









-
In flowering plants, cross-pollination is a common mode of reproduction and promotes genetic variation, gene exchange, and species adaptation. Plants have evolved a variety of reproductive systems involving cross-pollination at the individual and population levels, such as monoecy, dioecy, andromonoecy, gynomonoecy, and androdioecy[1].
Based on studies of Arabidopsis thaliana and snapdragon mutants, Coen & Meyerowitz and Causiera & Meyerowitz proposed the well-known ABC model, which suggests that the formation of the four whorls of floral organs is a result of the combined action of class A, B, and C genes[2,3]. Subsequent studies identified class D and E genes, and the flower organ development model gradually developed into an ABCDE model[4−6], with MADS-box genes accounting for the majority of genes in the model[7]. In core dicotyledons, A and E functional genes determine the development of the calyx; A, B, and E functional genes determine the development of petals; B, C, and E functional genes determine the development of stamens; and C and E functional genes determine the development of carpels[7].
Studies have shown that the class C MADS-box gene AGAMOUS (AG)[8−11] and the non-MADS-box gene CRABS CLAW (CRC)[12−14] play important roles in carpel development. AG regulates the differentiation of carpels and interacts with other genes to control floral determinacy. In Arabidopsis, the loss of AG function usually results in the absence of carpels, and the carpels are replaced by indeterminate perianth whorls[15,16]. The loss-of-function of AG and class C homeotic genes shows similar phenotypes in other plants, such as Antirrhinum[17], petunia[18], opium poppy (Papaver somniferum)[19], Nicotiana benthamiana[20], and apple (Malus domestica)[21].
CRC regulates the development of tissues derived from the abaxial side of the carpel primordium[22]. In plants with mutations in CRC and CRC orthologs, the carpel is replaced with petals, stamens, or a flower[23,24]. CRC genes influence carpel development in various plants, such as A. thaliana[25,26], Eschscholzia californica[27], Pisum sativum[12], Petunia hybrid[13], and Physalis floridan[14]. CRC is a master regulator in the gene regulatory network determining floral morphology, particularly carpel formation[11]. Both CRC and AG are core transcription factors that can regulate each other to promote carpel development[28,29].
Sweet osmanthus (Osmanthus fragrans Lour.) is a well-known ornamental and aromatic plant in China. Preliminary studies have found that sweet osmanthus is an androdioecious breeding system; however, the molecular mechanism underlying the formation of the androdiecious reproductive system in the species has not been determined. In this study, morphology and molecular analyses were combined to investigate the mechanism underlying pistil sterility in androdiecious sweet osmanthus plants. The results of this study will provide a theoretical basis for the conservation of germplasm resources and the cultivation of new cultivars of sweet osmanthus.
-
According to the observations of paraffin sections, flower bud differentiation in sweet osmanthus can be divided into five stages: involucre differentiation, primordium differentiation, terminal flower perianth differentiation, stamen differentiation, and pistil differentiation. Involucre differentiation was completed at the end of June, when the growth cone gradually flattened and widened, producing two protrusions on its surface, the involucre primordium (Fig. 1a–f). As the cells grew, the involucre and primordia became larger and bent upward until the two primordia were close together (Fig. 1g). At the beginning of July, the growth points inside the involucre gradually expanded into a hemispherical shape, and the central tip and both sides of the lower part showed darker staining. The growth points gradually expanded and separated to form multiple raised semicircles. At this time, the rudimentary structure of cymose inflorescence (Fig. 1h) was already visible. The protrusions on both sides of the base of the involucre formed the lateral flower primordium (Fig. 1i), while the central protrusion formed the terminal flower primordium. During the perianth differentiation stage, the terminal flower primordium gradually flattened, and two small protrusions were formed on both sides, corresponding to the calyx primordia (Fig. 1j−l). Since the calyx of sweet osmanthus is very short, the calyx primordium stopped growing after a short period, and two protrusions formed at the center. These protrusions were the petal primordia and continued to grow and bend into a curved petal shape (Fig. 1m, n). This period proceeded rapidly, and the differentiation of the perianth primordia was completed in less than a month. At the time of the formation of petal primordia, the stamen primordia began to appear, and two small protrusions (Fig. 1o) appeared on the inner side of petals at the end of July; these developed gradually, enlarged, and finally formed the stamen primordia (Fig. 1p). At this time, the filaments and anthers also began to differentiate. At the beginning of August, after the stamen primordia of the fertile 'Huangchuan Jingui' matured, two small protrusions were formed on its inner side. With continuous growth, the protrusions moved towards each other and gradually fused to form a carpel, forming a slit in the center (Fig. 1q). The carpel primordia continued to develop, expanded into an ovary, and formed a style and a stigma (Fig. 1r & s). Two small protrusions (Fig. 1t) were also formed on the inner side of the stamens of sterile 'Chenghong Dangui'; however, the protrusions gradually elongated and did not fuse (Fig. 1u). Instead, two fronds were formed, and ovary development was not seen (Fig. 1v). The pistil of 'Huangchuan Jingui' was able to develop normally to form fruits after fertilization (Fig. 2a & b), while pistil abortion was observed in 'Chenghong Dangui', with no fruit formation (Fig. 2c & d).
