






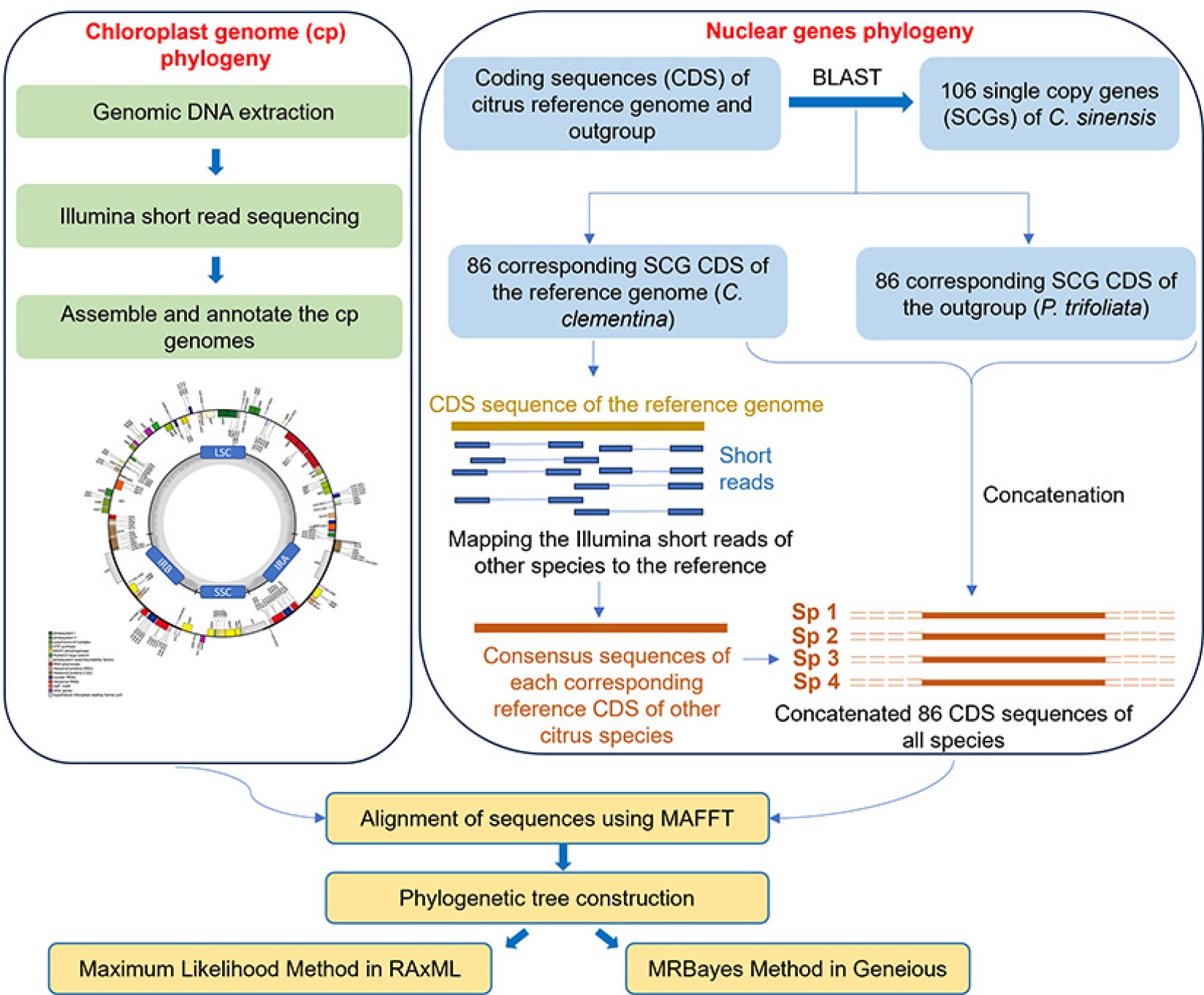



-
Citrus belongs to the family Rutaceae and subfamily Aurantioideae. The Aurantioideae subfamily is composed of 33 genera and 203 species[1]. The genus citrus has been described as comprising of 1−162 species in different classification systems[2]. Citrus is naturally distributed in a vast area covering the regions of Southeast Asia and Australia[3,4]. The progenitor species of Australian limes are thought to be derived from Southeast Asia and migrated to Australia where they evolved and diversified during the early Pliocene around 4 Ma[3]. The derivation of Australian citrus from Southeast Asia might have resulted from cross-oceanic dispersal by birds or ocean currents when the water level was very low between the two continents[4]. Phylogenies based on complete chloroplast genomes reveal two speciation events in history, one separating Australian citrus and citrons from all other citrus species 8.1 Ma, and a further divergence between Australian citrus and citrons at a later date[5]. There are six citrus species which are endemic to Australia. The evolution of these citrus has resulted in some species with adaptations to tropical rainforests (previously classified as Microcitrus (F.Muell.) Swingle) and one species adapted to semi-desert conditions (previously classified as Eremocitrus (Lindlay) Swingle. The geographical isolation of Australian species from all other citrus and the diversity of edaphic and climatic environments has resulted in them displaying very distinctive characteristics[4,6].
