









-
Finger lemon (Citrus australasica), also named finger orange, Australian finger lemon or lemon caviar, belongs to the family Rutaceae. It is well known for its caviar-like appearance of fruit flesh and is regarded as 'the caviar of fruit'[1]. Today Citrus australasica is mainly concentrated in a small region of northern New South Wales and tropical rainforest areas in southern Queensland[2]. Finger lemon is rich in citric acid, vitamin C and flavonoids. These natural antioxidants have great potential in healthcare[3]. Its unique characteristics and pulp flavor are often used in food processing and timber production, and have a high economic value[4]. Therefore, it has been widely noticed and loved by people[5]. At present, the demand of the global finger lemon market is growing exponentially, and the planting area of finger lemon is increasing year by year. Therefore, many countries have introduced cultivation[6]. China was first introduced by the United States in 1977 and achieved success in Guangxi, and now there are related artificial planting studies in Yunnan, Guangdong, Hainan, and Hubei province[7]. The species prefers warm climates, and the abundant sunshine in Hainan gives the planting of finger lemon a unique growing environment. At the same time, as one of Hainan's rare and characteristic varieties, the finger lemon has been effective in enhancing the core competitiveness of Hainan's fruit products, and has gradually become a well-known regional brand and geographical indications products in Hainan province[8].
In the evolution of the genus Citrus, the finger lemon has been used as a citrus hybrid parent to produce new varieties. It belongs to the same group as kumquat (Citrus hindsii) in the genus, and other citrus species in Australia and Papua New Guinea[9]. However, the genetic relationship between finger lemon cultivars is not clear, which makes it impossible to explore its evolution more accurately. At the same time, higher-quality finger lemon genomes are needed to study the origin and evolution of finger lemons. However, the genome of finger lemon has only been reported by Nakandala et al.[10]. Nevertheless, there is only one report on the genome of finger lemon, and most of the studies on finger lemon mainly focus on its chemical composition, especially citric acid, and vitamin C, as well as the observation and evaluation of varietal resources and cultivation[11]. In addition, a few studies on the genomic resources of the plant have been reported. Such as the preliminary identification and quantification of the physicochemical properties, antioxidant capacity, and phenolic composition of two finger lemon varieties, 'XiangBin' and 'LiSiKe' were reported by Wang et al.[6]. The volatile components of the rind about three finger lemon varieties 'Alstonville', 'Judy's Everbearing' and 'Durham's Emerald' was reported by Delort et al.[12]. Furthermore, purple finger lemon types were not recorded in this research, and the majority of the finger lemon varieties that were used had rinds that were green, red, and so on.
However, compared to other citrus plants, the finger lemon's peel color and texture, pulp flavor, fruit yield and quality, and plant disease resistance differs significantly from other species in the same family. Among citrus plants, Finger Lemon has the most varied rind color, which is usually lime green, but also yellow, red, purple, black and other colors. However, this characteristic of this species has not yet been thoroughly studied, and the intrinsic molecular mechanisms and key genes regulating rind color traits are unknown. Therefore, sequencing and analyzing the transcriptome of finger lemon, which is a non-model plant without complete genome information, using next-generation high-throughput sequencing technology is important in explaining this characteristic of finger lemon. In this study, we used Nanopore sequencing and second-generation sequencing to assemble the genome of the finger lemon cultivar 'YaoJi', to explore its evolutionary relationship, and combined with the transcriptome data analysis to investigate the mechanism of the differences affecting the color of the rind of the finger lemon, to investigate the intrinsic molecular mechanism, providing data for the law of evolutionary inheritance, and providing a reference for future selection and cultivation. The purpose of this study is to investigate the molecular mechanism and provide data for the evolutionary genetic pattern, to provide a reference value for the selection and cultivation in the future.
-
The experimental materials for this study were collected from Hainan Grand Agricultural Development Co., and the fully expanded leaves were collected from the top of a healthy and vigorous tree of Finger Lemon 'YaoJi' for genome sequencing library preparation, and the fruits of another variety of lemon 'JiaLiHong' and 'YaoJi' were collected from the same developmental period and the same growth state for RNA sequencing (RNAseq) and transcriptome analysis. The samples were snap-frozen in liquid nitrogen immediately after collection and then stored in a refrigerator at −80 °C.
High-quality genomic DNA of finger lemon was extracted using fresh leaf and root DNA extraction kits, and its concentration was measured by NanoDrop concentration detector and Qubit kit respectively, and after confirming that the DNA quality, degradation, and integrity met the requirements of purity and concentration, the finger lemon sequencing library was constructed. Double-end sequencing was performed on an Illumina HiseqXten sequencing platform[13] to obtain whole genome sequencing data of finger lemon for subsequent analysis of predicted genome size, heterozygosity, and GC content. Three-generation library building sequencing was performed on an Oxford Nanopore instrument to obtain fastq data for finger lemon genome assembly. The extracted RNA samples were interrupted, end-repaired, and configured for up-sampling libraries after PCR reaction, and high-throughput sequencing was carried out on the UW Genetics DNBSEQ sequencing platform for transcriptome analysis, with three replicates set up for each group.
