









-
Auxin, an important plant hormone, plays crucial roles in various aspects of plant growth and development processes, such as cell division, expansion, and differentiation[1], vascular tissue formation[2], apical dominance[3], and flower and fruit development[4,5]. During the initial stages of auxin signal transduction, specific gene families, including Auxin/IAA, GH3 (Gretchen Hagen3), and SAUR (small auxin up RNA), exhibit high responsiveness to fluctuations in auxin levels. The Auxin/IAA gene family was initially identified by Walker & Key in soybean[6]. This family consists of members with four highly conserved domains: domains I, II, III, and IV. Each domain has a distinct function: domain I is responsible for transcriptional repression, domain II is essential for auxin signal transduction, and domains III and IV enable proteins to form homo- and heterodimers. Aux/IAA proteins regulate auxin-responsive genes by interacting with ARFs (Auxin Response Factors), rather than directly binding to AuxREs (auxin-responsive cis-elements). So far, the Aux/IAA gene family has been identified in various plant species. For example, there are 23 members in Prunus persica[7], 29 in Arabidopsis[8], 35 in Populus euphratica[9], 42 in Malus domestica[10], and 44 in Musa nana[11].
Several studies have reported the crucial roles of Auxin/IAA family genes in various aspects of plant growth and development. In Arabidopsis, AtIAA6, 9, and 17 inhibit root initiation by interacting with AtARF6 and AtARF8[12]. The TIR1/AFB-Aux/IAA module regulates hypocotyl growth[13]. Similarly, PpIAA19, which is mainly expressed in peach (Prunus persica) fruit, has been demonstrated to control lateral root number, stem elongation, parthenocarpy, and fruit shape[14]. In poplar (Populus tomentosa), PtIAA9 acts as a negative regulator of secondary xylem development and controls wood formation through the PtIAA9-PtARF5 module[15]. Additionally, the Auxin/IAA gene family significantly influences cell proliferation, and cell expansion, and plays a vital role in flower development and the opening process[16]. In waterlily (Nymphaeale), Auxin/IAA genes induce the constitutive flower opening and are involved in floral movement[17]. In Rosa hybrida, the suppression of RhIAA14 and RhIAA16 expression leads to reduced flower cell expansion and smaller petals[18,19]. Moreover, accumulating evidence suggests that the Aux/IAA gene family also participates in plant stress responses and defense. In Arabidopsis, the auxin-sensitive genes AtIAA5, 6, and 19 have been found to enhance drought tolerance by regulating glucosinolate levels, which protect plants from herbivory and pathogen attack[20]. In Medicago falcata, MfAIR12 (Auxin induced in root culture 12) contributes to cold tolerance by regulating the expression level of cold-responsive genes and ascorbate synthesis and redox state[21]. In rice, OsIAA20 plays an important role in drought and salt stress responses through the ABA-dependent pathway[22]. On the other hand, OsIAA6 enhances drought tolerance by regulating the expression of auxin biosynthesis genes[23]. Additionally, the overexpression of MdIAA9 from cultivated apple (Malus × domestica) significantly improved osmotic stress tolerance in transgenic tobacco (Nicotiana tabacum L.)[24].
Osmanthus fragrans, one of the ten most traditional flowers in China, is widely cultivated as a garden tree in many countries because of its remarkable ornamental value and delightful fragrance. Although genome-wide analysis of the Aux/IAA gene family has been performed in various species, the identification, characterization, and functional analysis of Aux/IAA family genes in O. fragrans have remained unexplored. In this study, a thorough genome-wide identification of Aux/IAA family members in the O. fragrans genome was conducted. Subsequently, their chromosomal locations, domains, phylogenetic relationships, gene duplications, gene structures, motifs, and cis-regulatory elements (CREs) were analyzed. Additionally, transcriptome sequencing and quantitative real-time PCR (qRT-PCR) were used to investigate the involvement of Aux/IAA family genes in regulating flower opening processes and responding to abiotic stress in O. fragrans. The aim of these results is to provide a comprehensive understanding of the Auxin/IAA gene family in O. fragrans.