Australian native limes, including Citrus australasica F.Muell (Australian finger lime), C. australis (Mudie) Planchon (Gympie lime), C. glauca (Lindlay) Burkill (Desert lime), C. inodora F.M. Bailey (Russell River lime) and their hybrids have shown useful levels of resistance to Huanglongbing (HLB), a disease which continues to be highly detrimental to citrus production in many parts of the world[7]. Studies with C. australasica have revealed many genes involved in immune related signalling pathways and the regeneration of cell wall and phloem which provides HLB resistance and a fast recovery from infection[8,9]. Furthermore, recent genomic approaches using C. australis have led to the identification of defence related genes with possible involvement in HLB resistance[10]. Aside from disease resistance, the wide morphological and fruit compositional differences between C. australasica accessions has led to their use in breeding[11]. Fruits of C. australasica and hybrids with this species are popular as commercial foods due to their unique appearance and taste, whilst the tree itself has significant ornamental appeal[11]. C. glauca is another important candidate for use in citrus breeding, showing important characteristics such as tolerance to drought, salt, nematodes and viroids[12,13]. These traits make the Australian citrus species important genetic resources in citrus breeding. A better understanding of the genetic relationships of these species may facilitate improved utilization in rootstock and scion breeding programs.
The chloroplast genome is a highly conserved structure among angiosperms with the four main components in the most common quadripartite structure: a large single copy (LSC) region, a small single copy (SSC) region, and two inverted repeat (IR) regions[14]. Chloroplasts are primarily maternally inherited units in plants (biparental and paternal inheritance are also known) with a highly conserved structure in terms of gene content, gene sequence and order. They have a simple structure, small size, high copy number, lack of recombination and low nucleotide substitution rates thus making them the primary source for determination of maternal parentage, and the evolutionary relationships among species[15−18]. Complete chloroplast genome sequences provide overwhelmingly more accurate information about chloroplast evolutionary relationships than a selected few chloroplast DNA sequences, especially between closely related species[19]. An extensive number of chloroplast genomes of different citrus species have been assembled and annotated using short read technology and thus have been used in phylogenies to decipher the relationships, discriminate taxa between and/or within genera/species and to redefine taxonomic boundaries[3,20−24].
Nuclear genomes have higher evolutionary rates than mitochondrial and chloroplast genomes. Consequently, they encompass larger variation within a unit of sequence than in uniparental genomes, which is of great importance in achieving better resolved phylogenies, particularly at low taxonomic levels. The evolutionary independence and biparental inheritance of low copy nuclear genes make them particularly useful in the accurate assessment of taxa[18,25]. Numerous single copy genes have been used to better understand the relationships among species[26]. Polymorphisms in nuclear loci have been used to infer the relationships among citrus taxa using six nuclear genes[27], 27 nuclear genes[28], 80 single copy nuclear genes[29] and 103 single copy genes[30] in previous studies.
Although there are a growing number of phylogenetic studies for citrus, there are few which have detailed the evolutionary relationships among Australian citrus species with respect to other widely cultivated citrus species. Three Australian taxa (C. australis, C. australasica and C. glauca) were included in an evolutionary and phylogenetic assessment based on complete chloroplast genome sequences[5], polymorphic cp DNA markers[15], and polymorphisms in non-genic and non-pericentromeric genomic regions[3]. The only known molecular phylogeny study involving all six Australian endemic citrus is that of Rich (2004 unpublished) in which ten additional citrus species were compared using three introns and intergenic spacer regions . However, the deduced evolutionary relationships among Australian wild taxa in previous studies have limitations due to the use of a small number of taxa and/or phylogenies based on single copy genes and partial cp DNA sequences.
The objective of this study was to better understand the relationships among all six Australian endemic citrus species (C. australasica, C. australis, C. garrawayi F.M. Bailey, C. glauca, C. gracilis Mabb., and C. inodora), and how the other 13 main cultivated citrus [C. × aurantifolia (Christm.) Swingle, C. × aurantium L., C. cavaleriei H.Lév. ex Cavalerie, C. × clementina hort. ex Tanaka (Mabberly[31] suggests this should be included under C. × aurantium), C. × limon (L.) Burm. f., C. × limonia Osbeck (This is considered as C. × otaitensis based on recent revisions by Mabberley[31]), C. maxima (Burm.) Merr., C. medica L., C. micrantha Wester (This is considered as C. hystrix based on recent revisions by Mabberley[31]), C. reshni hort. ex Tanaka (Plants of the world online (POWO) suggests this should be included under C. × aurantium), C. reticulata Blanco, Fortunella hindsii (C. hindsii) (POWO suggests this is a synonym of C. japonica), and Poncirus trifoliata a (L.) Raf (C. trifoliata)] are related to Australian citrus based on both complete chloroplast genomes and 86 single copy nuclear genes. Another objective was to infer the relationships between an unknown citrus accession which was collected from Cape York Peninsula (Citrus sp.) and the other Australian citrus species. The results were used to assess the congruence between the chloroplast and nuclear phylogenies and the previous relationships among Australian taxa with the results derived from this study. Improved understanding of genetic relatedness may help explain the evolutionary drivers that have created such a unique clade of citrus species on the Australian continent.
-
Leaf samples of six native citrus species [five cultivars of C. australasica (cultivar names and their descriptions are given in Supplemental Table S1), C. australis, C. garrawayi, C. glauca, C. gracilis, C.inodora), two commercial mandarin cultivars (C. reticulata admixtures); Murcott and Ellendale and three seedlings of C. sp. Unknown (similar to, but morphologically distinct from C. garrawayi) were collected under dry ice for DNA extraction. The characteristics, geographical location and potential uses of the species/cultivars are indicated in the Supplemental Table S2. Materials of C. inodora, C. australasica, C. glauca, and three seedlings of the C. sp. were collected from Herbalistics Pty Ltd private orchard located in Maroochy River, Queensland, Australia. Leaves of C. garrawayi and C. australis were collected from plants maintained at the Glasshouse, University of Queensland, Australia. Leaf material of the two mandarin cultivars was obtained from the arboretum, Department of Agriculture and Fisheries (DAF), Bundaberg, Queensland, Australia. C. gracilis was obtained from Litchfield, South of Darwin, Northern Territory, Australia.
