









-
Pineapple (Ananas comosus) is one of the three major tropical fruits in the world, and is also an important ornamental plant and a valuable source of fiber. In the past few decades, traditional breeding techniques such as cross breeding and mutation breeding have played an important role in improving pineapple varieties. However, pineapple is an asexual reproductive plant with a narrow genetic basis and low genetic variation rate. It is thus difficult to obtain stable genetic traits or genotypes. Meanwhile, pineapple is a strict gametophytic self-incompatible plant, which means that homozygotes cannot be obtained by selfing, causing great challenges to gene mapping and genetic analysis[1].
Due to the high heterozygosity of the pineapple genome and strong gene linkage, coupled with its vegetative period of more than three years, even if the bud body is used for reproduction, it takes two to four years until first fruit development. This makes pineapple breeding a slow process, and it is difficult to create new disruptive varieties using conventional breeding methods. Breeding practices have revealed that in order to effectively integrate desired traits of the parents, four generations of backcross are generally needed to cultivate new varieties suitable for commercial application. Therefore, the breeding of an excellent new variety of pineapple often takes 25 years from the initial hybridization before commercial production[2,3].
Recently, some innovative molecular biology methods and bioengineering techniques have also been applied to the pineapple. In particular, the continuous improvement of pineapple genome sequencing has greatly promoted the study of pineapple genomics (
http://phytozome.jgi.doe.gov/pz/portal.html ). Using molecular markers, BSA analysis, genome-wide association analysis, and transgenic and gene editing methods, researchers have attempted to transfer constructs containing target genes such as leaf margin serration, leaf color, resistance and fruit quality into pineapple cells via Agrobacterium tumefaciens, and then obtain transgenic materials by tissue culture technology to identify gene function and cultivate new transgenic varieties[4−6].The genetic transformation of pineapple began in the late last century. Firoozabady et al. used embryogenic callus as the receptor material for transformation, and introduced the GUS gene into pineapple plants mediated by Agrobacterium tumefaciens[7]. Since then, genetic transformation of pineapple has been continuously reported, and the transgenic red flesh commercial variety 'Rosé' has emerged from these endeavors[7−13].
Although the in vitro regeneration technology of pineapple is relatively mature[8−10,14], it is still challenging to establish a stable pineapple transformation system due to the uniqueness of pineapple. There are very few transgenic materials that can change genetic traits. Since we began to engage in pineapple genetic transformation in 2001, we have carried out a series of explorations, accumulated more experience, learnt lessons, and established a relatively stable pineapple transformation technology system that has been stably applied to pineapple gene function identification. In this paper, we summarize some of the bottlenecks often encountered in the genetic transformation of pineapple, and present answers to several technical problems, with the ultimate aim of providing a useful reference for the large-scale identification of pineapple gene function and genetic improvement.
-
The explant materials of pineapple tissue culture include the stem tip, leaf base and regenerated plants from sterile tissue culture (Fig. 1). Shoots with a stem base diameter of 0.5 cm or more and normal development should be harvested on sunny days. Senescent leaves at the base are then stripped off, the buds rinsed with tap water, and all leaves severed about 0.5 cm above the growth point. The buds are pre-disinfected with 2% NaClO for 10 min and rinsed with sterile water three times. The buds are then disinfected with NaClO (or 0.1% HgCl) for 8 min and rinsed with sterile water again three times. Cut the white leaf base with growing points, and the leaf base is disinfected with 0.1% NaClO for 5 min, rinsed with sterile water three times, and then inoculated on MS + 2.0 mg/L BA + 2.5 mg/L NAA for callus induction. After 4 weeks, the callus is cut and transferred to proliferation medium (MS + 3 mg/L BA + 2 mg/L NAA), and subculture is performed every four weeks[15,16].