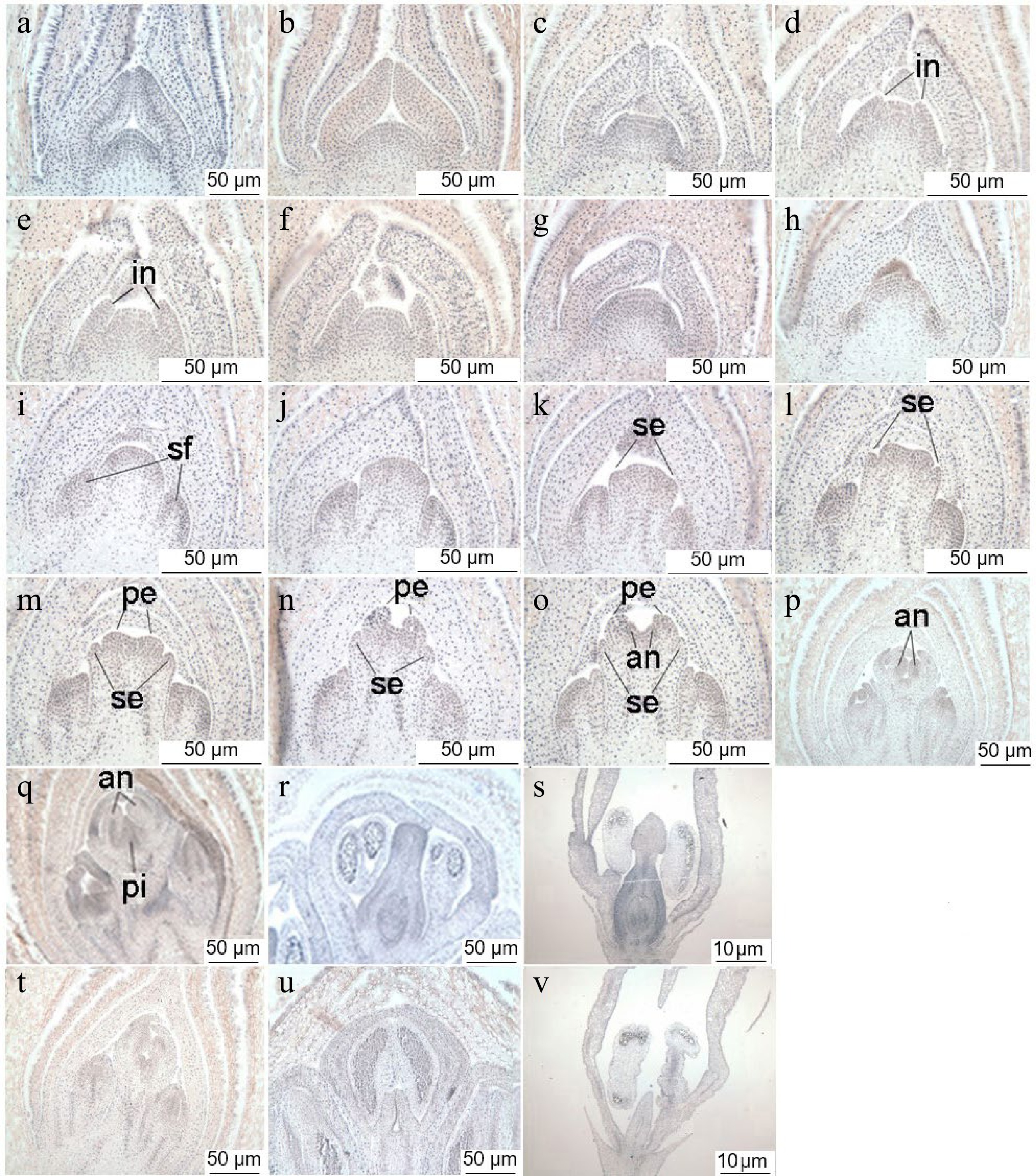
Figure 1.
Floral bud differentiation of sweet osmanthus. (a) − (g) Involucre differentiation. (h), (i) flower primordium differentiation. (j) − (n) Terminal flower perianth differentiation. (o), (p) Stamen differentiation. (q) − (s) Pistil differentiation. (t) − (v) Pistil differentiation (sterile). in: Involucre primordium, sf: Side flower primordium, se: Sepal primordium, pe: Petal primordium, an: Stamen primordium, pi: Pistil primordium.

Figure 2.
Development of the pistil of the two cultivars. (a) The full flowering stage of 'Huangchun Jingui' cultivar. (b) Fruit formation of 'Huangchun Jingui' cultivar. (c) The full flowering stage of 'Chenghong Dangui' cultivar. (d) Pistil abortion and no fruit formation in 'Chenghong Dangui' cultivar.