DNA extraction and sequencing
-
Total genomic DNA was extracted from pulverized leaf tissues using CTAB (cetyltrimethyl ammonium bromide) DNA extraction protocol[32]. The quality of DNA was evaluated using nanodrop spectrophotometer with A260/280 and A260/230 absorbance ratios and by agarose gel electrophoresis. The concentrations of genomic DNA were measured by SYBR-safe fluorescence in gel electrophoresis compared to lambda DNA concentration standards. A ratio of 1.8–2.0 for A260/280 and a ratio of 2–2.2 for A260/230 were considered pure for DNA. No shearing indicated high quality genomic DNA. DNA of C. australis, C. garrawayi, C. glauca, C. sp., Murcott, and Ellendale were sequenced at the Ramaciotti Centre, University of New South Wales, NSW, Australia, and C. australasica, C. inodora and C. gracilis species were sequenced at Australian Genome Research Facility (AGRF), Melbourne, Australia.
Nuclear gene assembly
-
Coding sequences (CDS) were extracted for C. clementina from the Citrus Genome database (
www.citrusgenomedb.org ) and were subjected to BLAST against 106 single copy genes, which were previously identified from C. sinensis[26] (Supplemental Table S3) to extract the corresponding CDS in CLC Genomics Workbench v23,0.4 (Qiagen, USA). CDS of the outgroup P. trifoliata were subjected to BLAST independently to get the corresponding CDS of P. trifoliata. The BLAST hits were manually checked to select only the single copy genes in C. clementina and P. trifoliata genomes with correct protein description with respect to C. sinensis. The same 86 CDS were selected from the reference species (C. clementina) and the outgroup (P. trifoliata). Then the paired end Illumina reads trimmed using 0.01 quality threshold of all the samples were independently mapped to all the CDS of C. clementina using the mapping setting LF (Length fraction) 0.5 and SF (similarity fraction) 0.8. in CLC Genomics Workbench v23,0.4 (Qiagen, USA). Mapping consensus sequences for 86 CDS of each species were extracted and joined independently to form one concatenated sequence per each species and the concatenated nuclear genes were used in the phylogeny.Chloroplast genome assembly, annotation and variant analysis
-
Raw Illumina paired-end reads were imported to CLC Genomics Workbench v23,0.4 (Qiagen, USA) and trimmed at 0.01 quality limit. A subset (3–4 GB) of quality trimmed Illumina reads were subjected to de novo chloroplast (cp) genome assembly using GetOrganelle toolkit v.1.7.5[33]. The parameters used for GetOrganelle assembly are given in Supplemental Table S4. For C. australis, the individual scaffolds obtained from GetOrganelle were imported to Clone Manager and were aligned with a reference cp genome (C. aurantium) to get the final complete cp genome. The cp genome of P. trifoliata which has previously been assembled by GetOrganelle toolkit[34] was used for our phylogenetic analysis. In addition, 12 other citrus species were obtained from the SRA database, NCBI (
www.ncbi.nlm.nih.gov/sra ) (Supplemental Table S2), and quality trimmed at the 0.01 quality limit. Subsequently, a subset of the trimmed sequences was assembled using the GetOrganelle toolkit.The assembled cp genomes were annotated by Geseq tool using the reference genome; C. aurantium (
https://chlorobox.mpimp-golm.mpg.de/geseq.html )[35] with the following settings: Annotation-BLAT search annotation with plastid IR, annotate plastid trans-spliced rps12, protein search identity – 25, rRNA, tRNA, DNA search identity – 85, HMMER profile search –chloroplast land plants (CDS + rRNA), 3rd party tRNA annotators – ARAGORN v1.2.38, tRNAscan-SE v2.0.7, BLAT reference sequence: MPI-MP reference set – chloroplast land plants (CDS + rRNA), 3rd Party Stand-Alone Annotators – Chloë v0.1.0 (CDS + tRNA + rRNA).Chloroplast genome sequences were imported to Geneious prime V2021.2, Biomatters Ltd (
www.geneious.com ) to perform the variant analysis. Two sequences to be compared were subjected to pairwise alignment using MAFFT alignment tool[36] in Geneious prime V2021.2, Biomatters Ltd with default parameters. The species within one subclade were mapped against another species within the same sub clade to identify the total number of variations in terms of SNPs (transitions and transversions), insertions and deletions existing among them.Phylogenetic analysis of native and domesticated citrus using chloroplast genome and nuclear gene assemblies
-
The phylogenetic relationships among the species were inferred from the assembled complete chloroplast genomes and 86 nuclear genes. Complete chloroplast genome sequences and 86 concatenated nuclear genes were independently subjected to multiple sequence alignment using the multiple alignment with fast Fourier transform (MAFFT) alignment in Geneious prime V2021.2, Biomatters Ltd with default parameters. Maximum likelihood (ML) tree construction was performed using RAxML (Randomized Axelerated Maximum Likelihood) method employing generalized time reversible (GTR) GAMMA nucleotide substitutional model[37]. A rapid bootstrap analysis and a search for a best-scoring ML tree was performed with 1,000 bootstrap replicates. MrBayes v. 3.2[38] was performed with GTR substitution model, gamma rate variation and other default parameters in Geneious prime V2021.2, Biomatters Ltd [MCMC settings (Chain length: 1,100,000, Heated Chains: 4, Heated Chain: Temp 0.2, subsampling Freq: 200, Burn-in Length: 100,000, Random seed: 16,907)]. The final tree outputs were further modified in iTOL version 6.5.2[39].