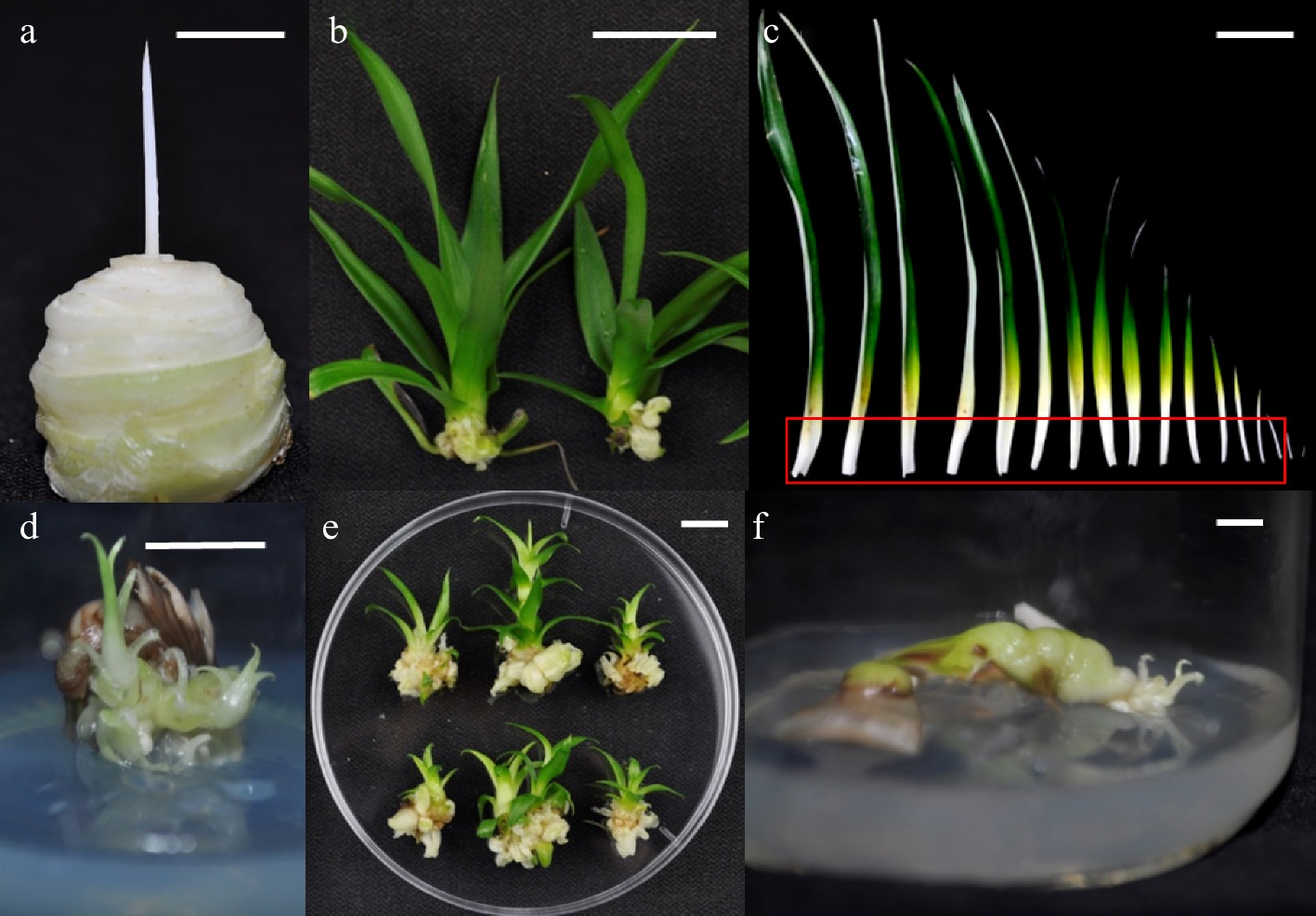
Figure 1.
Explant materials of pineapple in vitro culture. (a) Shoot apex after leaf removal; (d) Callus and adventitious buds observed after two generations in callus induction culture after vertical cutting of buds; (b) Adventitious buds cultured by tissue culture; (e) At the first generation of induction culture, callus is produced at the base; (c) Leaves, the white part in the red frame is the leaf base; (f) Leaf-based induction for two generations. Bar = 1 cm.
Pineapple plants are rosette, tissue culture seedlings, and hybrid seedlings need more than six months and a seedling height of about 25 cm to form a clear fleshy stem. Therefore, the stem tip culture is collected from the field, and the stem tip is cut into four pieces, but the number of explants is small, and the redifferentiation difficult. It usually takes two to three generations to obtain callus (Fig. 1a, d). When using sterile seedlings as explants, the disinfection process can be omitted, and redifferentiation is easier (Fig. 1b, e); In particular, the leaf base of sterile seedlings has a large number and is suitable for large-scale transformation. (Fig. 1c, f). When the materials are collected from the field as explants, they are only suitable for the initial establishment of the in vitro culture system. The callus obtained from explants is usually not more than eight generations when used as receptor material, otherwise a certain proportion of mutations will occur, which will affect the judgment of the function of the target gene in the T0 generation[17].
