









-
Grapevine belongs to the Vitaceae. Because of its wide planting area, high yield, rich nutritional value, and wide range of uses, the grapevine is one of the most economically valuable fruit trees in the world[1]. For hundreds of years, breeders have been working to select higher-yielding, disease-resistant, high-quality grape varieties. The traditional method of grape breeding is hybrid and superior line selection. The characteristic of perennation and high heterozygosity causing new cultivar breeding by the traditional method usually takes 15−20 years[2], which is inefficient and gradually replaced by molecular marker-assisted selection (MAS). MAS can screen progeny at the seedling stage or even before germination, and can select a variety of target characters at the same time, which has many advantages, such as short cycle, low labor cost, and progeny which can achieve multi-character pyramiding breeding[3−5].
Sex is one of the most important traits in grapevine breeding. There are some hypotheses to explain the dioecy of wild grapevines and its evolutionary origin, but the specific mechanism is still not clear[6,7]. Different sex types have different roles in breeding. Such as hermaphroditic vines (complete stamen and pistils can be seen at the flowering stage) are beneficial as cultivars because their self-pollinating characteristics ensure high yields. Female vines (stamens wilt and abortion at the flowering stage) are excellent pistillate parents, and the omission of stamen removal in the cross-operation makes breeding less costly and more efficient. Male vines (pistils absent or poorly developed at the flowering stage) are widespread in the wild and can be used as pollinators for female vine pairings in breeding. Therefore, mapping the locus of sex and developing the linkage identification markers can promote the selection of sex at the seedling stage and greatly improve breeding efficiency.
The marker types and detection techniques used by MAS have undergone many iterations, from the first generation of restriction fragment length polymorphism (RFLP), random amplified polymorphic DNA (RAPD), and amplified fragment length polymorphism (AFLP) markers to the subsequent second generation of simple sequence repeat (SSR) marker[8−11], and then updated to the third generation of SNP molecular marker[12,13]. However, the commonly used high-throughput SNP sequencing methods (genotyping by sequencing, solid gene chip) have many shortcomings[14−16]. Genotyping by target sequencing (GBTS) developed in recent years provides a new solution to this problem. This technology has the advantages of high efficiency, wide applicability, and economy[17,18]. It can detect a large number of targets designed at one time, and sequence thousands of known SNP sites covering the whole genome, so comprehensive genome information could be obtained at a lower cost. Therefore, GBTS can be used in many ways, such as genetic resource evaluation, genetic map construction, quantitative trait locus (QTL) mapping, target gene cloning, and so on. And because the tested SNP can be designed based on the mutation rate of the whole species, it has a high degree of versatility in this species. At present, GBTS technology has been successfully applied to many crops besides grapevine.
Flower sex locus were located from the American grape and the Eurasian grape, but there was still no sex locus from the East Asian grape. It was not clear whether the sex locus in the East Asian grape was still in the same position, which was important for understanding the evolutionary analysis of grape populations. In the present study, a total of 20,597 probes covering the whole grape genome were developed for GBTS. This array was used to sequence the hybrid population of 'Cabernet Sauvignon' × 'Huadong1058', construct the genetic map, detect flower sex determination locus by mapping and GWAS, and develop sex identification markers combination for MAS.
-
A total of 165 individual grapevines were used in this study, including 34 varieties/accessions and 131 progenies from the crosses of V. vinifera 'Cabernet Sauvignon' × V. pseudoreticulata 'Huadong1058'. All progenies were identified as true hybrids using 10 SSR markers (Supplemental Table S1). The vines were planted in the Center for Viticulture and Enology, Shanghai Jiao Tong University (Minhang District, Shanghai City, China).
DNA extraction
-
Genomic DNA was extracted from the young leaves of the all grapevines using CTAB methods as described by Qu et al [19]. DNA quality was estimated by 1% agarose gel electrophoresis with a λ-DNA ladder and the DNA concentration was evaluated using NanoDrop 2000 (Thermo Fisher Scientific, Waltham, MA, USA).