Genomic heterozygosity and structural assessment
-
Second-generation sequencing data with Fastp[14] (parameters: -n 0 -f 5 -F 5 -t 5 -T 5) to filter the raw data to obtain the final fastq file. The data were analyzed by K-mer[15] to estimate the genome size. The optimal K-mer value of 21 was first obtained by kmergenie (
https://github.com/habibr/kmergenie.git , V1.7051) software, and then utilized Jellyfish[16] (https://github.com/gmarcais/Jellyfish.git , V2.3.0) to obtain K-mer information, and then used the GenomeScope online tool with the parameter '-kmer length 21 -Ploidy 2' to predict and evaluate the genome size and heterozygosity of finger lemons.Genome assembly and annotation
-
Assembling of off-board triple data using Nextdenovo (
https://github.com/Nextomics/NextDenovo.git , V2.5.2)[17] Assembled with Nextpolish (https://github.com/Nextomics/NextPolish.git , v1.4.0) software[18]. The Contig version of the genome was error-corrected, and then the genome was mounted using RagTag (https://github.com/malonge/RagTag.git , v2.1.0)[19] , to get the final chromosome-level genome. Transcriptome assembly was performed using Trinity (V2.8.5), and proteins were constructed using finger lemon homologous species sweet orange and Cremantine red orange, and then annotated with MAKER[20]. Functional annotation was performed on Eggnog-mapper (http://eggnog-mapper.embl.de ) and the results were visualized with TBtools[21].Phylogenetic evolution
-
Phylogenetic evolutionary tree construction using Orthfinder (
https://github.com/davidemms/OrthoFinder.git , V2.5.5) using single-copy genes from 14 species[22], which are Vitis vinifera L., Arabidopsis thaliana (L.) Heynh., Elaeis guineensis Jacq, Oryza sativa, Zea mays, Setaria italica, Sorghum bicolor (L.) Moench, Mangifera indica L., Acer saccharum; Citrus sinensis (L.) Osbeck , Citrus × clementina , Amborella trichopoda and Nymphaea L.[23]. Then the hypermetric tree (number of times) was constructed using r8s (http://loco.biosci.arizona.edu/r8s/r8s.dist.tgz , V1.81), and then the genes for expansion and contraction were computed using CAFE (V4.2.1), after which the phylogenetic evolutionary tree was drawn using iTOL (https://itol.embl.de ). Additionly TBtools were used to calculate Ka and Ks values and plot ks distributions for finger lemons, grapes, sweet oranges and red oranges, to infer genome-wide replication events Finger lemon-specific gene families were enriched and analyzed in KOBAS (http://bioinfo.org/kobas/genelist/ ) and the results were visualized in R.Comparative analysis of finger lemon peel colors
-
Building indexes with Hisat2 (
https://github.com/DaehwanKimLab/hisat2.git , v2.2.0)[24], and compare the bipartite sequencing data to the reference genome, after Samtools (V1.18) to convert the format and sort, use Stringtie (https://github.com/gpertea/stringtie.git , V2.1.1) to calculate the expression quantification and extract the expression amount information[25]. After integration, inter-sample PCA analysis was performed based on expression quantities and visualized with R. Differential gene analysis was performed on R using the 'DESeq' package to analyze the expression (FPKM) of 'JiaLiHong' and 'YaoJi', and up- and down-regulated genes were screened, extracted, and visualized in R. The results were summarized in the following table. R was used for visualization. We searched for transcription factors related to their functions on PlantTFDB (https://planttfdb.gao-lab.org/index.php ), and investigated the mechanism of the difference in pericarp color. -
In this study, the genome survey of finger lemon 'YaoJi' was performed by the whole genome birdshot (WGS) strategy using second-generation sequencing technology, and a second-generation small fragment DNA library was constructed according to the genome characteristics, and 35.95 Gb of data were obtained by filtering the original data, and the genome size of finger lemon 'YaoJi' was predicted by using the K-mer method based on Illumina's short read lengths. K-mer method based on Illumina short read length was used to predict the genome size of finger lemon 'YaoJi', and the K-mer (K = 21) analysis showed that the genome size of finger lemon was about 290,100,953 bp, and the heterozygosity was 1.02% (Supplemental Fig. S1).
As shown by the genome survey, finger lemon is a slightly heterozygous species. Therefore, we obtained 13.69 Gb of long read and long sequence data by nanopore sequencing for ab initio assembly of finger lemon. The total genome size was about 313.91 Mb, including 175 Contigs, with Contig N50 and GC contents of 6.64 Mb and 34.38%, respectively. Busco's assessment of genome assembly quality and completeness showed that the plant had 96.4% single-copy homozygous gene integrity, 93.3% single-copy integrity, and 3.1% multi-copy gene integrity. The initially assembled finger lemon genome was of high quality and coverage. Construction of the initially assembled finger lemon genome to the chromosome level yielded a high-density genome map with a genome size of 314,637,567 bp, a scaffold N50 of 32.81 Mb, and nine chromosomes (Fig. 1; Table 1). The quality and integrity of the genome assembly was assessed using BUSCO[26], 98.4% of plants had intact single-copy homologous genes and 95.8% of intact single-copy genes, and 2.6% of multicopy genes. The LTR Assembly Index (LAI) is an index for assessing the quality of genome assemblies, and it is an index that evaluates genome assembly coherence in terms of the percentage of intact LTR-RTs over all LTR-RTs. In general, LAI between 10 and 20 is considered to be the reference genome standard[27]. The LAI index of this genome is 15.19, which shows that this genome can be used as a reference genome for lemon fingers because of its good assembly integrity and coherence.
