural Science (2022-BS-347), and Doctoral Research Launch Foundation of 2022 Science and education integration (Grant No. BS202204). The role of the funding body in the design of the study, the collection, analysis, and interpretation of data and in writing the manuscript is management and supervision.
-
The authors declare that they have no conflict of interest.
-
# These authors contributed equally: Xiaojuan Yin, Zhenhua Gao
- Supplemental Fig. S1 The enrichment analysis of the target genes A: The GO enrichment analysis of the target genes. B: KEGG Pathway Enrichment of the target genes.
- Supplemental Table S1 The primers information of miRNAs in this study.
- Supplemental Table S2 The primers information of target genes in this study.
- Copyright: © 2023 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Yin X, Gao Z, Sun S, Zhang L, Irfan M, et al. 2023. Insights into microRNA regulation of flower coloration in a lily cultivar Vivian petal. Ornamental Plant Research 3:9 doi: 10.48130/OPR-2023-0009 

Insights into microRNA regulation of flower coloration in a lily cultivar Vivian petal
- Received: 28 September 2022
- Accepted: 06 March 2023
- Published online: 24 April 2023
Abstract: MicroRNAs (miRNAs) are a class of non-coding small RNAs involved in the negative regulation of gene expression, which plays critical roles in developmental and metabolic pathways. However, it is not well understood how miRNA regulate the anthocyanin biosynthesis pathway in lily flowers. Using miRNA sequencing and target gene expression analysis, we explored the regulatory networks of miRNAs and their target-related flower coloration in lily petals. A total of 326 miRNAs were obtained by miRNA sequencing, including 285 known miRNAs and 41 new miRNAs. According to the psRNATarget prediction, there were a total of 75 differentially expressed miRNAs (DEMs) that target 898 potential genes. We also screened the target genes including LvSPL, LvMYB5, LvWD, Lv3GT, LvGRF, LvARF, LvNAC, and LvMADS, which were targeted by LvmiR156, LvmiR828, LvmiR166, LvmiR396, LvmiR160, LvmiR167, LvmiR164, and LvmiR5179. These genes may be involved in regulating other secondary metabolic pathways, and forming a complex regulatory network of anthocyanin biosynthesis. We therefore proposed a putative miRNA-target module associated with anthocyanin biosynthesis. In addition, we predicted the binding site of LvMYB5, the target gene of miR828, and speculated that miR828 targets regulate LvMYB5 transcriptional translation through a cleavage site, which then inhibits anthocyanin synthesis. Our findings contribute to an understanding of the functional characterization of miRNAs and their targets in controlling anthocyanin production in plants and may lead to future identification and characterization of miRNAs in lilies.
-
Key words:
- MicroRNA /
- Regulations /
- Coloration /
- Lily