Target location selection for GBTS and hybridization probe design
-
The target locations were selected from the Illuminar 20K Chip and previous GBS sequencing data[20−22]. A total of 20,597 locations were selected according to the following criteria: minor allele frequency (MAF) > 0.1, the proportion of the missing data < 5%, and loci evenly distributed on the genome. For each target location, a 110 bp probe covering the target location was designed using GenoBaits Designer software (MolBreeding Biotechnology Co., Ltd., Shijiazhuang, China).
Library construction, probe hybridization, and sequencing
-
GBTS library construction and probe hybridization was conducted as described by Guo et al. [23] In brief, library construction consists of four steps: 1) DNA was fragmentated by ultrasonic; 2) Fragmented DNA was end-repaired and added with an A tail; 3) Adapters with barcode sequences were ligated to the A-tailed segments; 4) Library was amplificated by PCR. After the library construction was done, probe hybridization was performed through library mixture, library hybridization, target capture, library amplified, purification, and library control. All processes were accomplished by kinds of instruments automatically for labor-saving and time-saving.
Qubit 2.0 Fluorometer (Thermo Fisher Scientific, CA, USA) was used to assess the quality of enriched libraries. Samples that passed quality control were loaded onto the flow cell, and sequenced with PE150 on the MGISEQ-2000 platform (MGI, Shenzhen, China).
The multiple SNPs developed from a single amplicon (including target SNP and adjacent regions) are called mSNPs. To maximize the use of sequencing data, mSNPs that might exist in each amplicon were detected. The method of mSNP development according to the report by Guo et al. [23]
Phenotyping of flower sex
-
Sex phenotype (male, female, and hermaphrodite) was evaluated by flower morphology observation according to the evaluation criteria of the International Organization of Vine and Wine descriptors (No. OIV-151)[24]. The evaluation was repeated in 2015 and 2019.
Map construction
-
The mSNP markers detected from the two parents were classified into eight segregation types: 'aa × bb', 'ab × cc', 'cc × ab', 'ab × cd', 'ef × eg', 'hk × hk', 'lm × ll' and 'nn × np'. Heterozygous markers in the parents were used to construct the genetic map. mSNPs with integrity > 0.9 were retained for further analysis. Because the chromosome information was carried on the mSNPs, markers from the same chromosome were assigned directly to the same group to reduce the computational complexity of JointMap 4.0 software. A LOD (log of odds) score of 6 was taken as the linked markers threshold. Markers that significantly affected the marker order of the linkage group were discarded. The 'Individual genot freq' function was used to discard individuals that had too many missing genotypes. The 'locus genot. Freq.' function was used to discard the markers with segregation distortion exceeding the threshold (p < 0.05) or abnormal segregation ratios. The 'similarity of Loci' was used to discard the markers with similarity equal to 1. The marker order was calculated by 'regression mapping' function and the distance between markers was calculated by Kosambi's function. Finally, the genetic map was drawn using MapChart software.
Locus mapping
-
Sex determination locus mapping was performed using MapQTL 6.0 software. The files of phenotypic (.qua), map (.map), and loci (.loc) were imported into MapQTL 6.0. Interval mapping (IM) was used to detect putative loci related to the flower sex in a step size of 0.5 cM. MQM (Multiple QTL mapping) was used to accurately calculate the loci detected by IM combined with the cofactor in step size of 0.5 cM. The cofactor was selected from the marker closest to the position with the highest LOD value. The LOD threshold (α = 0.05) was calculated by 1,000 permutation tests.
GWAS analysis
-
The GWAS was conducted by GAPIT (version 3)[25]. GLM (Generalized Linear Model), MLM (Mixed Linear Model), SUPER (Settlement of MLM Under Progressively Exclusion Relationship), FarmCPU (Fixed and random model Circulating Probability Unification), and BLINK (Bayesian-information and Linkage-disequilibrium Iteratively Nested Keyway) algorithms were tested. The GWAS algorithm performances were evaluated through quantile-quantile (QQ) plots. A conservative threshold for assessing SNP significance was calculated based on the Bonferroni correction for a type I error rate of 0.05.