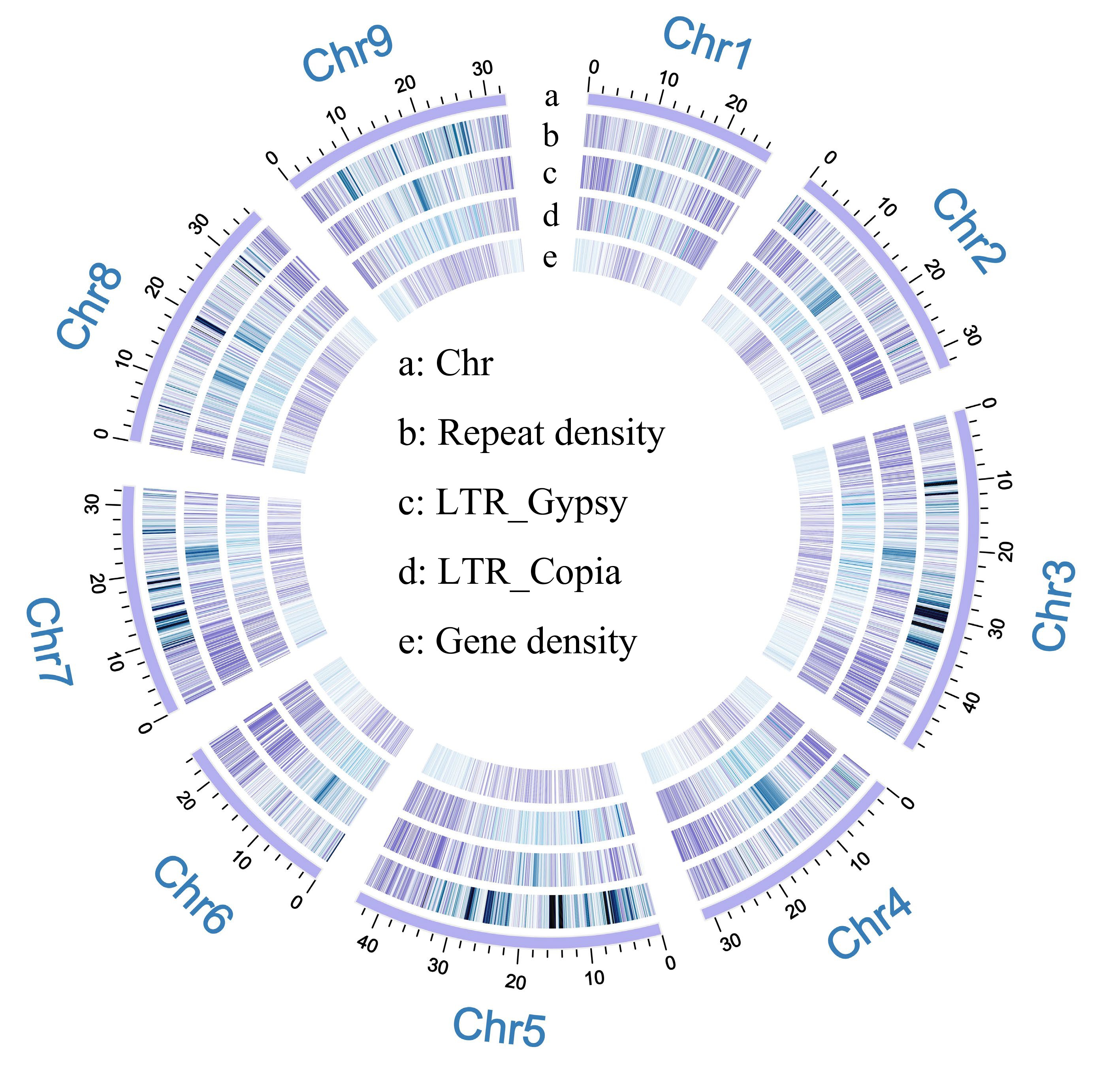
Figure 1.
Genome mapping of finger lemon. (a) Chromosomes. (b) Repeat density. (c) LTR_Gypsy (LTRs). (d) LTR_Copia (LTRs). (e) Gene density.
Table 1. Features of Citrus australasica genome assembly.
Genomic feature Value Genome size (Mb) 313,916,163 Chromosome-scale scaffolds 314,637,567 Total number of chromosomes 9 Number of contigs 175 Number of contigs N50 15 N50 of contigs (bp) 6,642,737 Number of contigs N80 37 Number of contigs N90 57 Total number of scaffolds 28 N50 of scaffold (bp) 32,805,073 N60 of scaffold (bp) 32,750,318 N70 of scaffold (bp) 32,749,230 N80 of scaffold (bp) 31,439,114 N90 of scaffold (bp) 27,021,073 N100 of scaffold (bp) 20,247 SD length of scaffold (bp) 313,375,644 GC content of the genome (%) 34.26% Complete BUSCOs 98.4% Quality value (QV) 27.68 LTR Assembly Index (LAI) 15.19 Repeat sequences (Gb) 164,723,365 (52.35%) Total number of protein-coding genes 21,154 Homology and de novo prediction were used to characterize the genome of the finger lemon. The genome was annotated for repetitive elements and protein-coding genes. Among them, about 164.7 Mb (52.35%) of the genome was identified as repetitive regions. Among these regions, long terminal repeat sequences (46.79 Mb, 14.91% of the genome) were identified as major repeat sequences. Reverse transposons were dominated by Copia (4.01%) and Gypsy (4.12%), and protein-coding genes were predicted and annotated by protein matching for finger lemons, resulting in 21,154 protein-coding genes.
Phylogenetic relationships and WGD analysis
-
A gene family is a group of genes originating from the same ancestor and consisting of two or more copies of a single gene through gene duplication, which shares significant structural and functional similarities and encode similar protein products[28].
To investigate the evolution and divergence of finger lemons, a phylogenetic tree was constructed for comparative genomic analysis using single-copy genes from 14 species to assess the expansion and contraction of the finger lemon gene family. These species included seven monocotyledons and seven dicotyledons. The phylogenetic evolutionary tree utilizes tree-branching graphs to represent the affinities between species or genes (Fig. 2a)[29]. The phylogenetic evolutionary tree shows that finger lemons diverged from sweet oranges and cremandine red oranges at about 10 Mya. The differentiation time of Rutaceae and Lacertidae (sugar maple) and Sapindaceae (mango) is around 60 Mya and 70 Mya, respectively. The differentiation time of Sapotaceae and Vitis vinifera (grape) is about 90 Mya ago. Expansion or contraction of gene families is an important feature of selective evolution[30]. In plant genomes, class I transposon factors, LTR-RT (LTR retrotransposons) are the main cause of genome expansion[31], the complete LTR length ranges from 85 to 5000 bp. In finger lemons, 721 gene families underwent an expansion and 5,600 gene families underwent contraction. The synonymous substitution rate (Ks), non-synonymous substitution rate (Ka), and the ratio between them were calculated for the homologous genes of finger lemon and sweet orange based on the expansion and contraction of the gene families, and the WGD events were inferred, and the results were presented in a Ks distribution plot. As can be seen from the Ks distribution (Fig. 2b), there is a peak in the KS plot of finger lemon, from which it is inferred that a WGD evolutionary event occurred in finger lemon.