-
The phylogeny based on 86 single copy nuclear genes derived from MrBayes (Supplemental Fig. S1) and RAxML methods (Fig. 1) were congruent. The posterior probability values were high for all the nodes (1/0.99) indicating high supports for all the relationships. The ML tree (Fig. 1) revealed two major clusters separating Australian wild limes from the rest of the citrus cultivars. The Australian clade (Clade I) had two distinct sub-clades in which C. inodora was more closely related to C. australasica (sub-clade A) and the other citrus species were grouped into another sub-clade (sub-clade B). In sub-clade A, the grouping of C. inodora with C. australasica had highly significant node support. Within this sub-clade, C. australasica cv 5 and cv 3 were more closely related to each other than with the other four cultivars of C. australasica. C. australasica cv 4 was more closely related to C. australasica cv 1 and that sister group was nested with C. australasica cv 2 revealing their close relationships. The relationships within C. australasica were well supported by high bootstrap values (Fig. 1). Within the sub-clade B, C. glauca was the most distantly related species and the grouping of C. glauca with other native limes had reasonably high node support. C. gracilis is the next most distant in this sub-clade containing C. australis, C. garrawayi and C. sp., C. garrawayi was closely related to C. sp. with high bootstrap support (Fig. 1).
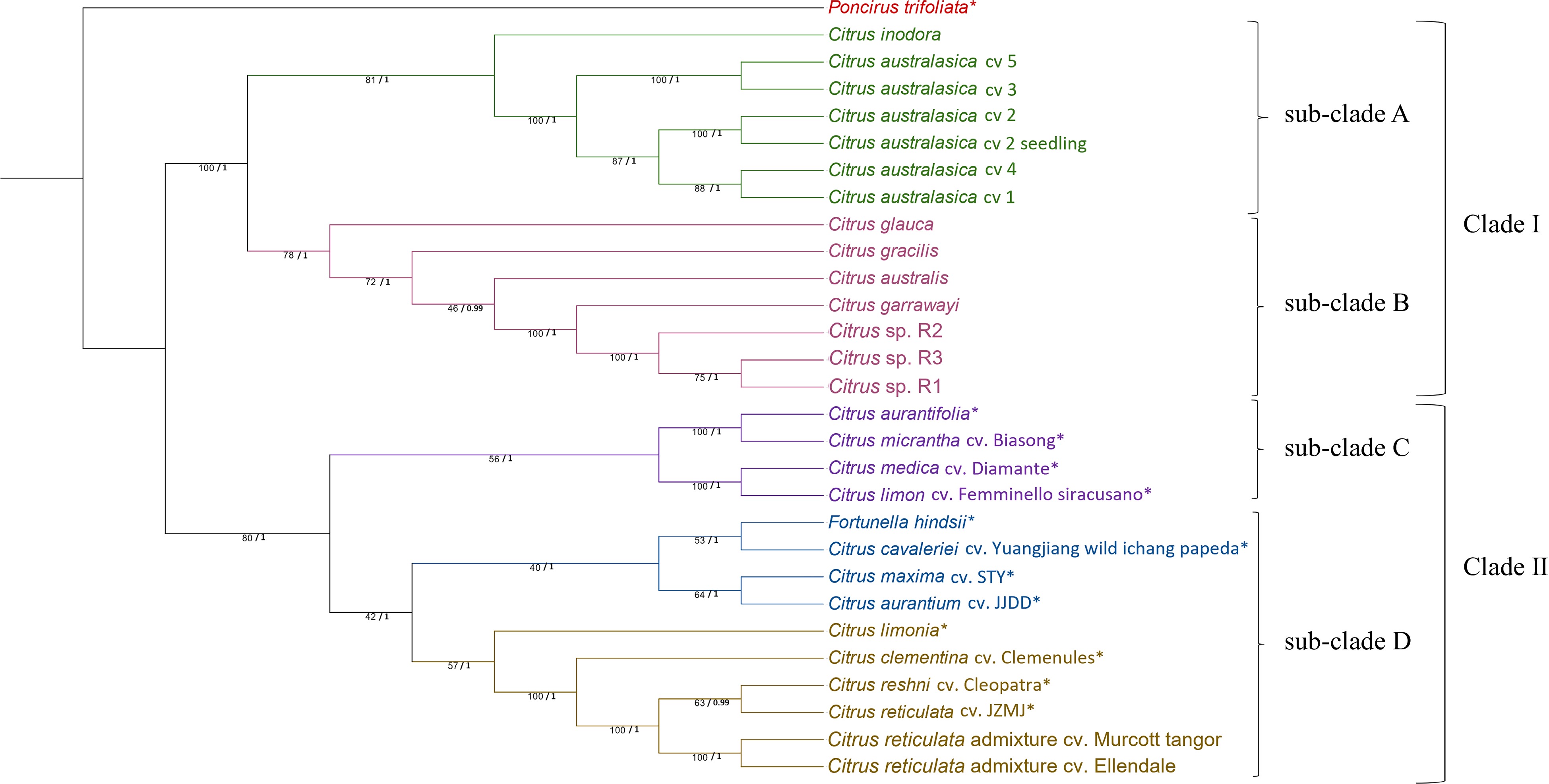
Figure 1.
Phylogenetic tree generated from 86 nuclear genes sequences of 29 citrus samples (species/cultivars) with Poncirus trifoliata as the outgroup. The tree was generated using Maximum Likelihood (ML) method in RAxML with 1,000 bootstrap replicates. ML bootstrap values from 1,000 replicates (/100) and posterior probability values are indicated on each node respectively. * SRA data obtained from National Centre for Biotechnology Information for nuclear genes assembly. Citrus sp. R1, R2 and R3 refers to three different seedlings of the Citrus sp.