-
Among the reported transformation receptors of pineapple, the stem apex (Fig. 1a), leaf base (Fig. 1b), callus (Fig. 2a, b), suspension cell line (Fig. 2c, d) and somatic embryo (Fig. 2k, l) are considered the most useful[14,18−20]. At present, compared with plants with high transgenic efficiency such as model crops Arabidopsis and tobacco, the transformation efficiency of pineapple is still very low. Therefore, the receptor material must be a tissue that can be obtained in large quantities, has a strong plant regeneration ability, and can be easily induced to accept the introduction of T-DNA. As mentioned above, the stem tip should not be directly used as transgenic receptor material because of the small number, requirement for disinfection, and that plant redifferentiation is more difficult. The conversion efficiency of the other materials is not much different, but each has its own technical characteristics and requirements. For example, the leaf base must be derived from adventitious buds produced by tissue culture. It can be obtained in large quantities, and is sterile, tender, and easy to redifferentiate. It can be used as the preferred receptor material for researchers with less experience in pineapple in vitro culture.

Figure 2.
In vitro culture methods are commonly used to produce receptor material. (a) Common callus; (b) Embryogenic callus; (c) Embryogenic suspension cell lines; (d) Embryogenic suspension cell lines cultured on solid culture for 30 d; (e) Non-embryogenic callus sections; (f) Embryogenic callus section. Circle indicates a large nucleus of small individual embryonic cells; (g) Embryogenic suspension cell lines under stereomicroscope; (h) Embryogenic suspension cell lines cultured on solid culture for 30 d under stereomicroscope; (i) Callus subcultured several times on differentiation medium; (j) Differentiated young adventitious buds differentiated on the callus of multiple subcultures; (k) Globular somatic embryo (indicated by the arrow); (l) Mature embryo stage. Bar = 0.5 cm in (a)−(d), (i); bar = 20 μm in (e), (f); Bar = 700 μm in (g), (h), (j), (l); Bar = 300 μm in (k).
When in vitro cultures such as callus, suspension cell lines and somatic embryos are used as receptors, there are obvious advantages in that they are easy to obtain in large quantities, are sterile, and do not need to undergo dedifferentiation. However, because it is difficult to accurately assign their growth stage, transformation efficiency can be highly variable, and often no transformants are produced. The basic reason is that the callus formed by dedifferentiation of pineapple will produce a large number of bulbous adventitious buds with a diameter of about 1 mm when subcultured more than three times on proliferation medium ( MS + 3.0 mg/L BA + 2.0 mg/L NAA ) (Fig. 2i, j). Sections observed under the stereoscope will reveal that these small bulbs are actually rosette-like: the outer layer is one to many numbers of young leaves, the middle layer is empty, and the top growth point is extremely small. If this bulblet is not cut from the growth point, the possibility of T-DNA entering the recipient cell is minimal. The suspension cell line just taken from the liquid medium is too dispersed (Fig. 2c), and it is not easy to cut out the wound. And it needs to be cultured on solid medium for one generation (Fig. 2d). Somatic embryos need to be embryogenic callus at the early stage of somatic embryo induction (Fig. 2k), and the material (Fig. 2l) that has entered the middle and late stage of somatic embryo differentiation has a low conversion rate. Therefore, the use of callus obtained from explants within three generations that have not yet entered the redifferentiation stage (Fig. 2a, b), or adventitious bud leaf base, as receptors is key to successful transformation. Our pineapple transgenic work chose the pineapple cultivar ‘Shenwan’ as the material.
-
Like other plants, pineapple genetic transformation uses protease genes that can inactivate antibiotics as selectable marker genes, such as the kanamycin resistance gene nptII, tetracycline resistance gene tetR, glyphosate resistance gene aroA, glufosinate Ammonium (Basta) resistance gene bar, bromobenzonitrile resistance gene bxn, and the chlorsulfuron resistance gene csrl. The chloramphenicol acetyltransferase gene cat, the luciferase gene luc, the β-glucuronidase gene gus, and the green fluorescent protein gene gfp are also used as reporter genes. However, the results of comparative experiments in our laboratory have shown that nptII is the most valuable marker gene for pineapple transformation. Specifically, when Km is used as the screening agent, the buds with green spear leaf can be directly selected from the candidate materials according to the color of the buds as the candidate transformation buds for the next round of screening. It is easy to identify the resistant buds with the naked eye, and this approach has the advantages of being fast, sensitive, low cost and simple. The other selection marker genes are more difficult to detect, because of positive transformants, or difficult to test, making them problematic to use.
Pineapple is sensitive to Km but also has a certain tolerance. We previously inoculated callus into the differentiation medium containing different concentrations of Km (with agar as a coagulant). When the concentration of Km in the medium was 10 mg/L, the albino buds in the differentiated adventitious buds only accounted for 27.6%, and 72.4% of the differentiated adventitious buds showed green. However, when the Km concentration increased to 15 mg/L, the differentiated adventitious buds were all albino buds (Fig. 3).