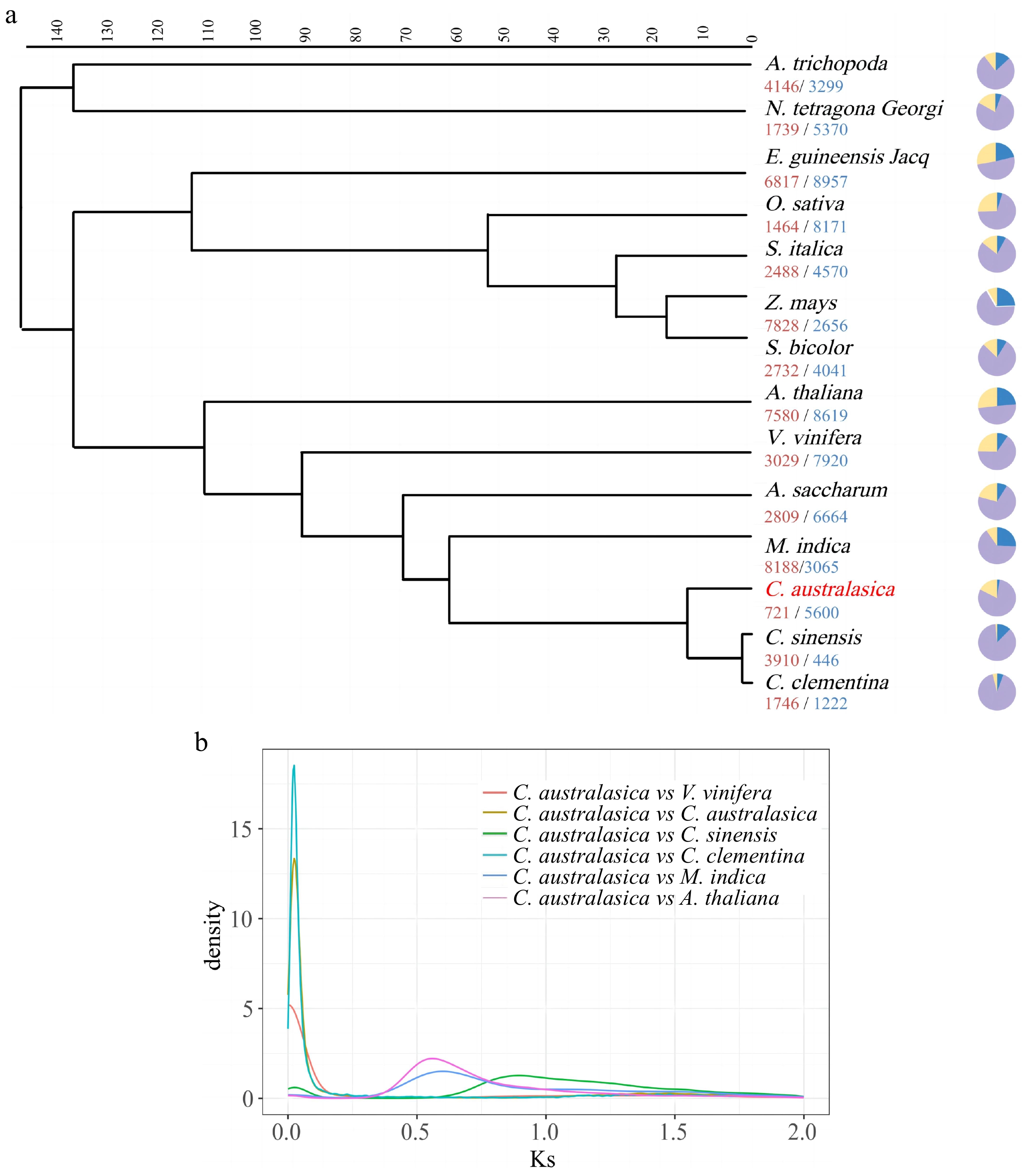
Figure 2.
Phylogeny and evolution of Citrus australasica. (a) Phylogenetic analysis based on homologous genes from 14 plant species, including five Saprotaceae species and two outgroups (Amborella trichopoda and Nymphaea L.), using the Orthfinder module. Pie charts represent the number of gene family expansions and contractions. (b) Frequency distribution of synonymous substitution rates (Ks) between homologous gene pairs in the syntenic blocks of C. australasica vs V. vinifera, C. australasica vs C. australasica, C. australasica vs C. sinensis, C. australasica vs C. clementina, C. australasica vs M. indica, C. australasica vs A. thaliana.