Development of sex linkage marker combination
-
According to the co-analysis results of GWAS and flower sex phenotyping, the marker 'chr2_4,825,970' co-segregated with male/non-male phenotypes and was named 'SLS1' (Sex Linkage SNP1). Further correlation analysis was performed between all the makers in the sex determination locus with hermaphrodite or female individuals. A marker 'chr2_4,758,220' was identified upstream of 'SLS1', which co-segregated with the female/hermaphrodite phenotype under a non-male condition and was named as 'SLS2'.
-
With a large amount of previously known resequencing data, 20,597 core SNPs (cSNPs) with high detection rates, homogeneity, and repeatability were screened out. By designing probes covering cSNPs and high-throughput detection of their captured fragments, these cSNPs and 76,856 other SNPs in these regions were identified. All these 97,453 SNPs are called multiple SNPs (mSNPs).
These markers were evenly distributed on 19 chromosomes (chr), covered 457,925,245 bp of the genome (Fig. 1a, Table 1). Among them, Chr18, the longest chromosome, was covered by 1,408 cSNPs/6,587 mSNPs, and the shortest chr17 was covered by 1,013 cSNPs/4,808 mSNPs. The average distance between cSNPs was 22,233 bp. By comparison with the reference genome annotation (VCost. v3), the coverage length of markers was more than 99% in each chromosome of the grapevine genome (Fig. 1b). By analyzing the location of cSNPs in the genome, the three largest classes were: 7,199 cSNPs in introns, 4,659 cSNPs in intergenic regions, and 4,165 cSNPs in exons, respectively (Fig. 1c). That implied that a large number of markers were located in gene regions.

Figure 1.
Characteristics of GBTS array. (a) Distribution of cSNPs on each chromosome. Color indicates the number of cSNPs within 1 Mbp window size. (b) Coverage length of markers compared with reference genome (VCost. v3). (c) Annotation of the location of the cSNPs. (d) Number and proportion of MAF for all cSNPs.
Table 1. Characteristics of SNPs distributed on 19 grape chromosomes.
Chr. SNP no. mSNP no. Distance
(bp)Average SNP
interval (bp)mSNP/
SNP1 1,198 5,357 24,200,107 20,200 4.47 2 1,015 4,619 18,860,487 18,582 4.55 3 944 4,408 20,668,111 21,894 4.67 4 1,175 5,347 24,682,346 21,006 4.55 5 1,134 5,229 25,554,074 22,534 4.61 6 1,038 4,838 22,638,265 21,810 4.66 7 1,328 5,447 27,330,303 20,580 4.10 8 754 3,067 22,542,685 29,897 4.07 9 735 3,464 22,840,396 31,075 4.71 10 1,230 5,487 23,441,644 19,058 4.46 11 958 4,390 20,025,463 20,903 4.58 12 1,156 6,032 24,240,562 20,969 5.22 13 1,313 6,880 29,056,180 22,130 5.24 14 1,262 6,200 30,244,820 23,966 4.91 15 878 4,276 20,254,536 23,069 4.87 16 1,048 5,447 23,491,226 22,415 5.20 17 1,013 4,808 18,650,226 18,411 4.75 18 1,408 6,587 34,516,940 24,515 4.68 19 1,010 5,570 24,686,874 24,442 5.51 Total 20,597 97,453 457,925,245 22,233 4.73 Minor allele frequency (MAF) is an important indicator to evaluate the diversity of markers. In this array, the cSNP with MAF > 0.1 was 8,727, accounting for 42% of all cSNPs (Fig. 1d). A sufficient number of cSNPs with high MAF means that this array has the ability to detect different grapevine cultivars or lines.
Sex segregation in the hybrid population
-
Flower sex is closely related to cultivar selection, cultivation management, and the yield of grapes. Identification of sex related locus can quickly determine the sex of progenies in juvenile when wild grapes, usually male or female unisexual flowers, are used for making crosses. The hybrid population of V. vinifera 'Cabernet Sauvignon' (hermaphrodite flower – female parent) and V. pseudoreticulata 'Huadong1058' (male flower) was separated into flower types (Fig. 2a). The phenotypes of flower sex among the 131 F1 hybrids were collected in 2015 and 2019, respectively. In 2015, among the 83 seedlings bloomed, there were 58 males, 16 females, and 9 hermaphrodites. In 2019, all 131 progenies bloomed with 70 males, 47 females, and 14 hermaphrodites (Fig. 2b).