Morphological characteristics and activity of pollen grains of 'Huangchuan Jingui' and 'Chenghong Dangui'
-
Scanning electron microscopy results showed that the pollen grains of both cultivars ('Huangchuan Jingui' and 'Chenghong Dangui') were oblong and round with three lobes in polar view (Fig. 3a & b); they were classified as N3P4C5 according to the NPC classification system. The outer wall of the pollen had a reticulate pattern, with fine and small reticulation. The germination pore was a 3-pore groove, and the poles of the pore groove were narrow and became wide in the middle (Fig. 3c−h). The mean polar axis length of 'Huangchuan Jingui' was 18.0 μm, while that of 'Chenghong Dangui' was 18.6 μm. Both cultivars had small pollen with significant differences in polar axis length (Table 1). In addition, the P/E, pore groove length (L), and L/P values differed significantly between the two cultivars (Table 1). After triphenyltetrazolium chloride (TTC) staining and observing under a microscope, the pollen grains of both 'Huangchuan Jinghui' and 'Chenghong Dangui' were stained red or light red (Fig. 3i,j). After fluorescein diacetate (FDA) staining and observing under fluorescence microscope, the pollen grains of both 'Huangchuan Jingui' and 'Chenghong Dangui' exhibited green fluorescence (Fig. 3k,l), indicating that the pollen grains of both cultivars were active. The results of aniline blue staining showed that regardless of whether the pollen of 'Huangchuan Jingui' or 'Chenghong Dangui' was used to pollinate the stigma of 'Huangchuan Jingui', the germinated pollen tubes could be seen on the stigma of the pistil style of the 'Huangchuan Jingui' (Fig. 3m,n). These results indicated that pollen grains of both male and bisexual flowers were active on the stigma of the pistil.
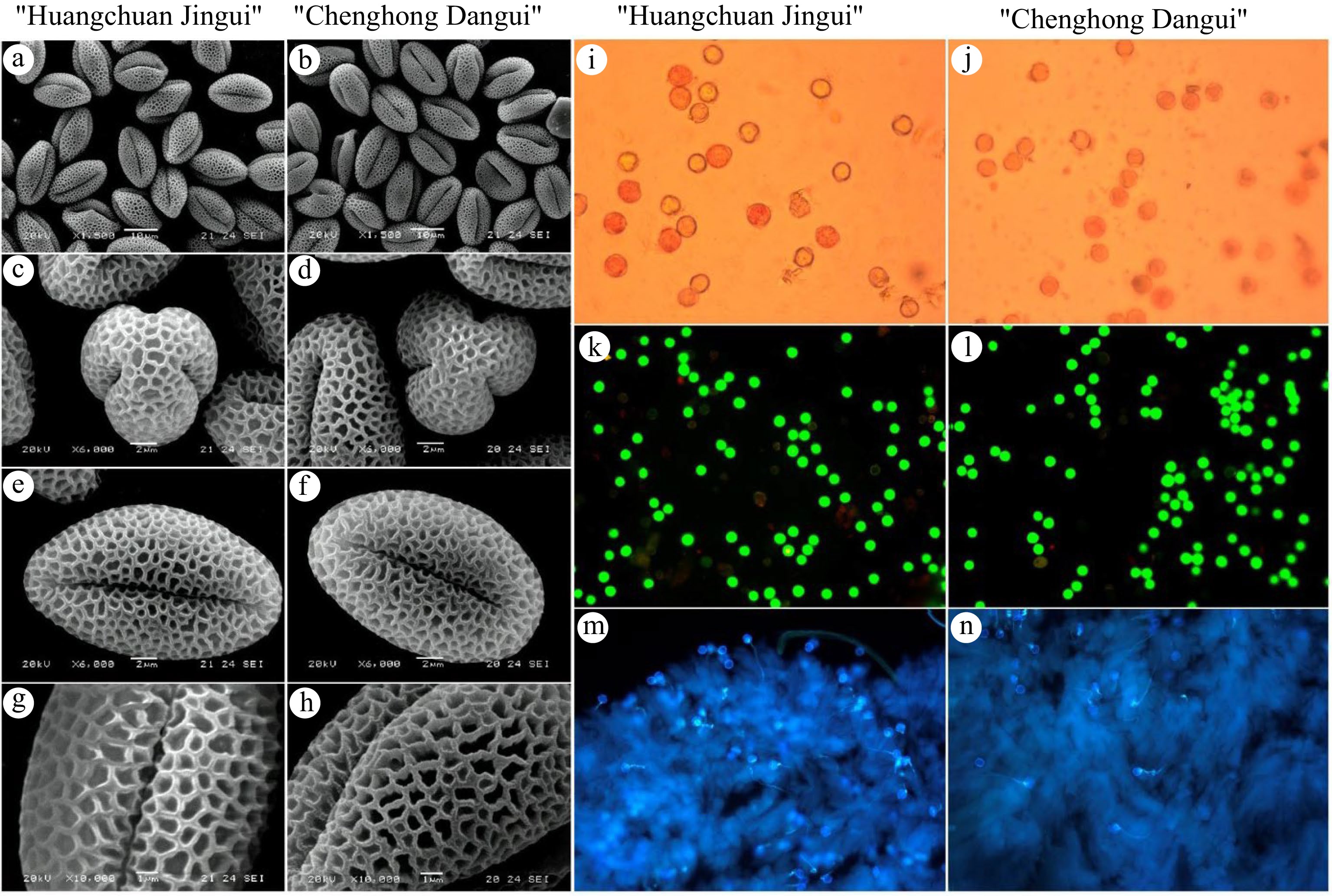
Figure 3.