KEGG database[32] analysis of specific gene families in finger lemons showed that these gene families were mainly focused on metabolism-related biological functions such as metabolic pathways, carbon metabolism, phenylalanine biosynthesis, and phytohormone signaling (Supplemental Fig. S2). In addition, the GO database[33] enrichment analysis showed that these gene families mainly functioned in cellular components, such as nucleus, chloroplast, cytoplasm, and cytosol (Supplemental Fig. S3)
Comparative analysis of finger lemon peel colors
-
To better investigate the mechanism of the differences in the rind color of finger lemons, we sampled and analyzed the transcriptome data of different finger lemon varieties at the same developmental period (Fig. 3a). Three replicates were set up for each sample. After removing low-quality reads and reads with junctions, high-quality reads were obtained. In both samples, Q30 (%) accounted for more than 96% of the total high-quality reads, Q20 (%) accounted for more than 99% of the total high-quality reads, and GC (%) accounted for an average of 44.68% of the total. Sample expression was calculated using sequencing data, and the FPKM values of the genes were used to express the sample gene expression. The data were downscaled using principal component analysis (PCA) to see the correlation between the samples[31] (Supplemental Fig. S4). The PCA results showed that the two samples were far apart, which indicated that the samples were highly differentiated from each other and the two samples could be distinguished from each other. To further understand the changes of gene expression during pericarp development, the two samples were subjected to DEGs difference analysis and screened for differential genes, and p < 0.05 and Log2FC > 1 were used as the screening criteria for differential genes[34]. A total of 2,552 differential genes was obtained in the two samples, and 2,480 up-regulated genes and 45 down-regulated genes were detected in the control 'JiaLiHong' (Fig. 3b).
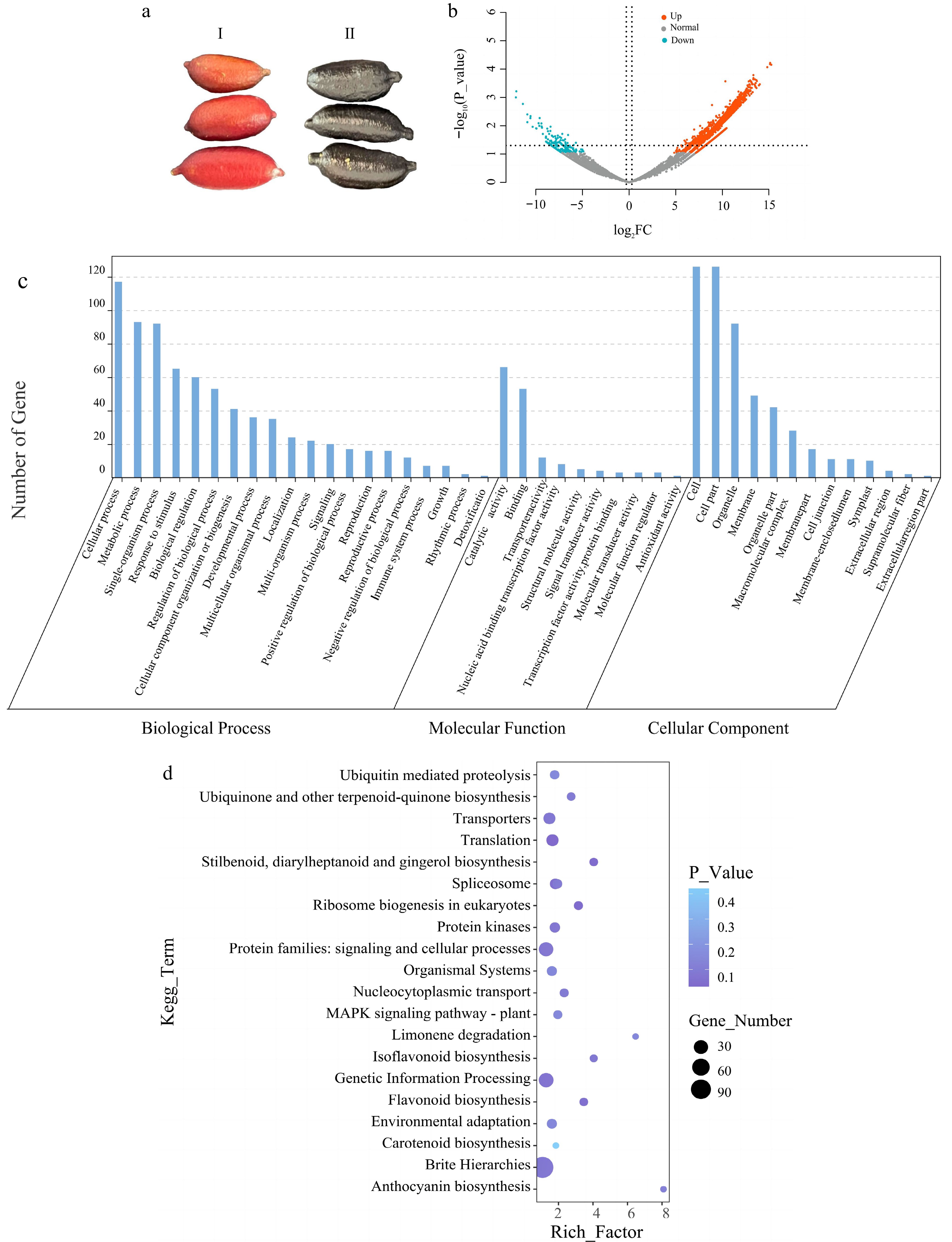
Figure 3.
Differential gene analysis of finger lemons. (a) Photographs of two finger lemon samples of different varieties 'JiaLiHong' and 'YaoJi' used for transcriptome analysis; I: 'JiaLiHong', II: 'YaoJi'. (b) Volcano plot of the expression of two different varieties of finger lemons after differential gene analysis; red indicates up-regulation; blue indicates down-regulation. (c) Enrichment results of up-regulated genes in the GO database. (d) Enrichment results of up-regulated genes in the KEGG database.