Figure 2.
The characteristics of flower sex. (a) Flower types among the individuals in the mapping population. (b) Distributions of flower types among the F1 hybrids in 2015 and 2019.
Mapping of sex determination locus
-
From the sequencing data, 27,647 polymorphic mSNPs were identified and divided into eight types: 'aa × bb', 'ab × cc', 'cc × ab', 'ab × cd', 'ef × eg', 'hk × hk', 'lm × ll' and 'nn × np' (Table 2). 'aa × bb' type markers were filtered out because they were not separated in the progeny. 'ab × cc', 'cc × ab', 'ab × cd' and 'ef × eg' types were filtered out because they were too less to have significant influence. Finally, only 'hk × hk', 'lm × ll' and 'nn × np' types of mSNPs were used to construct the genetic linkage map.
Table 2. Marker types distribution.
Marker types Cabernet Sauvignon Huadong
1058Marker number Percentage (%) aa × bb aa bb 10,022 36.25 ab × cc ab cc 84 0.30 cc × ab cc ab 65 0.24 ab × cd ab cd 0 0.00 ef × eg ef eg 86 0.31 hk × hk hk hk 883 3.19 lm × ll lm ll 12,225 44.22 nn × np nn np 4,282 15.49 Total − − 27,647 100.00 The remaining markers were used for further analysis. In segregation distortion analysis, the markers with p > 0.05 were retained. From a similarity analysis, the markers with similarity equal to 1 were filtered out. Then the LOD score of 6 was taken as the threshold for deciding whether loci were linked and the markers were discarded which significantly affected the linkage group marker order. These measures conduced to enhance the accuracy of genetic map and reduce the computational complexity. After that, the genetic linkage map with 422 mSNPs was constructed (Fig. 3). The map contained 19 linkage groups (LGs) and spanned 2,351.71 cM, with an average inter-SNP distance of 5.57 cM. The number of mSNPs on each LG ranged from 16 to 30. The LG8 had the longest length with 177.65 cM, and the LG19 had the shortest length with 80.38 cM (Supplemental Table S2).

Figure 3.
Genetic map of hybrid population crosses from Vitis vinifera 'Cabernet Sauvignon' × Vitis pseudoreticulata 'Huadong1058'. LG1 to LG19 represents 19 linkage groups respectively, and each bar represents a SNP marker. The ruler on the left is the genetic distance (cM).
There was one sex determination locus was identified on the linkage group (Fig. 4). This locus was located on LG2, between 54.74 and 58.80 cM. The physical position was from 3.29 to 5.78 Mbp and the LOD peak was located at 4.83 Mbp. The locus could be detected in 2015 and 2019 by interval mapping and MQM mapping which showed good repeatability, and the PVE (phenotypic variance explained) up to 98.6%.

Figure 4.
Mapping of sex determination locus. (a) Interval mapping of sex determination locus on chr2. (b) Multiple-QTL mapping of sex determination locus on chr2. (c) The overlap region of IM and MQM on chromosome 2, and the markers on the peak. The boundaries of locus were determined by the markers closest to the threshold (LOD = 30) on the flanks.
GWAS analysis of floral sex and linkage locus
-
GWAS approach was conducted by GAPIT (Version 3) using Blink, FarmCPU, SUPER, MLM, and GLM models (Fig. 5a). A locus on chr2 was obtained from different models based on the Manhattan plots constructed using data from both years. This locus had different boundaries among the five models, but its peak was consistently at 4.85 Mbp.