-
A total of 44 potential Aux/IAA gene sequences were identified in the O. fragrans genome. Five sequences were excluded from further analysis due to the presence of open reading frame (ORF). Subsequently, 39 OfAux/IAA (OfIAA) genes with a typical Aux/IAA domain were obtained and designated as OfIAA1 to OfIAA39. More details about these 39 OfIAAs are presented in Supplemental Table S1, including protein length, MW (molecular weight), pI (isoelectric point), instability index, aliphatic index, grand average of hydropathicity, and subcellular localization prediction. The identified OfIAAs have protein lengths ranging from 142 amino acids (OfIAA14) to 392 amino acids (OfIAA1), with MWs ranging from 15.5 to 42.5 kDa. The pI of the 39 OfIAA genes varies from 4.94 (OfIAA14) to 9.37 (OfIAA20). Additionally, chromosome mapping revealed that the 39 OfIAA genes are unevenly distributed across 19 chromosomes. Chromosome 4 contains the largest number of OfIAA genes (five members) (Fig. 1).
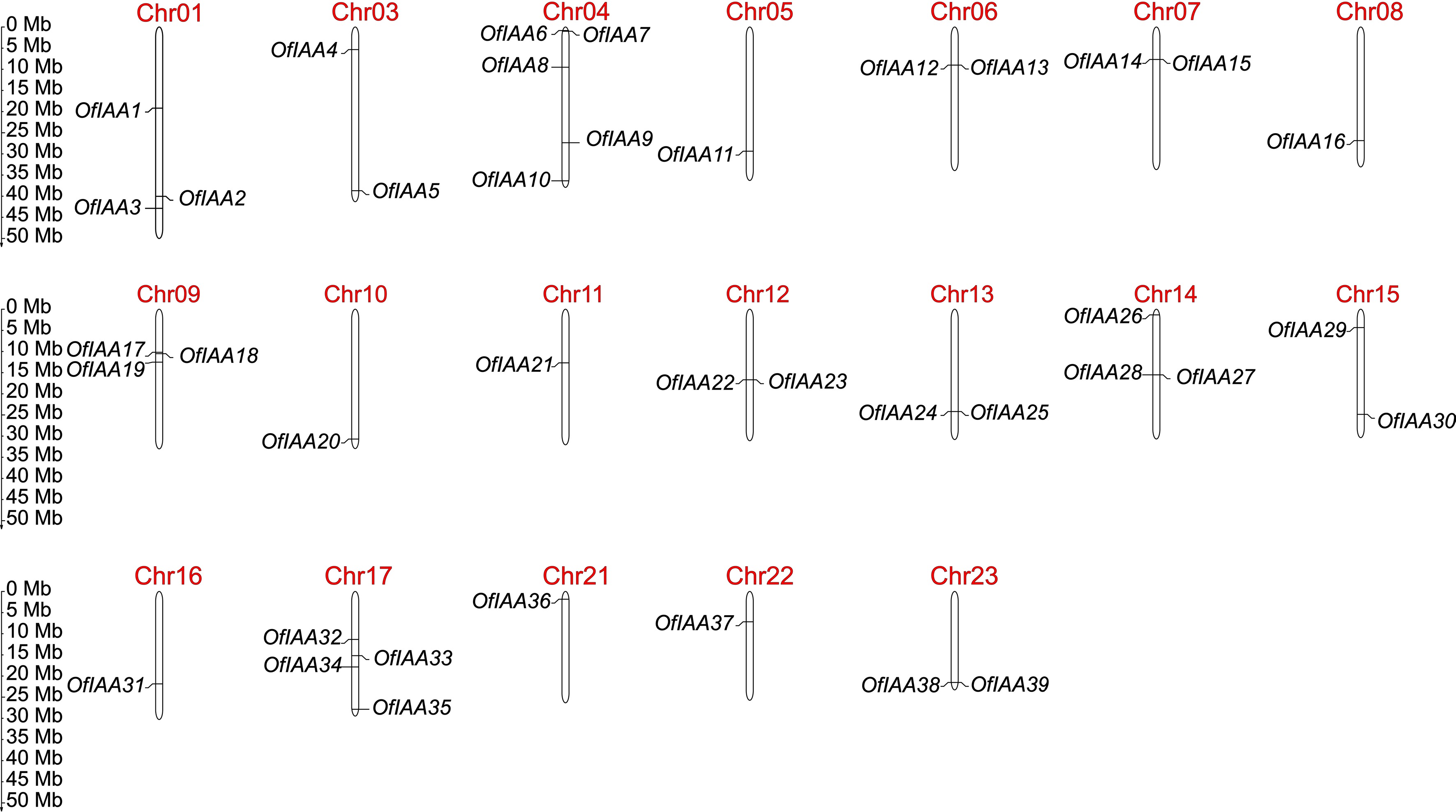
Figure 1.
Chromosomal distribution of OfIAA genes in O. fragrans.