Morphological characteristics and activity of pollen grains of the two cultivars. (a) − (h) Morphological characteristics of pollen grains. (i) − (n) Activity of pollen grains of the two cultivars.
Table 1. Measurement results of pollen.
Cultivars Polar axis (P) (μm) Equator axis (E) (μm) P/E Pore groove length (L) (μm) L/P 'Huangchuan Jingui' 18.0 ± 1.01 10.8 ± 1.05 1.63 ± 0.11 12.4 ± 1.52 0.70 ± 0.08 'Chenghong Dangui' 18.6 ± 0.66* 10.9 ± 0.54 1.70 ± 0.12* 13.9 ± 1.21* 0.77 ± 0.05* * P < 0.05 Differential expression of genes related to flower organ development in 'Huangchuan Jingui' and 'Chenghong Dangui'
-
To investigate the differential expression of genes related to floral organ development in 'Huangchuan Jingui' and 'Chenghong Dangui' and their effects on the development of carpels and pistils, a transcriptome sequencing analysis of the floral organs of 'Huangchuan Jingui' and 'Chenghong Dangui' at the linggeng stage, xingyan stage, initial flowering stage, full flowering stage, and late full flowering stage was performed. Compared with 'Chenghong Dangui', the numbers of upregulated differentially expressed genes (DEGs) were 4,937, 4,644, 3,981, 4,165, 4,910 during the linggeng stage, xingyan stage, initial flowering stage, full flowering stage and late full flowering stage, while the downregulated DGEs were 3,842, 3,341, 3,003, 3,462, 4,090 respectively in 'Huangchuan Jingui' (Supplemental Files 1−5). From the KEGG enrichment analysis results, we found that the most enriched DEGs exists in the plant-pathogen interaction pathway of the five flowering stages, with 209, 204, 178, 222, and 252 DEGs, respectively. The secondly DEGs enrichment pathway is plant hormone signal transduction, which contains 162, 163, 140, 190, and 226 DEGs in the five flowering stages (Supplemental Files 6−10). Thus, we presumed that the development of sweet osmanthus flower organs may be greatly influenced by hormones.
MADS-box genes are key genes for the development of floral organs, and the transcriptome sequencing results showed that, compared with levels in 'Chenghong Dangui', 7, 8, 6, 8, and 10 MADS-box genes were up-regulated in 'Huangchuan Jingui' during the linggeng stage, xingyan stage, initial flowering stage, full flowering stage, and late full flowering stage, respectively (Fig. 4). Among these, class A genes included AP1 and FUL, which were up-regulated in the linggeng stage and the xingyan stages of the flowering organs of 'Huangchuan Jingui'. Class B genes included GLOBOSA and PISTILLATA, which were up-regulated in the floral organs of 'Huangchuan Jingui' at both the full flowering and the late full flowering stages. The class C gene AGAMOUS (AG) was up-regulated in the floral organs of 'Huangchuan Jingui' at both the initial and full flowering stages. In addition, several AGAMOUS-like (AGL) genes, such as AGL11, AGL12, AGL81, AGL17, and AGL81, were up-regulated in the floral organs of 'Huangchuan Jingui' in different flowering stages (Fig. 4). These genes may also be involved in the development of floral organs and carpel formation. In particular, the expression of AGL11 was significantly up-regulated in all five periods of flowering organ development of 'Huangchuan Jingui'. In addition, the CRC gene, a non-MADS-box gene involved in carpel development, was significantly up-regulated in all five periods in the floral organs of 'Huangchuan Jingui'.