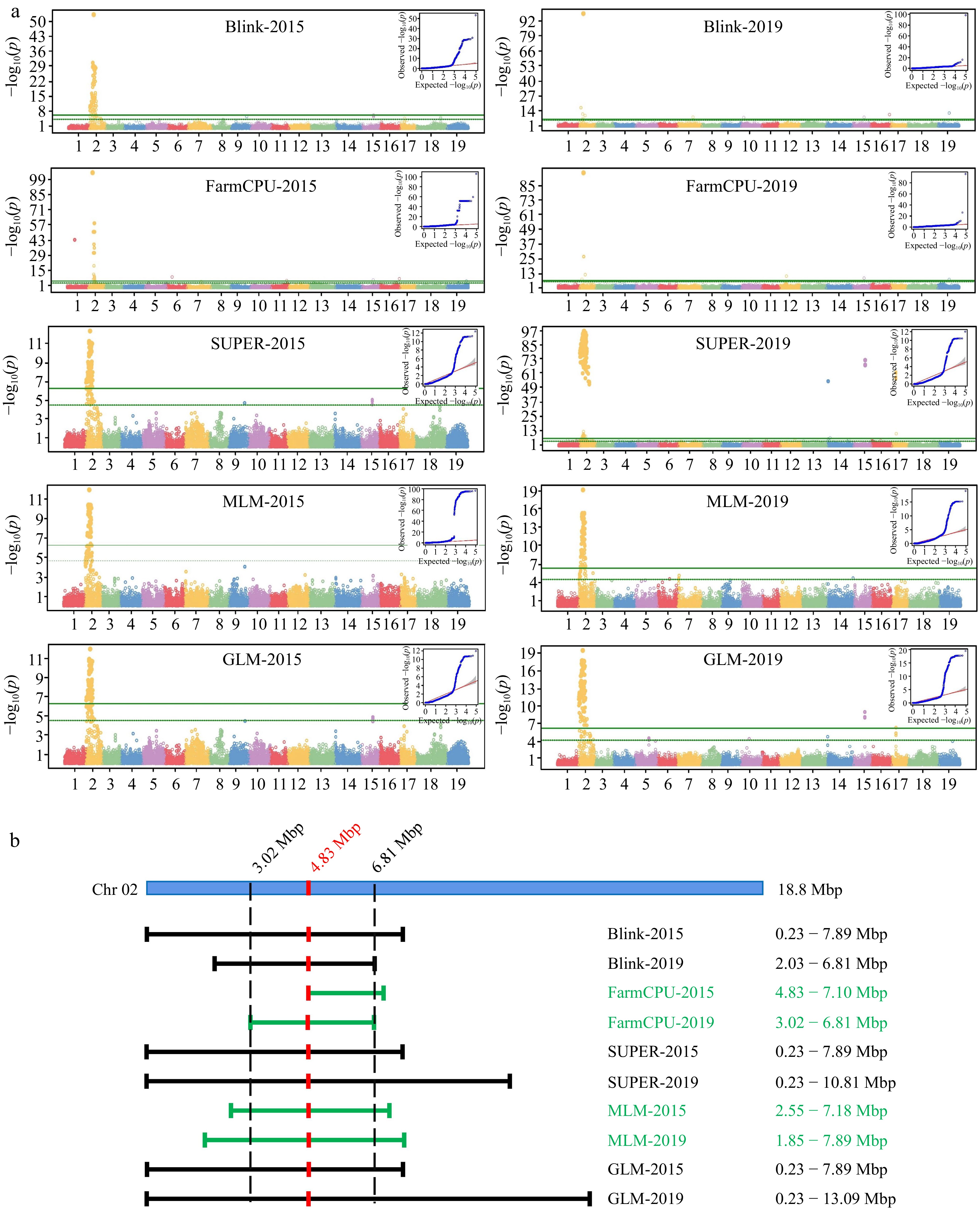
Figure 5.
GWAS Analysis of floral sex and linkage locus. (a) Manhattan plots and QQ plots of GWAS analysis based on five model (Blink, FarmCPU, SUPER, MLM and GLM). The vertical axis of the Manhattan map is the −log10(p) of each marker based on the analysis of different models. (b) Interval of sex determination locus on Chr2.
The boundary position of this locus was further determined using the 2019 segregation data, and the locus was confined between 3.02 Mbp and 6.81 Mbp in chromosome 2 (Fig. 5b). QQ plot of each Manhattan plot indicated that there were significant markers of deviation from random effects that were highly correlated with the phenotype in each group.