Figure 3.
Effect of Km on adventitious bud differentiation of pineapple callus. (a), (b) Tolerance of adventitious buds to different concentrations of Km during regeneration of pineapple callus. (c) The first generation of screening culture: red arrows indicate green resistant buds (20 mg/L Km). (d) The second generation was selected and cultured, and the red arrows indicating cut heads are green resistant buds (30 mg/L Km).
Km has an inhibitory effect on adventitious bud differentiation of pineapple callus, and it increases with increased concentration. The proliferation rate of adventitious buds in 20 mg/L Km + MS was about 200% (based on a height of more than 3 mm, which can clearly distinguish the individual adventitious buds); however, the differentiation of adventitious buds was basically inhibited after three generations of continuous culture in 50 mg/L Km + MS (Fig. 3a). In order to ensure a higher differentiation rate, reduce false positive plants and enhance the screening effect in selective culture, the optimal Km concentration was determined to be 20 mg/L when the transformants are first selected. From the second round, the concentration of Km in selective medium is increased to 30~50 mg/L with the growth of transformant buds. Km may also be degraded, leading to the albino leaves turning green again by the 7th week. Thus, the culture time of each generation is controlled to be about four weeks.
We also found that the tolerance of pineapple adventitious buds to Km was affected by the type of coagulant. In the medium with the same composition, when carrageenan was used as the medium coagulant, even if the concentration of Km reached 50 mg/L, the adventitious buds differentiated from callus were still 100% green, so that Km could not play a selective role. In addition, since the larger the pineapple plant, the stronger the resistance to Km, only the young green buds differentiated from the screening can be used as candidate transformants, and the false positive buds of the previous generation will differentiate into white heart leaves during subculture screening (Fig. 2d).
-
When the ordinary callus (Fig. 2a, e) was used as the transformation receptor and screened according to the organ regeneration pathway, although the Km concentration increased from the first generation to the fourth generation, the negative (albino buds and white heart buds) rate did not change significantly, at 50%−60% (Table 1). From the 5th generation to the 7th generation, the incidence of green shoots decreased significantly with the increase of Km concentration: to the 7th generation, the incidence of green shoots was only 8.57%, and the elimination rate increased significantly. Since the subculture is to cut the green buds on the previous generation together with the base callus to continue screening, the base of the adventitious buds will continue to differentiate into adventitious buds. The cumulative positive rates were 9.9%, 0.77% and 0.017% after 3, 5 and 7th generation screening, respectively. Using embryogenic callus (Fig. 2b, f) as the receptor material, the cumulative positive rates of the 3rd, 5th and 7th generations were 8.5%, 0.92% and 0.25%, respectively, and the positive rate from the 6th generation was basically stable at about 50%[17].
Table 1. Screening results of pineapple common callus as the transformation receptor.
Number of generations filtered Km
(mg/L)Number of
receptor materialsTotal number of adventitious buds Number of whitening buds Number of white cores Number of green buds Proportion of
green buds (%)1 20 200 357 68 97 192 53.78 2 30 192 358 102 88 168 46.93 3 50 168 383 82 151 150 39.16 4 50 150 197 26 90 81 41.12 5 60 81 251 81 122 48 19.12 6 60 48 84 20 42 22 26.19 7 70 22 35 6 26 3 8.57 Total 0.017 Cumulative green bud rate is the product of green bud rate from each generation. The above results show that receptors obtained from embryogenic callus and the somatic embryo regeneration pathway can significantly improve the screening efficiency. And that the number of screening cultures should be 3−5 generations. Too few screening alternative can increase the difficulty and cost of molecular detection. And too many screening alternatives also reduce screening efficiency.
-
The transgenic research of pineapple focuses on establishing a stable and efficient transformation system. The molecular biological identification of the transformed plants is similar to that of other plants. Under the current technical conditions, the pineapple conversion technology system we recommend is shown in Fig. 4. The adventitious bud leaf base produced by tissue culture (Fig. 4a) and undifferentiated calli within three generations are used as receptor materials for infection (Fig. 4b). Then, screening and plant regeneration are performed simultaneously through somatic embryogenesis (Fig. 4, step ②) to increase the transformation frequency and reduce the production of transgenic chimeras. The organogenesis pathway can also be used for simultaneous screening and plant regeneration (Fig.4, step ③). Although the conversion efficiency of this method is slightly lower, the technical difficulty is very low[21,22].

Figure 4.