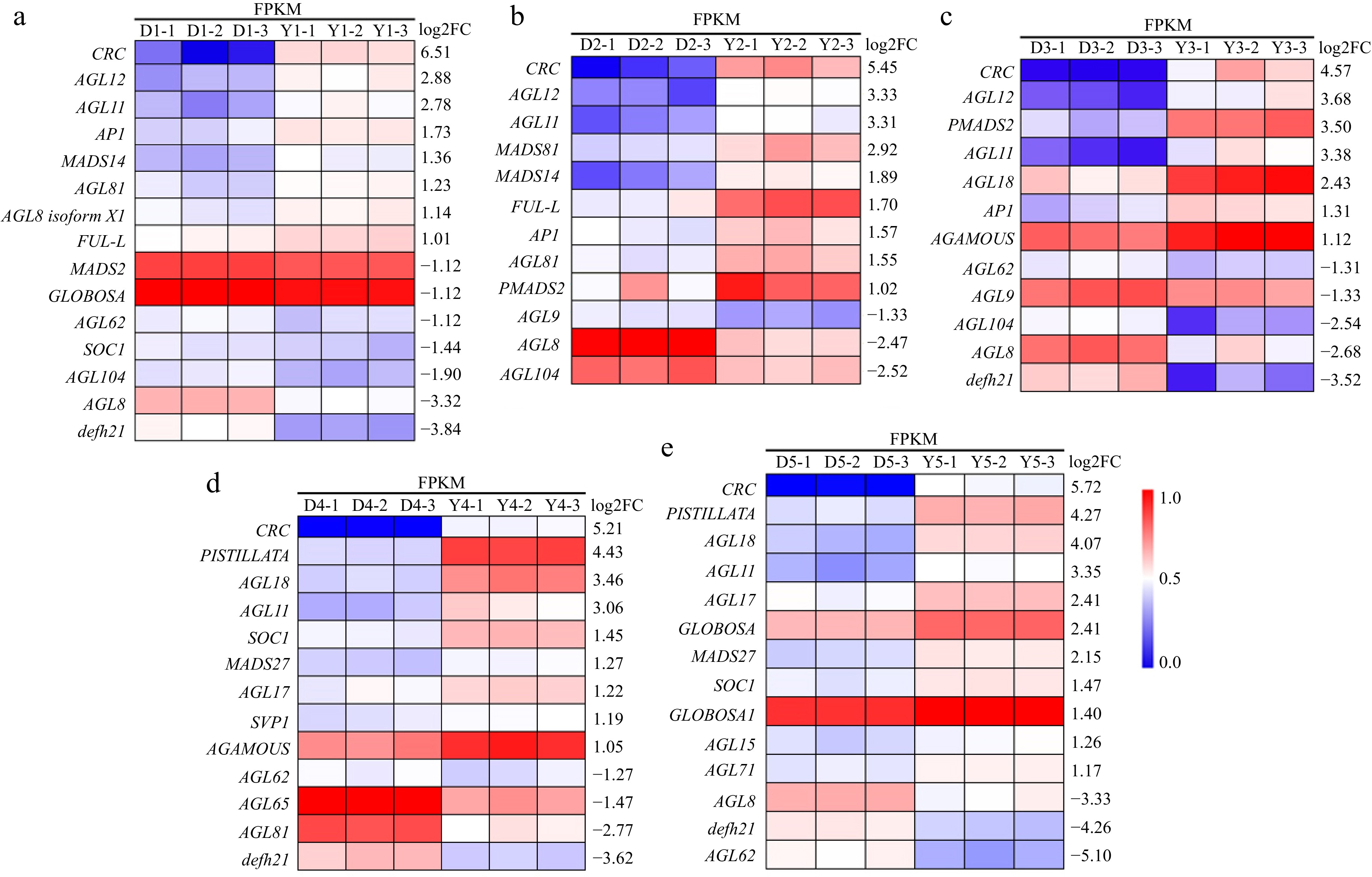
Figure 4.
Differential expression of genes related to flower organ development in the two cultivars. (a) Differential expression genes related to flower organ development in the linggeng stage. (b) Differential expression genes related to flower organ development in the xingyan stage. (c) Differential expression genes related to flower organ development in the initial flowering stage. (d) Differential expression genes related to flower organ development in the full flowering stage. (e) Differential expression genes related to flower organ development in the late full flowering stage. Y: 'Huangchuan Jingui', D: 'Chenghong Dangui'. Y1 and D1: Linggeng stage, Y2 and D2: Xingyan stage, Y3 and D3: Initial flowering stage, Y4 and D4: Full flowering stage, Y5 and D5: Late full flowering stage. Three replicates for each analysis.
CRC and AG transcription factors may regulate each other to promote the development of carpels in sweet osmanthus
-
A transcriptome sequencing analysis showed that the CRC gene was significantly up-regulated in the floral organs of 'Huangchuan Jingui' across all five periods, compared with levels in 'Chenghong Dangui', while the AG gene was up-regulated in the floral organs of 'Huangchuan Jingui' at the initial and full flowering stages. Previous studies have shown that CRC and AG are two key genes that determine carpel formation[10,11,13,14]. A dual luciferase assay showed that the activity of the AG gene promoter was 1.43-fold higher in tobacco leaves co-transformed with 35S::CRC and AGpro::LUC plasmids as compared to that in control tobacco leaves transformed with only AGpro::LUC plasmids (Fig. 5a). Compared with activity in control tobacco leaves transformed with only CRCpro::LUC plasmids, the activity of the CRC gene promoter was up-regulated by 1.45-fold in tobacco leaves co-transformed with 35S::AG and CRCpro::LUC plasmids (Fig. 5b).