The identification of sex identification marker combination for MAS
-
In order to find SNP markers closely associated with flower sex types that could be used for MAS, two SNPs were identified within this locus based on the QTL mapping and GWAS analysis. The first marker with T/C substitution localized at 4,825,970 bp of chr2 was named as SLS1. The second marker localized at 4,758,220 bp near the peak position with C/T substitution, was named as SLS2. The SLS1 are 'CC' in 'Cabernet Sauvignon' and 'TC' in 'Huadong1058', and in SLS2, they are 'AG' in 'Cabernet Sauvignon' and 'GG' in 'Huadong1058', respectively. Individual genotypes and flower phenotypes in this locus were analyzed, and results showed that progenies with 'TC' in SLS1 were always male, regardless the genotypes in SLS2. When progenies carried 'CC' in SLS1, their flower types were determined by the alleles in SLS2, in which progenies with 'GG' are female while those carrying 'AG' were hermaphrodite. In summary, 'T' in SLS1 was tag SNP of male and 'A' in SLS2 was tag SNP of hermaphrodite. The genotypes of SLS1 and SLS2 can always accurately predict the flower types of grapevines, and those progenies carrying 'TC-XX' was male, while the progenies carrying 'CC-GG' was female and the progenies carrying 'CC-AX' was hermaphrodite ('X'represents any nucleotide type). We proved that the accuracy of flower type estimation was 100% in these hybrid populations (Fig. 6, Supplemental Table S3).

Figure 6.
Model of sex identification marker combination on chromosome 2. 'A', 'T', 'C' and 'G' represent four nucleotide type respectively, 'X' represents any nucleotide type. Two dashed lines of each sex type represent the two sister chromatids of chromosome 2.
To determine whether the SLS1-SLS2 combination can be used for prediction of flower types in different grapevines varieties/accessions, a total of 34 wine, table, juice, and rootstock grape varieties of which six are male, 27 are hermaphrodite, and one is female, were used for the validation study. Based on the results obtained from the segregating populations. All sex types were predicted accurately, which also confirms the accuracy of sex identification markers combination in this study (Supplemental Table S4).
-
SNP marker development usually adopts whole WGS (whole genome sequencing)[15], GBS, solid chip, and GBTS. WGS can detect each possible SNP marker by sequencing all regions of the genome. But the high cost limited the application of WGS in the larger number of plant materials, and breeding does not need all the nucleotide information. GBS technology reduced the difficulty of data analysis and cost by selecting DNA fragments for sequencing and SNP development, and a large number of SNP markers throughout the genome also meet the needs of marker development and breeding[14,26,27]. The GBS technology has been successfully applied in a variety of crops[28]. However, the instability of SNP markers caused by the limitations of species, materials, and platforms seriously affects the comparison and utilization of GBS data in different studies. Solid chip can efficiently detect predefined target SNPs, which has been widely used extensively in horticultural crop research, but it was too costly to be economical in breeding. The GBTS technology, which selects fixed targets for sequencing, combines the advantages of GBS and solid chip to meet both fixed sites and low cost[29,30]. The makers obtained by GBTS have stable repeatability through different platforms, and this integration can provide markers across the board for different usages. Compared with traditional genotyping chip (solid gene chip), the GBTS can detect more abundant SNPs, the mSNPs are distributed in clusters, and it has universality in different studies (Supplemental Figs S1, S2).
The GBTS has a broad application prospect in breeding because of its advantages, and it has been widely used in many crops such as maize[23,31,32], wheat[33,34], rice[35], pepper[36], broccoli[37], etc., but there is no report on grapevine. The SNP array developed in the present study is the first application of GBTS on grapevine. and its reliability in locus mining and breeding marker development have been explored and verified.
The sex determination loci and its mechanism are consistent
-
Grapevine with different flower sex have different uses in breeding. The hermaphrodite varieties can self-pollinate due to their own pistils and stamens, which faciliate higher yields and regular fruit production[38]. The female varieties are convenient material for genetic research because of no artificial stamen removal. This can greatly reduce the effort of artificial pollination operations and avoid false hybrids caused by own pollen.