The non-Australian citrus species (Clade II) formed two major and distinct clades. The first major clade (sub-clade C) was split into two sub-clades. The first sub-clade has two sister-clades, one grouping C. aurantifolia and C. micrantha together and the other sister-clade grouping C. medica and C. limon together with high node support. The second major clade (sub-clade D) had two sub-lineages. The first sub-lineage split into two sister groups with F. hindsii and C. cavaleriei in one group and C. maxima and C. aurantium in the other group. The second sub-lineage was comprised of mandarins and related hybrid cultivars (mandarin × sweet orange and mandarin × citrons). Within the mandarin clade, C. limonia (Rangpur lime) (mandarin × citron) was distantly related to mandarins and mandarin × sweet orange hybrids. C. clementina was closely related with the other four mandarin cultivars. C. reticulata (cv. JZMJ) and C. reshni (cv. Cleopatra mandarin) were grouped in one sub-clade and Murcott and Ellendale were grouped in another sub-clade (Fig. 1).
Chloroplast genome assembly and annotation
-
Chloroplast genomes of all the species exhibited the typical quadripartite structure of most angiosperms’ chloroplast genomes (Supplemental Fig. S2) with the total genome size ranging from 159,882−160,997 bp. The cp genomes contained a large single-copy region (LSC: 87,137−87,945 bp), small single-copy region (SSC: 18,385−18,801 bp), and a pair of inverted repeat regions (IR: 26,944−27,300 bp). 113−115 functional genes were encoded by all the species. The total number of genes included 78 or 79 CDS, 31 or 32 tRNA genes, and 4 rRNA genes. (Table 1). The species with 78 CDS were found to have two missing genes with compared to those having 79 CDS. The CDS infA was found to be missing in all C. australasica cultivars, C. inodora, and C. glauca and the CDS rps12 was missing in C. australis, giving rise to 78 total number of CDS in them. The gene encoding trnS-CGA tRNA was found to be missing in the species with 31 tRNA.
Table 1. Characteristics of the chloroplast genomes of 28 citrus samples.
Species Cultivar Genome size (bp) LSC (bp) SSC (bp) IR (bp) Genes CDS tRNA genes rRNA genes C. australasica cv 1 160,400 87,686 18,757 26,952 113 78 31 4 C. australasica cv 2 160,400 87,723 18,759 26,959 113 78 31 4 C. australasica cv 3 160,335 87,677 18,754 26,952 113 78 31 4 C. australasica cv 4 160,365 87,692 18,755 26,959 113 78 31 4 C. australasica cv 5 160,335 87,677 18,754 26,952 113 78 31 4 C. australis N/A 160,530 87,882 18,760 26,944 114 78 32 4 C. garrawayi N/A 160,495 87,780 18,769 26,973 115 79 32 4 C. glauca N/A 160,570 87,849 18,763 26,979 114 78 32 4 C. gracilis N/A 160,372 87,652 18,752 26,984 115 79 32 4 C. inodora N/A 160,669 87,945 18,728 26,998 114 78 32 4 C. sp. R1 N/A 160,585 87,866 18,787 26,966 115 79 32 4 C. sp. R2 N/A 160,572 87,853 18,787 26,966 115 79 32 4 C. sp. R3 N/A 160,572 87,853 18,787 26,966 115 79 32 4 C. aurantifolia − 159,882 87,137 18,763 26,991 115 79 32 4 C. aurantium JJDD 160,140 87,755 18,385 27,000 115 79 32 4 C. cavaleriei Yuangjiang wild
ichang papeda160,997 87,634 18,763 27,300 115 79 32 4 C. clementina Clemenules 160,722 87,941 18,801 26,990 115 79 32 4 C. limon Femminello
siracusano160,141 87,754 18,385 27,001 115 79 32 4 C. limonia − 160,715 87,910 18,789 27,008 115 79 32 4 C. maxima STY 160,186 87,791 18,395 27,000 115 79 32 4 C. medica Diamante 160,048 87,490 18,576 26,991 115 79 32 4 C. micrantha Biasong 159,923 87,178 18,763 26,991 115 79 32 4 C. reshni Cleopatra 160,666 87,866 18,784 27,008 115 79 32 4 C. reticulata JZMJ 160,699 87,918 18,801 26,990 115 79 32 4 C. reticulata admixture Murcott 160,699 87,918 18,801 26,990 115 79 32 4 C. reticulata admixture Ellendale 160,699 87,918 18,801 26,990 115 79 32 4 F. hindsii − 160,265 87,587 18,734 26,972 115 79 32 4 P. trifoliata − 160,260 87,442 18,760 27,029 115 79 32 4 Chloroplast genome phylogeny
-
The trees generated by MrBayes (Supplemental Fig. S3), and ML approaches (Fig. 2) were topologically similar. The Mr Bayes tree had high node support for all nodes. We discuss the RA×ML tree for both nuclear and chloroplast phylogenies for ease of comparison. The RA×ML tree (Fig. 2) displayed two major lineages for the citrus species giving rise to two major clades. The first major clade (Clade I) was a cluster grouping all the Australian citrus species and C. medica. In this clade, the sub-clade containing the Australian wild limes was further split into two sub-clusters (sub-clade A and sub-clade B) with high bootstrap support. In sub-clade A, the five different cultivars of C. australasica were grouped into two sub-clusters, while the other wild limes were grouped separately in sub-clade B. In sub-clade A, C. australasica, cv 2 and cv 4 clustered separately from cv 1, cv 3 and cv 5. The sub-clade B, containing other Australian citrus had two sub-lineages separating C. inodora, C. glauca and the C. australis group from the rest. The other sub-lineage clustered C. gracilis, C. garrawayi and the three seedlings of C. sp. together in one sub-clade. The second major clade (Clade II) contained all the domesticated citrus species used in this study and formed two sub-clades. The first sub-clade (sub-clade C) was further divided into two sister groups where C. aurantifolia and C. micrantha formed one group, while C. maxima, C. limon and C. aurantium formed another group. In the second sub-clade (sub-clade D), C. cavaleriei was sistered to the other species. C. limonia and C. reshni formed a sister group and C. clementina and C. reticulata cultivars including the two commercial cultivars (Murcott and Ellendale) formed another sister group.