Flow chart of the pineapple transformation system. (a) Leaf base (Bar = 0.5 cm). (b) Non-embryogenic cells (scanning electron microscopy, SEM, Bar = 20 μm ). (c) Embryogenic cells (SEM, Bar = 50 μm). (d) Somatic embryogenesis, where the red arrows indicate the original embryo (SEM, Bar = 20 μm). (e) Mature somatic embryos (Bar = 500 μm). (f) Adventitious shoot regeneration from callus (Bar = 700 μm). (g) After three consecutive generations of screening, the regenerated plants are green for Km-resistant transformed buds (Bar = 0.5 cm). (h) Somatic embryos produced from embryogenic callus (Bar = 700 μm ). (i) Km-negative plants (Bar = 0.5 cm). (j) Transforming buds (Bar = 0.5 cm). ① Dedifferentiation culture; ② Somatic embryogenesis pathway; ③ Organogenesis pathway; ④ Rooting culture.
Pineapple genetic transformation requires the use of medium and antibiotics. The specific process includes the following steps. First, receptor materials are co-cultured on MS + 3.0 mg/L BA + 2.0 mg/L NAA + 100 μmol/L AS + 8 g/L agar for 3 d. Then they are transferred to selective medium (MS + 3.0 mg/L BA + 2.0 mg/L NAA + 20 mg/L Km + 400 mg/L Carb + 8 g/L agar). This process lasts about 10 d when adventitious buds begin to differentiate. After 28 d, the green Km-resistant adventitious buds are selected and transferred to the second round of screening medium MS + 2.0 mg/L NAA + 30 mg/L Km + 400 mg/L Carb + 8 g/L agar. The third screening and strong seedlings are carried out on the medium MS + 1.0 mg/L NAA + 50 mg/L Km + 400 mg/L Carb + 8 g/L agar. Finally, the green buds are transferred into rooting medium MS + 1.0 mg/L IBA + 50 mg/L Km + 8 g/L agar for culture. It is worth emphasizing that pineapple is a monocotyledonous plant, lacking phenolic substances which are important signalling compounds that induce T-DNA to enter the recipient cells. Therefore, 100 μmol/L Acetosyringone (AS) must be added to the medium during the co-culture stage.
This work was supported by the National Key R & D Program of China (2018YFD1000500) and National Natural Science Foundation of China (No. 32272677). The authors gratefully acknowledge experimental material support from National Tropical Plants Germplasm Resource Center.
-
The authors declare that they have no conflict of interest.
-
Received 3 December 2022; Accepted 18 April 2023; Published online 29 May 2023
-
Undifferentiated calli within three generations are used as receptor materials for infection.
The optimal kanamycin concentration of first selection was 20 mg/L, then the concentration is to 30~50 mg/L. The number of screening cultures should be 3−5 generations.
Screening and plant regeneration are performed through somatic embryogenesis pathway.
- Copyright: © 2023 by the author(s). Published by Maximum Academic Press on behalf of Hainan University. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
He Y, Luan A, Wu J, Zhang W, Lin W. 2023. Overcoming key technical challenges in the genetic transformation of pineapple. Tropical Plants 2:6 doi: 10.48130/TP-2023-0006 

Overcoming key technical challenges in the genetic transformation of pineapple
- Received: 03 December 2022
- Accepted: 18 April 2023
- Published online: 29 May 2023
Abstract: In recent years, transgenic technology has become the most important tool for molecular breeding. An efficient genetic transformation system is the key to improving the efficiency of biological breeding, and Agrobacterium-mediated genetic transformation is the common method used in plant genetic transformation experiments. Pineapple is an important tropical horticultural plant, but it has a very narrow genetic base, high genome heterozygosity, and strict self-incompatibility, thus limiting the value of conventional breeding techniques. To shorten the breeding cycle and create new subversive varieties, transgenic research of pineapple is imperative. Due to the characteristics of pineapple, in vitro regeneration technology is relatively straightforward, but it can still be very difficult to obtain pineapple transgenic materials. Over more than 20 years of research on pineapple genetic transformation, we have explored, continuously improved and now established a set of transformation tools for the simple and effective transformation of pineapple genes. The basic premise of our approach is a straightforward redifferentiation of pineapple suckers as explants. Specifically, the receptor material that is the basis for the successful transformation of pineapple is the in vitro culture of callus, which is a tissue that has not yet entered the organ differentiation stage. The nptII gene was selected as the optimal selection marker gene and the somatic embryogenesis pathway is used for screening and regeneration.
-
Key words:
- Pineapple /
- genetic transformation /
- explants /
- receptor material /
- regeneration pathways