The flower sex and related markers have been reported many times in previous studies. In 2000, the sex locus had been identified which located at 3.7 Mbp on chromosome 2 from a hybrid population of the cross 'Horizon' ('Seyval' × 'Schuyler') × Illinois 547-1 (V. cinerea B9 × V. rupestris B38[39]. After that, the sex locus had been located between markers VVIB23 and VVMD34, the population used was from the cross V. rupestris and V. arizonica[40]. In 2012, the sex locus had been located between 4.92−5.05 Mbp on chromosome 2. Sequencing and gene annotation of the target region were performed to reveal several potential candidate gene with flower sex. The population material derived from a cross of V. vinifera background variety and a rootstock variety (V. riparia × V. cinerea)[41]. Two years later, sex locus had been revealed that which located between 4.921−5.010 Mbp on chromosome 2. Furthermore, the results that H alleles were more closely related to M than to F alleles was revealed by both diversity and network analysis[42].
Flower sex locus were located from the American grape and the Eurasian grape, but there was still no sex locus from the East Asian grape. It was not clear whether the sex locus in the East Asian grape was still in the same position, which was important for understanding the evolutionary analysis of grape populations. In the present study, flower sex locus was located in an approximate position compared to previous results. It was also the first time that variety of V. pseudoreticulata was used to build the hybrid population for sex locus mapping, and confirmed the flower sex locus was unique in different populations. The similar results indicate the data from GBTS SNP array we designed is reliable for genetic map construction and QTL mapping.
The mechanism of sex determination in grapevine flowers has been partially revealed
-
As with many important crops, grapevine in the wild is usually dioecious. The two-locus model is a hypothesis for the origin of dioecy, which states that dioecy evolved from a hermaphroditic ancestor and involved two stages[6,7,43]. The first stage is to generate gynodioecy, which caused by a male-sterility mutation. The individuals with this homozygous mutation have decayed stamens and retain only female function. The second stage is to generate male individuals, which caused by a dominant female-sterility mutation. The individuals with this mutation suppress female function and retain male characteristics. In this hypothesis, the recombination between the two loci would lead to the restoration of hermaphrodites[6,44].
The Vitis genus contains dozens of dioecious wild species, a rare occurrence in plant[45,46]. This observation suggests that dioecy originated once in the Vitis genus because the majority of its ancestors had hermaphroditic flowers and because dioecy is uncommon in flowering plants. Previous study has identified the male (M) and female (f) haplotypes of the sex-determining region (SDR) in the wild grapevine species V. cinerea, which confirmed the boundaries of the SDR. Based on the whole-genome shotgun sequences of 556 accessions, the sex-determining locus was considered conservative in Vitis genus[47]. In breeding, the markers co-separated with SDR can be used to screen the sex of grapevines at seedling stage. The hybrid populations from 'Cabernet Sauvignon' and 'Huadong 1058' used in this study can be used to breed excellent progenies with both flavor quality from V. vinifera and disease resistance V. pseudoreticulata. On the other hand, given the separation of its gender, it can be used to explore the markings that are separated from the SDR.
In this research, compared with SLS2, the SLS1 was a dominant SNP. SLS1 heterozygous resulted in male individuals regardless of SLS2 genotype, which was consistent to the two-locus model. We hypothesized that SLS1 is closely linked to the female-sterility mutation. Previous study has shown that a transcription factor VviYABBY3 (4.81 Mbp) which had potential female-sterility function was located near SLS1[43]. This result supported our hypothesis.
The genotypes of SLS2 in individuals crossed by 'Cabernet Sauvignon' × 'Huadong1058' were only 'GG' and 'AG'. However, among the 34 cultivated varieties/accessions, that showed 'GG', 'AG' and 'AA'. When SLS1 was 'CC' homozygous, individuals with 'AG' and 'AA' SLS2 genotypes were hermaphrodites. We also hypothesized that the 'A' in SLS2 might be linked to stamen development locus, which remained to be proven.
In results related to male-sterility locus, a candidate mutation in the VviINP1 had been identified, that revealed an INDEL in VviINP1 was conserved in all female haplotypes[43]. Recently research indicated that VviPLATZ1 is a key regulator of female flower formation in grapevine[48]. Functional analysis in the rapid cycling hermaphrodite microvine utilizing the CRISPR/Cas9 gene-editing method revealed that deletion of VviPLATZ1 is a crucial component that governs reflex stamen development during female flower production.