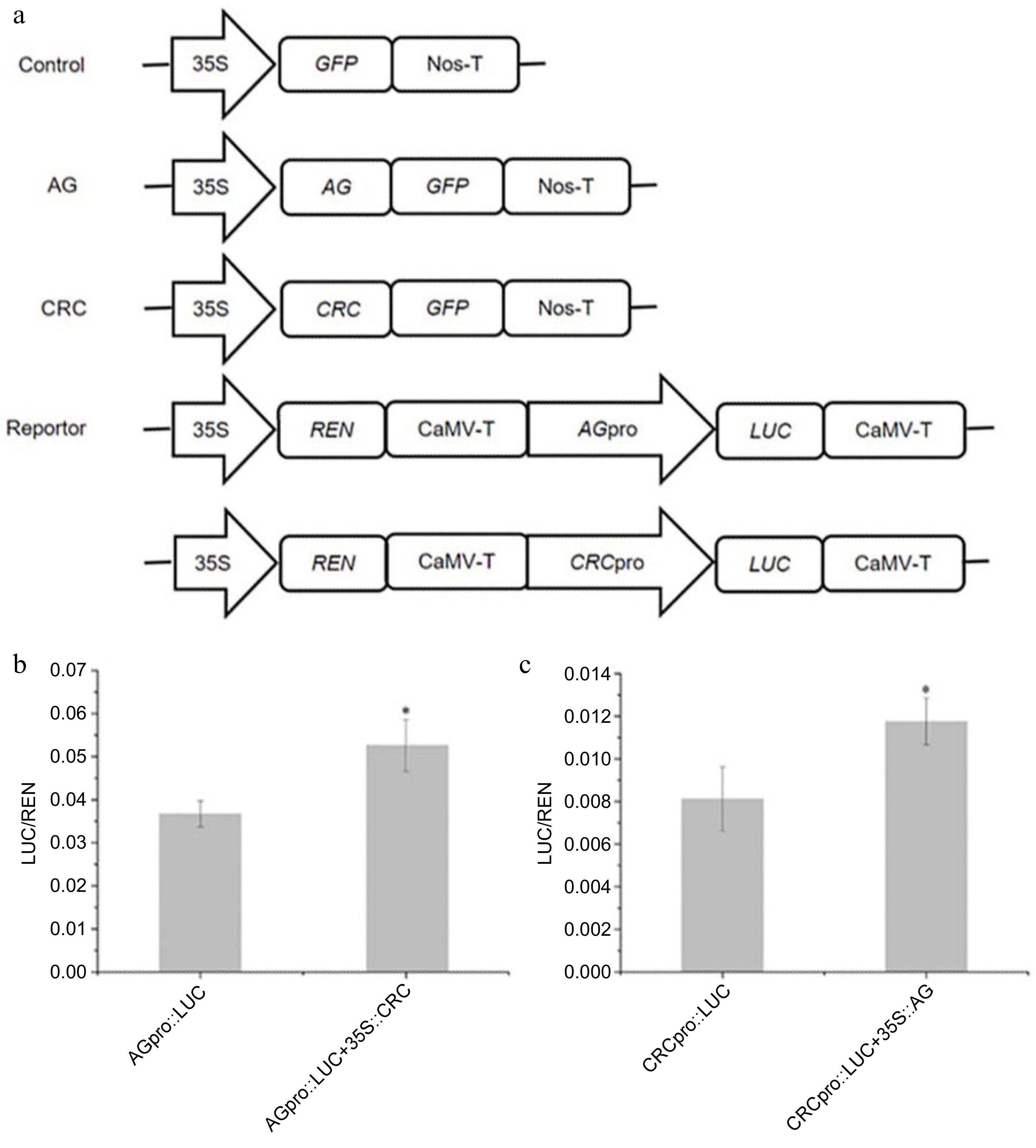
Figure 5.
Transcriptional regulation of CRC and AG genes. (a) The effector and reporter plasmids used in dual-LUC assays. REN: Renilla luciferase, LUC: Firefly luciferase. (b) The AG promoter activity (LUC/REN ratio) of tobacco leaves coinfiltration with Agrobacteria carrying effector and reporter. (c) The CRC promoter activity (LUC/REN ratio) of tobacco leaves coinfiltration with Agrobacteria carrying effector and reporter. The data represent the means ± SD of three replicates from three independent experiments. * P < 0.05.
-
Flower bud differentiation is an important process in the transition of plants from the vegetative to the reproductive stage[30]. Understanding the mechanism underlying flower bud differentiation in plants is important for formulating reasonable cultivation measures for flowering regulation, implementing them to improve annual production of ornamental plants, and clarifying the genetic regulation of plant traits[31]. In this study, the leaf-like structure surrounded by each inflorescence of sweet osmanthus was considered as the involucre; accordingly, the bract differentiation period could be more appropriately termed as the involucre differentiation period. The period from the emergence of the inflorescence primordium to the differentiation of the calyx primordium involves the emergence of the embryonic cyme of sweet osmanthus, and the terminal flowers and lateral flowers differentiate later. Therefore, the inflorescence primordium and flower primordium emerge simultaneously at this stage, which is more appropriately referred to as the flower primordium differentiation period. The emergence of the flower primordium and the formation of the perianth primordium are short and continuous processes; accordingly, the sepal and petal differentiation stages should be combined into the perianth differentiation stage. Therefore, in this study, flower bud differentiation in sweet osmanthus was divided into five periods: involucre differentiation, floral primordium differentiation, terminal perianth differentiation, stamen differentiation, and pistil differentiation.