Eggnog functional annotation of screened differential genes[35]. GO enrichment analysis showed that these genes were mainly enriched in biological processes, followed by molecular functions, and to a lesser extent in cellular components (Fig. 3c). KEGG enrichment analysis showed that these genes were significantly enriched in the pathways of genetic information, metabolism, and biological systems[36]. Following the combination of the up-regulated gene screen, it was hypothesized that flavonoid biosynthesis and carotenoid synthesis, two of the enriched genes, may be involved in the variations in the color of finger lemon peel. Therefore, based on p_value values, 20 paths were chosen for visualization that were deemed to be reasonably significant (Fig. 3d). Subsequently, we searched for genes associated with the manufacture of flavonoids and carotenoids and identified this pathway. From the heat map in Fig. 4, it can be seen that compared with 'JiaLiHong', the expression of PAL and 4CL genes was high in the flavonoid synthesis pathway, which resulted in the black color of the epidermis of the finger lemon (Fig. 4a, b); and the expression of VDE and PSY genes were high in the carotenoid synthesis pathway (Fig. 4c, d). All these genes were localized on the chromosomes of finger lemon (Supplemental Figs S5 & S6). Meanwhile, we found the transcription factors related to the regulation of flavonoid synthesis: bHLH, MYB, WD40, and NAC, which were more highly expressed in 'YaoJi' compared to 'JiaLiHong' (Supplemental Fig. S7).
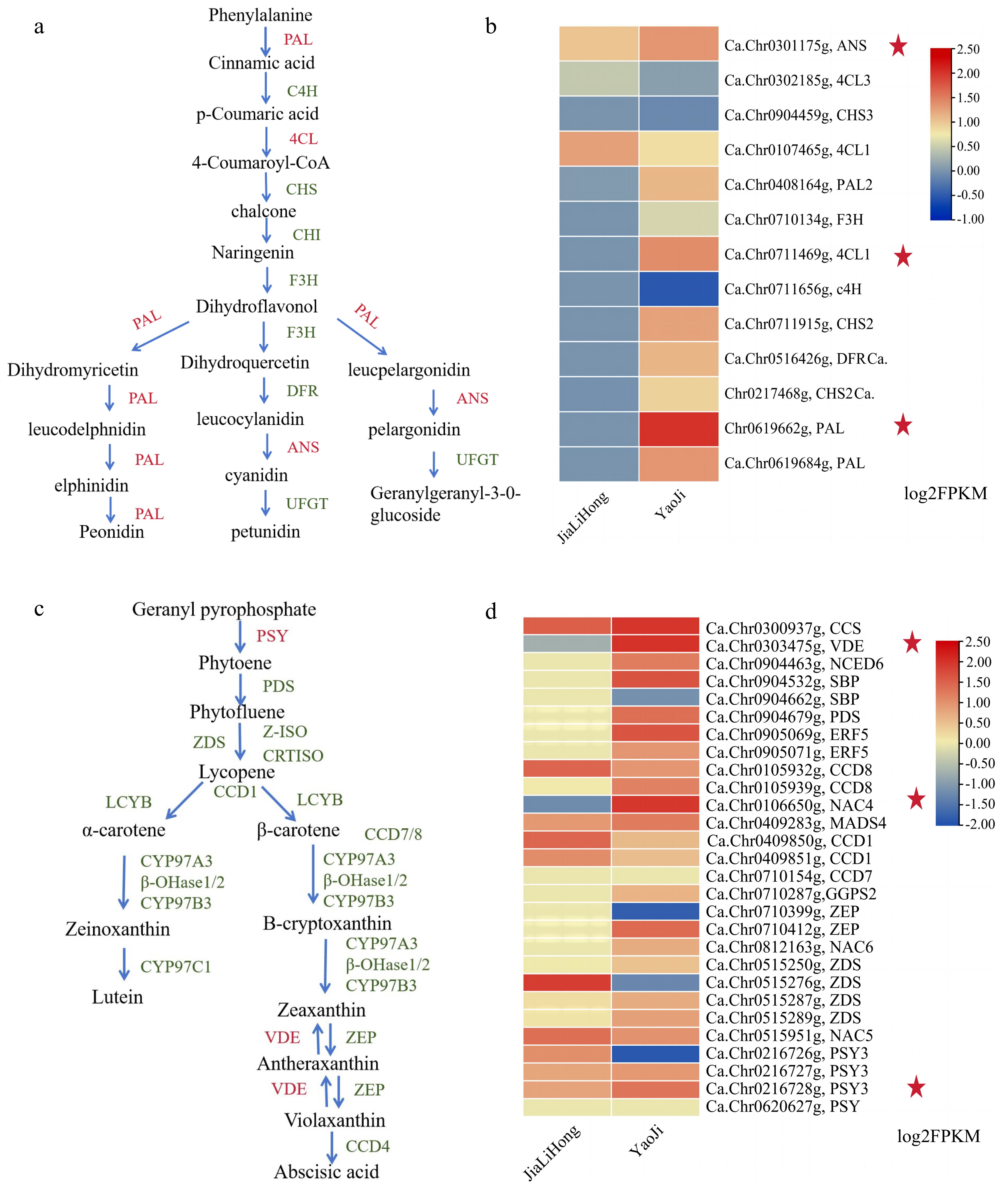
Figure 4.
Biosynthetic pathways of flavonoids and carotenoids. Normalized FPKM values were expressed using 'JiaLiHong' as control. (a) Flavonoid biosynthetic pathway. (b) Heat map of highly expressed genes for flavonoid biosynthesis in 'YaoJi'. (c) Carotenoid biosynthetic pathway d. Heat map of highly expressed genes for carotenoid biosynthesis in 'YaoJi'.
These results suggest that peel color is differentially affected by some highly expressed genes and transcription factors for flavonoid synthesis and carotenoid synthesis during finger lemon growth and development.