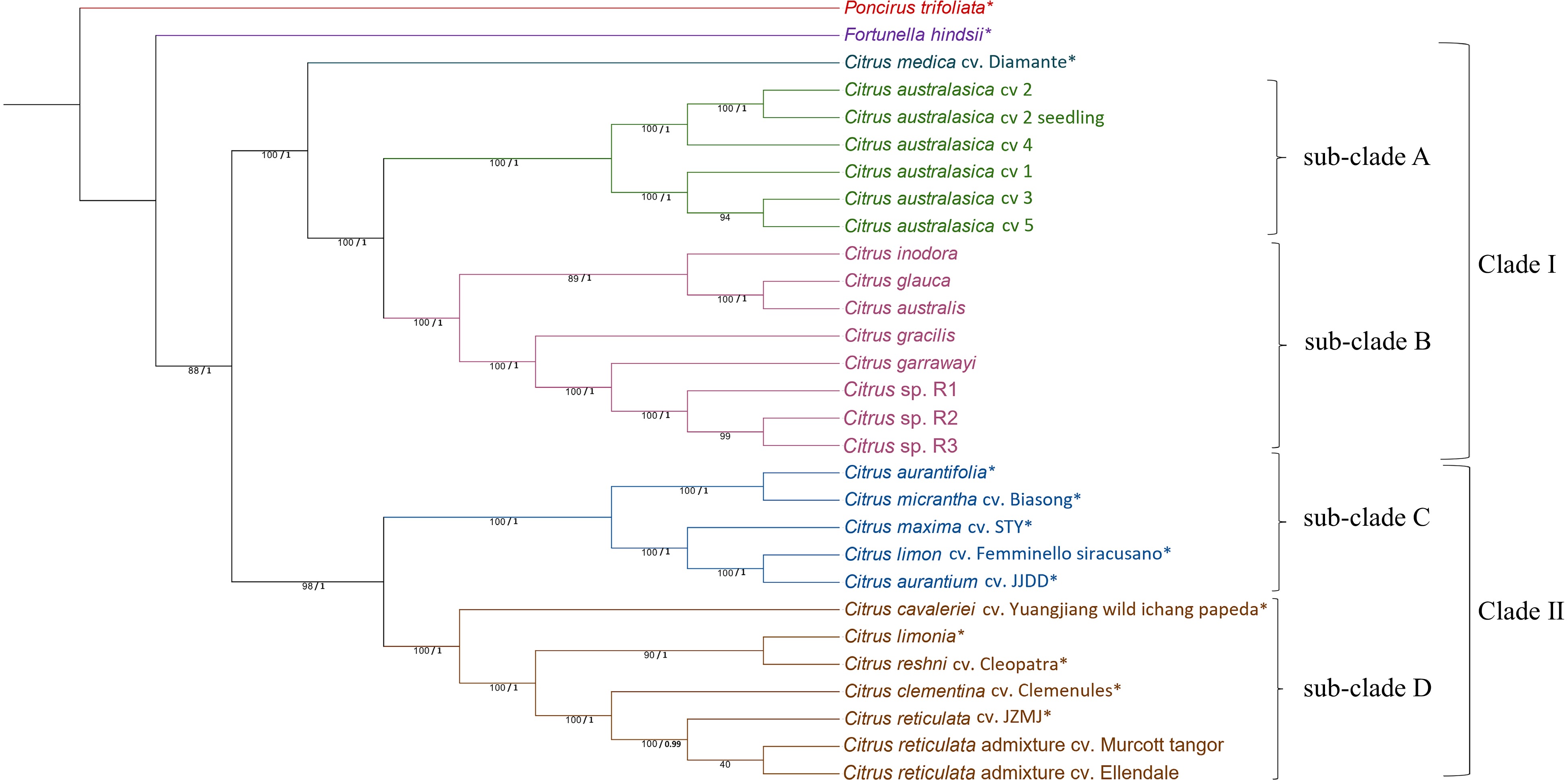
Figure 2.
Phylogenetic tree generated from complete chloroplast genome sequences of 29 citrus samples (species/cultivars) with Poncirus trifoliata as the outgroup. The tree was generated using Maximum Likelihood (ML) method in RAxML with 1,000 bootstrap replicates. ML bootstrap values from 1,000 replicates (/100) and posterior probability values are indicated on each node respectively. * SRA data obtained from National Centre for Biotechnology Information for chloroplast genome assembly. Chloroplast genome for P. trifoliata which was assembled by GetOrganelle toolkit was obtained from[34]. Citrus sp. R1, R2 and R3 refers to three different seedlings of the Citrus sp.
Analysis of the Australian clade showed that the C. garrawayi chloroplast had a total of 192−198 variants [SNPs (transitions and transversions), insertions and deletions] when compared to the C. sp. The chloroplast sequences of C. sp. R2 and R3 were identical with each other. C. glauca and C. australis formed a sister group with 688 variants between them. In C. australasica cultivars, cv 3 and cv 5 had no variants in their chloroplasts. There were six variants between cv 1 and cv 3 / cv 5 and 38 variants between cv 4 and cv 2.