Previous studies have shown that when the pistil of fertile varieties differentiate, the carpel primordium first appears as a protrusion and develops gradually, forming a small hole in the center, which extends and fuses to form the carpel primordium. In sterile varieties, two protrusions form on the inner side of the stamen, and these do not fuse at the end but form two fronds[32]. In this study, regardless of whether the variety was fertile or sterile, two protrusions formed initially on the inner side of the stamen primordia. However, in the later growth period of the fertile 'Huangchuan Jingui', the two protrusions fused together and gradually developed into the ovary, style, and stigma. The 'small hole' would be the slit between the two protrusions when they fuse.
The shape and size of plant pollen, the type of germination pore, and surface characteristics of plant pollen are important features for evolutionary analyses and plant classification. Previous studies have shown that a longer pollen is related to a smaller surface-to-volume ratio and more derived taxa, while the most derived pollen shows reticulate, striped reticulate, or fine reticulate outer wall characteristics[33,34]. The results of this study revealed that the surface of the pollen grains of the two varieties of sweet osmanthus had reticulate patterns, indicating that their pollen was highly evolved. Among the two varieties, the P/E value of 'Huangchuan Jingui' was smaller than that of 'Chenghong Dangui', indicating that the pollen grains of 'Huangchuan Jingui' were rounder while those of 'Chenghong Dangui' were longer. Hence, 'Chenghong Dangui' may be more evolved than 'Huangchuan Jingui'.
Androdioecy is an extremely rare plant reproductive system where in both male and hermaphroditic plants are present. It has been reported in only a few plants (<0.005%) and is usually found in Oleaceae[1,35]. Androdioecy is considered an intermediate state between monoecious and dioecious plants[36,37], probably resulting from the sterility of pistils in monoecious plants[36, 38−40]. In the ABCDE model of floral organ development, class C functional genes (e.g., AG) determine the development of pistils and carpels. Class C genes have been found in Zeamays[6], E. californica[9], P. sativum[41], Populus alba[10], A. thaliana[16,42,43] and other plants. In this study, the class C functional gene AG was up-regulated in both the initial and full flowering stages of 'Huangchuan Jingui', suggesting that these stages are critical periods for carpel development. The ectopic expression of the STK (AGL11) gene in ag-deficient mutants can promote carpel development[44]. Hence, AGL11 is an important gene for the development of carpels, ovules, and fruits[43−46]. In this study, the expression levels of AGL11 in 'Huangchuan Jingui' across five periods, including the ling geng stage, xingyan stage, initial flowering stage, full flowering stage, and late full flowering stage, were significantly higher than those of 'Chenghong Dangui', indicating that this gene is an important determinant of the development of carpels and ovules in sweet osmanthus. Previous studies have shown that the CRC gene contributes to carpel and fruit development. Arabidopsis CRC is primarily required for the elaboration of carpel morphology[25,28], and mutations in the CRC gene contribute to the female-to-male transition in melons[47]. In this study, the expression level of the CRC gene in all five stages was significantly higher in 'Huangchuan Jingui' than that in 'Chenghong Dangui', suggesting that this gene is involved in the development of carpels in sweet osmanthus.
Previous studies have shown that both CRC and AG are important genes for carpel development, and AG can promote the expression of CRC genes[13,48−53]. The results of this study showed that AG in sweet osmanthus can bind to the promoter and thereby regulate the expression of CRC, and CRC can bind to the promoter and regulate the expression of AG. The results of this study suggest that both genes, CRC and AG, may regulate each other and promote the expression of downstream genes, thereby determining carpel development in sweet osmanthus. Genes that function downstream of CRC and AG and their mechanisms of action needs further investigation.
-
The flower buds and flower organs at the linggeng stage, xingyan stage, initial flowering stage, full flowering stage, and late full flowering stage of both the 'Huangchuan Jingui' and the 'Chenghong Dangui' cultivars were harvested from the campus of Henan University (China).
Paraffin sectioning
-
The experimental materials were washed with water and fixed with formalin-acetic acid-alcohol (FAA) solution. Flower buds of different growth parts and sizes were collected and cut into slices according to the conventional paraffin sectioning method[54], with a thickness of 8 μm. Sections were stained with alum-hematoxylin, sealed with neutral gum, and observed and photographed under an OLYMPUS BX60 microscope.
Scanning electron microscopy
-
The anthers were peeled off and placed in a dry place to allow the pollen to disperse naturally. The pollen was spread on double-sided conductive adhesive, vacuum-dried, sprayed with gold coating, and observed and photographed under a JSM5600LV scanning electron microscope. The polar axis length (P), equatorial axis length (E), and germination groove length (L) were measured using a screen measurement tool, and the P/E and L/P values were calculated. In total, 30 pollen grains of each variety were measured and average values were obtained for each parameter. SPSS 22.0 statistical software was used to analyze the measurement data. Difference is considered significant at P < 0.05. There were three independent repeated experiments for the analysis.