-
The genome is the sum of all the genetic material (DNA or RNA) of an organism[37]. Genome size refers to the amount of DNA held in a genome. Genome size varies between species due to species differences[38]. For example, the plantain taro of the family Plantaginaceae[39] (Canna edulis Ker), the species of Myrtus communis in the family Hamamelidaceae, Adin maple[40](Altingia chinensis), the species Momordica of the genus Cucurbitaceae[41] (Siraitia grosvenorii) with genome sizes of 555.6, 650.77, and 344.95 Mb, respectively, the acquisition of chromosome-level reference genomes have become indispensable with the continuous development of sequencing technologies, the diversification of algorithms and techniques related to assembly, and the significant increase in accuracy. High-quality reference genomes are of great significance for the study of the origin and evolution of species and the mining of functional genes for further research.
Finger lemons are economically important globally and studies of their molecular mechanisms can accelerate variety selection. Provide new insights and genomic resources into the genetic systems and evolution of Brassicaceae. These require a high-quality genome sequence as a foundation. Genomes can be classified into two categories based on the degree of heterozygosity: micro-heterozygous (0.5% ≤ heterozygosity < 0.8%) and hyper-heterozygous (heterozygosity ≥ 0.8%)[42]. Combined with the results of the preliminary K-mer analysis, finger lemons belong to a highly heterozygous genome. Among the citrus species in the Brassicaceae family, the genomes of sweet orange[43], mandarin orange[9], and lemon[44] have been analyzed and published, with genome sizes of 367, 344.3, and 380.14 Mb. Respectively, it is inferred that the genome size of Citrus aurantium should be similar to them. This provides a basis for the subsequent development of sequencing and assembly strategies for finger lemon. Based on this, we sequenced the finger lemon 'YaoJi' and assembled a high-quality finger lemon genome with a size of 314.63 Mb (Contig N50, 6.60 Mb; Scaffold N50, 32.81 Mb) and 9 chromosomes. The genome was highly coherent and complete and could meet the standard of the reference genome. The high-quality reference genome is important for revealing the position of the family genus finger lemon in evolutionary history. Phylogenetic evolutionary analyses showed that the divergence of finger lemon from its close relative, sweet orange, occurred about 10 Mya ago; about 60 Mya ago, the Brassicaceae and Lacertaceae diverged into a separate family, which provides a basis for further study of the evolutionary relationships of the Sapindaceae. WGD events are a common type of paleopolyploidy events in plants, which indicate the development of new gene functions or the formation of new species. A polyploidization event is presumed to have occurred in finger lemon based on the KS distribution map. This genomic information provides important information for the study of gene content, repetitive elements, localization of genes on chromosomes and evolutionary relationships in finger lemons, which will help to elucidate the evolutionary process of Brassicaceae species and contribute to an improved understanding of the physiological and morphological diversity of Brassicaceae species.
To further investigate the mechanisms underlying the differences in the color of the rind of finger lemons of different varieties, transcriptome analysis was performed in this study on the rinds of finger lemons of different varieties with different colors at the same developmental period. The results showed that the differences in the color of finger lemon peels were associated with the high expression of genes involved in flavonoid biosynthesis and carotenoid synthesis. Flavonoids are a class of compounds commonly found in the biosynthetic pathway in plants, with a variety of biological activities and secondary metabolites involved in plant development and defense, which is one of the largest groups of secondary metabolites and the main source of plant pigments, which accumulate in all parts of the plant. Not only can they protect pigments and play a role in regulating the null of plant growth and development, but they can also protect plant cells from damage caused by oxidative stress[45]. The biosynthetic pathway of plant flavonoids is accomplished by a series of structural and regulatory genes[46]. The structural genes encode enzymes that catalyze the synthetic pathway, and these key structural enzymes include PAL, C4H, CHS, CHI, F3H, DFR, etc., and are regulated by the transcription factors MYB, bHLH, WD40, and NAC, as well as other transcription factors[47]. The regulatory network of flavonoid biosynthesis in different flower colors of Rhododendron pulchrum was constructed by the Woody Flower Research Team of Shanghai Botanical Garden[48]. Using Rhododendron pulchrum as experimental material, they identified different kinds of flavonoid metabolites by combining metabolomics and transcriptomics. The mechanism of sugar signaling in tea tree (Camellia sinensis (L.) O. Kuntze) involved in the regulation of tea tree flavonoid biosynthesis was revealed by Lv et al.[49], which provided new insights into the regulatory role of sugar signaling on flavonoid biosynthesis in tea tree. Transcription factors (TF) are proteins that assist in the transcription process in eukaryotes. The HSFB2b transcriptional gene can directly activate the flavonoid synthesis pathway, and with the high accumulation of flavonoid metabolites which in turn promotes the salt tolerance of soybeans (Glycine max (L.) Merr.) that phenomenon was discovered by Bian et al.[50]. The mechanism of dates (Ziziphus jujuba Mill.) pericarp color formation was revealed by Liu Ping using dates pericarp of three ripening periods[51]. A new NF-Y transcription factor-based mechanism affecting flavonoid biosynthesis and providing a new mechanism for tomato (Solanum lycopersicum L.) fruit color formation is described by Huazhong Agricultural University[52]. Carotenoids are not only light-trapping and photoprotective pigments for photosynthesis, but also precursors for the synthesis of phytohormones such as abscisic acid, which are essential for plant growth and development[53]. The phenomenon that carotenoid isomerase DN regulates rice (Oryza sativa) tillering and yield through the carotenoid biosynthesis pathway was reported by Ding et al.[54]. Key regulatory genes for capsaicin biosynthesis and the mechanism of carotenoid biosynthesis in chili (Capsicum annuum L.) fruits were revealed by the Institute of Modern Agriculture, Peking University. These bring some insights to this study[55].