-
The present study revealed a conflict in the topologies of nuclear and chloroplast phylogenies. Previous studies have explained similar conflicts as potential reticulate evolution, incomplete lineage sorting, or hybridization/introgression[40,41]. The nuclear phylogeny revealed the relationships among species. The Australian native citrus species are distinct from non-Australian citrus based on the nuclear phylogeny. The Australian species are thought to have evolved over millions of years from an ancestral species which originated in Southeast Asia and underwent speciation in Australia[13]. They have adapted to the prevailing edaphic and climatic variations in Australia and developed traits that do not exist or are rarely found in domesticated species[11,42]. C. australasica accessions are particularly interesting amongst the native Australian citrus because of their high morphological and genetic diversity, with wide variations in peel and pulp colours and flavours[11]. They possess a unique nuclear genome closely shared amongst the five accessions used in this study. The fact that C. inodora has a nuclear genome close to C. australasica but with a chloroplast similar to the other native limes suggest that hybridization/introgression and chloroplast capture might have occurred during their evolution.
Nuclear relationships further showed that C. glauca is relevantly distantly related to C. gracilis, C. australis, C. garrawayi and the C. sp. Our result is congruent with a previous phylogeny based on 27 nuclear genes in which it found a closer relationship between C. glauca and C. australis than with C. australasica[28]. However, this result differs from the finding of Wu et al.[3], which showed a closer relationship between C. australis and C. australasica, than with C. glauca, thus indicating the separation of C. glauca from these two species much earlier. The results of Wu et al.[3] were based on a large number of single nucleotide variations in non-genic and non-pericentromeric genomic regions, which might be a reason for the different topology of our phylogeny, which was based on the coding regions of 86 genes. The close relationship between C. garrawayi and the C. sp. support the view that C. sp. could be a distinct form of C. garrawayi. Morphological differences in terms of the size and shape of the fruit, leaves, (Supplemental Fig. S4) and the flavour of the fruit of the C. sp. support the possibility of extensive genetic variation in this species. Conclusions from future studies would be strengthened by the inclusion of multiple accessions of all six Australian species.
The availability of complete cp genomes and multiple taxa have improved the ability to define an evolutionary history of Australian native limes. Our results indicate that Australian citrus species have a similar cp sequence to citrons (C. medica), which might be explained by reticulate evolution. Previous studies showed that ancient citrus went through two events in history, one separating Australian citrus and citrons from all other citrus, followed by the separation of citrons from Australian citrus[5]. The origin of citrons and Australian citrus were known to be in Southeast Asia[3,43] and it is possible that the progenitor species of Australian citrus and C. medica might have shared a similar chloroplast through chloroplast capture during their evolution.
C. australasica forms a monophyletic clade which diverged from the rest of the Australian species and indicates that the five cultivars of this species are likely to share a distinct cp genome, different from all the other wild limes. C. inodora, C. australis and C. glauca have chloroplasts which are closer to the other native limes. Previous studies have suggested that chloroplast phylogenies are associated with the geographical distribution of species and that chloroplast capture can occur as a result of introgression and hybridization between species[44,45]. Intriguingly, our chloroplast phylogeny does not strongly support the geographical distribution of these six species within the Australian continent, nor does it correspond with differences in current edaphic and climatic conditions. The most incongruous sub-clade contains C. inodora, C. glauca and C. australis, representing species that are widely dispersed in their geographical locations, and climates that range from open semi-desert (~300 mm annual rainfall) to dense tropical rainforests (~8,000 mm annual rainfall). This may suggest that the evolutionary pressures that created six distinct and unique citrus species on the Australian continent were very different from those that prevail today.
The close relationship of C. australis and C. glauca chloroplast genomes is well established in many previous chloroplast phylogenies[2,3,5,15,46]. Some of those phylogenies have also revealed a clear separation of C. australasica from the sub-clade of C. australis and C. glauca indicating that these two latter species are likely to share similar chloroplast sequences. Our results further suggest that C. gracilis, C. garrawayi and the C. sp. might have shared a similar chloroplast during their evolution with a smaller number of variations between the chloroplasts of two forms of C. garrawayi.
-
The nuclear gene phylogeny has identified close relationships among the Australian wild citrus species and confirmed a monophyletic clade quite distinct from other species in this genus. Of the Australian citrus species, C. inodora and C. australasica were closely related and distinct from the other species. However, C. inodora had a chloroplast similar to the other Australian limes probably as a result of reticulate evolution. C. medica shares a chloroplast similar to that found in the Australian species, suggesting a common origin. The Australian clade represents a divergent group of species that is an important genetic resource for citrus[8,10]. This study has clarified their relationships and raised questions about the evolutionary pressures that resulted in such vast extremes of environmental adaptation within the clade.
-
The authors confirm contribution to the paper as follows: study conception and design: Henry R, Furtado A; data collection: Nakandala U; analysis and interpretation of results: Nakandala U, Henry R, Furtado A, Smith M, Williams D; draft manuscript preparation: Nakandala U. All authors reviewed the results and approved the final version of the manuscript.
-
Raw sequence data of Illumina short reads generated in this study have been deposited in NCBI Sequence Read Archive (SRA) under the BioProject PRJNA1010857 and BioSamples SAMN37202700, SAMN37203473, SAMN37203569, SAMN37212709, SAMN37217921, SAMN37217994, SAMN37218139, SAMN37218149, SAMN37218318, SAMN37218425, SAMN37218444, SAMN37282787, SAMN37282790, SAMN37282997 with SRA accession IDs of SRR25867464, SRR25888267, SRR25893096, SRR25899463, SRR25905657, SRR25919244, SRR25923911, SRR25933438, SRR25915022, SRR25948168, SRR25994105
This project was funded by the Hort Frontiers Advanced Production Systems Fund as part of the Hort Frontiers strategic partnership initiative developed by Hort Innovation, with co-investment from The University of Queensland, and contributions from the Australian Government and Bioplatforms Australia. We thank the Research Computing Centre (RCC), University of Queensland, for providing high performance computing facilities.