Phylogenetic classification and conserved domain analysis
-
Based on the phylogenetic analysis, the IAA proteins (39 members) were grouped into two major clades (clades A and B), similar to Arabidopsis and Oryza sativa. Clade A consisted of 27 OfIAA genes and clade B consisted of 12 (Fig. 2). Synteny analysis of the OfIAA genes was conducted and 34 pairs of segmental duplications were identified distributed across 19 chromosomes (Fig. 3a). The divergence time indicated that the duplications of the OfIAA genes commenced 75.90 million years ago (Mya) and continued until 0.56 Mya. All OfIAA genes evolved under purifying selection (Ka/Ks < 1) (Table 1). In addition, the synteny analyses revealed that 20 and five pairs of OfIAA homologous genes were identified in Arabidopsis and rice, respectively (Fig. 3b).
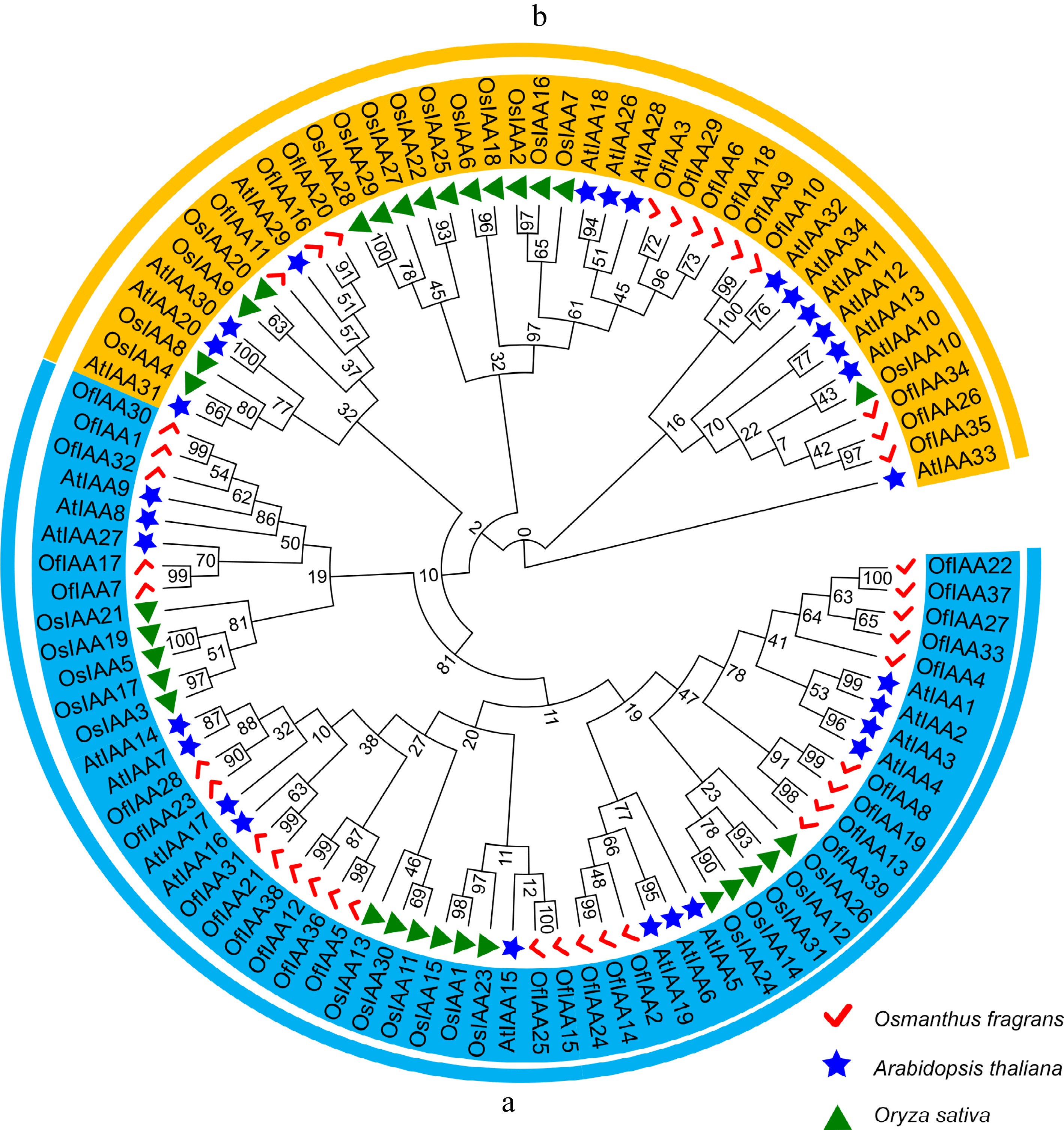
Figure 2.