However, there are also some shortcomings in this study. Compared with the Hifi data obtained by PacBio, the data obtained by Nanopore sequencing is not as accurate as the Hifi data obtained by PacBio[56]. Even though the second-generation data is corrected to ensure that the read lengths are sufficiently long, which also indirectly leads to poor genome integrity, it does not affect the subsequent analyses[57]. Meanwhile, in terms of assembly data and assembly strategy, compared with the analyzed species of the same family[58]. Finger lemon was assembled using ONT data only, while the others were assembled by combining the Hifi data from PacBio sequencing, which is a better result in comparison[59]. Meanwhile, we need to confirm the metabolomic data and experimentally verify the regulation of rind color differences[60].
-
In this study, we sequenced and assembled the genome of a typical purple variety of finger lemons—'YaoJi' by Nanopore sequencing technology, and investigated the mechanism of the differences in the color of finger lemon rinds by transcriptome analysis using the rinds of finger lemons of different varieties with different colors at the same developmental period. The results showed that the Finger Lemon genome was 314.63 Mb (Contig N50, 6.60 Mb; Scaffold N50, 32.81 Mb) and containing nine chromosomes. Both the integrity and continuity of the genome were high. Annotation yielded 21,154 protein-coding genes and 164.7 Mb (52.35%) of repetitive sequences. Phylogenetic evolutionary trees revealed the evolutionary relationships of finger lemons. Rutaceae diverged from Lacertaceae nearly 60 Mya ago, and finger lemon diverged from sweet orange and Cremantine red orange 10 Mya ago, which presumably underwent a WGD event based on KS. Transcriptome analysis showed that the differences in rind color of finger lemon were associated with higher expression of PAL and 4CL genes in flavonoid biosynthesis and VDE and PSY genes in carotenoid synthesis. Transcription factors regulating flavonoid biosynthesis, such as bHLH, MYB, WD40, and NAC were also identified, which provide insights into the origin and evolution of finger lemon species and functional gene mining.
-
The authors confirm contribution to the paper as follows: study conception and design: Tian Y, Xia Z, Zou M; data collection:Liang T, Chen B, Peng H, Wang Q; analysis and interpretation of results: Tian Y, Liang T, Zhao L, Xu R, Wang Z; draft manuscript preparation: Tian Y, Liang T, Luo X, Kumpeangkeaw A. All authors reviewed the results and approved the final version of the manuscript.
-
The whole genome sequence data reported in this paper have been deposited in the Genome Warehouse in China National Center for Bioinformation, under accession number PRJNA1092589 that is publicly accessible at
https://ngdc.cncb.ac.cn/gwh . This study was supported by the Hainan Province Science and Technology Special Fund (ZDYF2022XDNY149) and Hainan University Startup Fund (KYQD(ZR)-20101).
-
The authors declare that they have no conflict of interest.
-
accompanies this paper at (https://www.maxapress.com/article/doi/10.48130/tp-0024-0021)
-
Received 28 March 2024; Accepted 29 April 2024; Published online 21 May 2024
-
Constructed a high-quality reference genome of the finger lemon "YaoJi" by nanopore sequencing.
Comparison of protein sequences between Lemon Finger and 13 other species to understand its evolution and differentiation.
Combined with transcriptome analysis to investigate the mechanisms underlying the differences in rind color of finger lemons.
- Supplemental Fig. S1 Finger Lemon Surveys.
- Supplemental Fig. S2 KEGG enrichment plot of finger lemon.
- Supplemental Fig. S3 Finger Lemon GO Enrichment Map.
- Supplemental Fig. S4 PCA diagram.
- Supplemental Fig. S5 Chromosomal localization of flavonoid synthesis genes.
- Supplemental Fig. S6 Carotenoid synthesis chromosome localization map.
- Supplemental Fig. S7 Heat map of transcription factors.
- Copyright: © 2024 by the author(s). Published by Maximum Academic Press on behalf of Hainan University. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Tian Y, Liang T, Peng H, Wang Q, Luo X, et al. 2024. Chromosome-scale genome assembly provides insights into the evolution and color synthesis of finger lemon (Citrus australasica). Tropical Plants 3: e015 doi: 10.48130/tp-0024-0021 

Chromosome-scale genome assembly provides insights into the evolution and color synthesis of finger lemon (Citrus australasica)
- Received: 28 March 2024
- Revised: 27 April 2024
- Accepted: 29 April 2024
- Published online: 21 May 2024
Abstract: The Australian native plant finger lemon (Citrus australasica) is well-known for its distinct flavor and high economic and medicinal value. 'YaoJi' is a typical purple variety of finger lemon, but its genome has not been analyzed yet. In this study, we used three-generation nanopore sequencing technology to sequence and assemble the genome of the finger lemon and combined it with the transcriptome analysis of different varieties with different colors of rinds at the same developmental period to investigate the mechanism of the differences in the color of the rinds of finger lemons. According to the results, the finger lemon genome was built and has nine chromosomes and a size of 314.63 Mb (Contig N50, 6.60 Mb; Scaffold N50, 32.81 Mb). By using homologous protein comparison and gene annotation, a total of 21,154 protein-coding genes and 164.7 Mb (52.35%) of repetitive sequences have been identified. The phylogenetic evolutionary tree shows that 10 Mya years ago finger lemon diverged from sweet orange (Citrus sinensis (L.) Osbeck) and Cremantine red orange (Citrus × clementina). Presumably it's a whole genome duplication (WGD) event according to the synonymous substitution rates (KS). Transcriptome analysis showed that the differences in the rind color of finger lemon were associated with higher expression of PAL, 4CL, and ANS genes in flavonoid biosynthesis and VDE and PSY genes in carotenoid synthesis. Transcription factors regulating flavonoid biosynthesis, such as bHLH, MYB, WD40, and NAC were also identified, which provides insights into the origin and evolution of finger lemon species and functional gene mining.
-
Key words:
- Citrus australasica /
- Genome assembly /
- Color synthesis