-
In this study, the sex determination locus was mapped and sex identification marker combination was developed, using the sequencing data of 131 progenies from crosses of V. vinifera 'Cabernet Sauvignon' × V. pseudoreticulata 'Huadong1058' by GBTS. A total of 20,597 cSNP (97,453 mSNP) coving more than 99% of the genome were developed to construct SNP array, in which most of markers were located in gene regions and had sufficient diversity. In order to mapping the sex determination locus, sex types were surveyed in 2015 and 2019, genetic map construction and GWAS were performed using GBTS data. The sex determination locus was finally located at 54.74−58.80 cM by mapping and 3.02−6.81 Mbp by GWAS, with the common peak at 4.83 Mbp on chr2. In this locus, a marker combination of 'SLS1-SLS2' was identified, and 34 species/cultivars were used to evaluate the accuracy of the combination for identifying the sex type of grapevine.
This work was supported by Key Research and Development Program of Xinjiang Autonomous Region (Grant No. 2022B02045-1-2), Shanghai Municipal Agricultural Commission (Grant No. 2021-02-08-00-12-F00751), National Natural Science Foundation of China (Grant No. 32272652), The Special Fund for the Central Government Guides Local Science and Technology Development (Guike ZY21195039), State Key Laboratory of Crop Stress Biology for Arid Areas, NWAFU (CSBAA202201), and Digital agriculture and grape whole industry chain agriculture and tourism integration development project (Grant No. liangshanbei-2021).
-
The authors declare that they have no conflict of interest. Jiang Lu is the Editorial Board member of Fruit Research who was blinded from reviewing or making decisions on the manuscript. The article was subject to the journal's standard procedures, with peer-review handled independently of this Editorial Board member and the research groups.
- Supplemental Table S1 Primers for hybrid identification.
- Supplemental Table S2 Genetic map data of hybrid population crosses from Vitis vinifera 'Cabernet Sauvignon' × Vitis pseudoreticulata 'Huadong1058'.
- Supplemental Table S3 Detail of hybrid population sex type and identification marker combination
- Supplemental Table S4 Test of sex identification marker combination in 34 varieties/accessions.
- Supplemental Fig. S1 Flowchart for genotyping by target sequencing with GenoBaits.
- Supplemental Fig. S2 Comparison of GBTS and Solid Genotyping Chip. (a) Distribution of core SNP and mSNP which developed around them based on GBTS. (b) Distribution of SNP based on solid genotyping chip.
- Copyright: © 2023 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Yang B, Wu W, Lv J, Li J, Xu Y, et al. 2023. Identification of sex determination locus and development of marker combination in Vitis based on genotyping by target sequencing. Fruit Research 3:31 doi: 10.48130/FruRes-2023-0031 

Identification of sex determination locus and development of marker combination in Vitis based on genotyping by target sequencing
- Received: 21 April 2023
- Accepted: 11 August 2023
- Published online: 03 November 2023
Abstract: The grapevine is an important and economically valuable fruit crop, with flower sex being a key genetic trait that directly affects grapevine yield and quality. Despite its significance, there is a lack of studies on sex-linked molecular markers that can assist in grapevine breeding. In this study, we developed a grapevine single nucleotide polymorphism (SNP) marker array using a combination of genotyping by target sequencing (GBTS) and capture-in-solution technology and applied it to marker-assisted selection (MAS) of grapevine gender. The SNP array could detect a total of 20,597 core SNPs and 97,453 multiple SNPs (mSNPs), covering over 99% of the grapevine genome on each chromosome. A total of 131 progenies from a cross between Vitis vinifera 'Cabernet Sauvignon' and Vitis pseudoreticulata 'Huadong1058' that exhibited segregated sex phenotypes were sequenced using this array. Through locus mapping and a genome-wide association study (GWAS), a locus on chromosome 2 (54.74−58.80 cM) that explained 98.6% of the phenotypic variation was identified. To further utilize this locus, a sex prediction marker combination consisting of two SNPs was developed, which accurately predicted the sex of 34 natural grapevine varieties/accessions. This study demonstrates the application of GBTS in grapevine breeding and provides a reliable MAS marker set for early-stage sex selection.
-
Key words:
- Grapevine /
- GBTS technology /
- Sex determination locus /
- Genetic breeding /
- Marker-assisted selection