-
The authors declare no conflict of interest. Robert J. Henry is the Editorial Board member of Tropical Plants who was blinded from reviewing or making decisions on the manuscript. The article was subject to the journal's standard procedures, with peer-review handled independently of this Editorial Board member and the research groups.
-
accompanies this paper at (https://www.maxapress.com/article/doi/10.48130/TP-2023-0021)
-
Received 11 September 2023; Accepted 26 October 2023; Published online 21 December 2023
-
Australian citrus species formed a monophyletic clade which was separated from the non-Australian species, indicating their unique status within the genus Citrus.
Citrus inodora and Citrus australasica had a close relationship based on 86 single copy nuclear genes and were distinct from the other four Australian species.
Citrus australasica had a unique chloroplast which was distinct from all the other Australian species.
Citrus medica had a similar chloroplast to the Australian species but was more closely related to the Asian citrus species based on nuclear genes.
- Supplemental Table S1 Names and descriptions of five finger lime cultivars based on the colour of skin and juice vesicles.
- Supplemental Table S2 The collection sites, geographical locations, characteristics and uses of the citrus species used in this study.
- Supplemental Table S3 List of single copy nuclear genes used for nuclear gene analysis.
- Supplemental Table S4 Parameters used to get the complete chloroplast genomes from GetOrganelle.
- Supplemental Fig. S1 Phylogenetic tree generated from 86 nuclear genes sequences of 29 citrus samples (species/cultivars) with Poncirus trifoliata as the outgroup. The MrBayes tree was generated in Geneious. Posterior probability values are given on each node.
- Supplemental Fig. S2 Chloroplast genome map of citrus species. All the species showed the typical quadripartite structure of the chloroplast genome. The genes belonging to different functional groups are shown in different colours. The thicker black lines indicate the extent of the IR regions separating the LSC and SSC regions. Genes inside the circle are transcribed in clockwise direction whereas the genes outside the circle are transcribed in counter-clockwise direction. LSC: Large Single-Copy, SSC: Small Single-Copy, IR: Inverted Repeat regions.
- Supplemental Fig. S3 Phylogenetic tree generated from complete chloroplast genome sequences of 29 citrus samples (species/cultivars) with Poncirus trifoliata as the outgroup. The MrBayes tree generated in Geneious. Posterior probability values are given on each node. * SRA data obtained from National Centre for Biotechnology Information for chloroplast genome assembly. Chloroplast genome for P. trifoliata which was assembled by GetOrganelle toolkit was obtained from (He et al., 2020).
- Supplemental Fig. S4 The morphological variations of the leaves and fruit of Citrus garrawayi and Citrus sp. Unknown. (a) The difference of the leaves of the two accessions. C. garrawayi is on the left and C. sp. Unknown is on the right. C. garrawayi has lanceolate to obovate leaves and C. sp. Unknown has ovate to obovate leaves (b) The inner and outer appearance of the fruit of C. sp. Unknown. (c) Variation in fruit size and shape of C. garrawayi (top) and C. sp. Unknown (bottom) The fruit of C. sp. Unknown are not as long as C. garrawayi. The juice vesicles and the taste of the fruit are different between C. garrawayi and C. sp Unknown. They both have angular vesicles than C. australasica, however, those of C. sp Unknown are more like C. australis. The taste of the C. sp Unknown fruit is more like green apple (which C. garrawayi can have), however, it is less acidic than C. garrawayi.
- Copyright: © 2023 by the author(s). Published by Maximum Academic Press on behalf of Hainan University. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Nakandala U, Furtado A, Smith MW, Williams DC, Henry RJ. 2023. Phylogenetic relationships among Australian native citrus species based upon complete chloroplast genomes and single copy nuclear genes. Tropical Plants 2:21 doi: 10.48130/TP-2023-0021 

Phylogenetic relationships among Australian native citrus species based upon complete chloroplast genomes and single copy nuclear genes
- Received: 11 September 2023
- Accepted: 26 October 2023
- Published online: 21 December 2023
Abstract: Citrus is widely consumed across the world as a fruit crop. Despite many citrus species being extensively studied around the world, phylogenetic relationships among Australian native species remain unresolved. Here we present the phylogenetic relationships among six Australian native species, two domesticated citrus cultivars of commercial importance in Australia, and another 13 accessions cultivated internationally based on complete, de novo assembled chloroplast genomes and 86 single copy nuclear genes. The chloroplast and nuclear phylogenies were topologically different. The Australian species formed a monophyletic clade based on their nuclear genes. The nuclear phylogeny revealed a close relationship between Citrus inodora and Citrus australasica. These two species were distinct from the other four Australian limes that were more closely related to each other. Citrus australasica had a unique chloroplast which was distinct from all other Australian limes. Among the other Australian limes, Citrus glauca was the most distinct species based on nuclear genes, however, it had a similar chloroplast sequence to C. australis. The undetermined Citrus sp. was more closely related to Citrus garrawayi, indicating that it is a distinct form of Citrus garrawayi. Citrus medica, had a chloroplast similar to Australian species. However, the nuclear gene phylogeny analysis revealed that C. medica was more closely related to Asian citrus species. This study improves our understanding of phylogenetic relationships among Australian citrus species and confirms their unique status within the genus since it formed a monophyletic clade which was clearly separated from the other non-Australian species.
-
Key words:
- Phylogenetic /
- Relationships /
- Australian /
- Native /
- Species /
- Citrus