Pollen activity analysis
-
The pollen grains at the initial flowering stage were taken and stained with 2,3,5-triphenyltetrazolium chloride (TTC) staining solution and fluorescein diacetate (FDA) staining solution, and images were obtained under a light microscope and a fluorescence microscope, respectively. The viability of pollen was examined by in vitro culture, using aniline blue staining and fluorescence microscopy for observation and photography.
RNA sequencing
-
RNA sequencing (RNA-seq) was performed by Frasergen (Wuhan, China). Transcriptome datasets were generated using the Illumina HiSeq 2,500 sequencing platform (San Diego, CA, USA). Differential expression analysis was performed using the DESeq2R package[55]. Expression levels of differentially expressed genes were computed using the following formula: FPKM = cDNA fragments / [mapped fragments (millions) × transcript length (kb)]. Fold Change ≥ 2 and FDR (False Discovery Rate) < 0.01 were used as differential expression genes screening criteria.
Dual-luciferase reporter assay
-
The cDNA sequences of AG and CRC were amplified and inserted into the pHBT vector (effectors). The promoter regions of AG (2.07 kb) and CRC (2.00 kb) were amplified and ligated into the pGreenII 0800-Luc vector (reporters). The primers for amplification are listed in Supplemental Table S1. EHA105 strains containing effectors and reporters were cotransformed into 5-week-old N. benthamiana leaves. Agroinfiltration was carried out following the method described by Han et al.[56]. The infiltrated plants were cultured at 23 °C under continuous lighting for 72 h. The LUC and REN activities were detected using a dual-luciferase assay kit (Solarbio, China). There were three independent repeated experiments for the analysis. Difference is considered significant at P < 0.05.
This research were supported by the Henan Province Major Research Fund of Public Welfare (No. 201300110900), National Natural Science Fund in China (No. U1604114), Basic Research Project of Key Scientific Research Program of higher education institutions in Henan Province (No. 20zx015).
-
The authors declare that they have no conflict of interest.
-
# These authors contributed equally: Yuanji Han, Yanxia He
- Supplementary Table S1 Primers used in this study.
- Supplemental Files 1
- Supplemental File 1 Differential expression of genes in the linggeng stage of the 2 cultivars.
- 5
- Supplemental File 2 Differential expression of genes in the xiangyan stage of the 2 cultivars.
- Supplemental Files 6
- Supplemental File 3 Differential expression of genes in the initial flowering stage of the 2 cultivars.
- 10
- Supplemental File 4 Differential expression of genes in the full flowering stage of the 2 cultivars.
- Supplemental File 5 Differential expression of genes in the late full flowering stage of the 2 cultivars.
- Supplemental File 6 Enrichment of KEGG pathway in linggeng flowering stage.
- Supplemental File 7 Enrichment of KEGG pathway in xingyan flowering stage.
- Supplemental File 8 Enrichment of KEGG pathway in initial flowering stage.
- Supplemental File 9 Enrichment of KEGG pathway in full flowering stage.
- Supplemental File 10 Enrichment of KEGG pathway in late full flowering stage.
- Copyright: © 2023 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Han Y, He Y, Yue S, Guo B, Zhu Q, et al. 2023. Floral bud differentiation and mechanism underlying androdioecy of Osmanthus fragrans. Ornamental Plant Research 3:11 doi: 10.48130/OPR-2023-0011 

Floral bud differentiation and mechanism underlying androdioecy of Osmanthus fragrans
- Received: 01 February 2023
- Accepted: 12 May 2023
- Published online: 30 May 2023
Abstract: Sweet osmanthus is an androdioecious plant; however, the mechanism underlying pistil sterility in male plants is still unclear. Scanning electron microscopy showed that the structure of pollen grains in the stamens does not differ between the sterile cultivar 'Chenghong Dangui' and the fertile cultivar 'Huangchuan Jingui'. Triphenyltetrazolium chloride and fluorescein diacetate staining as well as in vitro culture experiments revealed that pollen grains were active in both cultivars, indicating that the stamens in both 'Chenghong Dangui' and 'Huangchuan Jingui' could develop normally. When the pistils of the fertile cultivar 'Huangchuan Jingui' differentiated, two protrusions formed on the inner side of the stamen primordium, and these gradually developed and fused together to form the ovary, style, and stigma. The pistil of the sterile cultivar 'Chenghong Dangui' also formed two protrusions on the inner side of the stamen during differentiation; however, instead of fusing, two fronds were formed. These results suggest that male sweet osmanthus are formed due to the abortion of pistils during the development of floral organs. Transcriptome sequencing revealed that the expression levels of carpel development gene CRC, AG, and AGL11 were significantly lower in 'Chenghong Dangui' compared with 'Huangchuan Jingui' at different flowering stages, which provide new insight in the molecular mechanism of pistil abortion in 'Chenghong Dangui'. CRC and AG may regulate each other to promote carpel development.
-
Key words:
- Sweet osmanthus /
- Androdioecy /
- Floral bud differentiation