Phylogenetic relationship of the IAAs among O. fragrans, A. thaliana, and O. sativa. The IAA proteins in O. fragrans are represented by the red ticks, the IAA proteins in A. thaliana are represented by the blue stars, the IAA proteins in O. sativa are represented by the green triangles. Two main groups (a) and (b) were displayed by colored arcs.
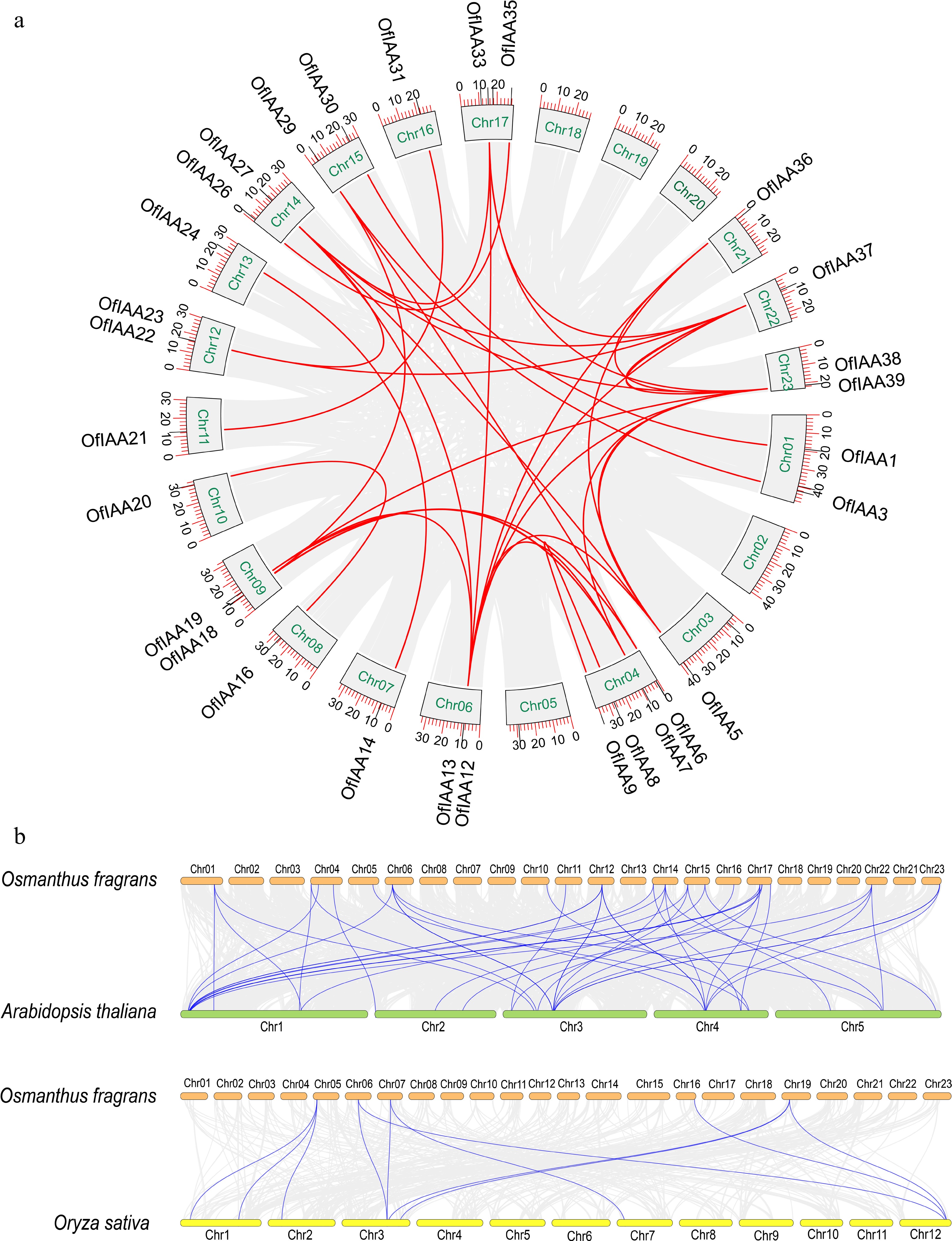
Figure 3.
Synteny analysis of the IAA genes. (a) Synteny analysis of the OfIAA genes in O. fragrans. (b) Synteny analysis of the OfIAA genes between A. thaliana and O. sativa.
Table 1. Ka/Ks analysis and estimated divergence time of OfIAA genes.
Duplicated gene pairs Ka Ks Ka/Ks Divergence time
(Mya)OfIAA1 & OfIAA30 0.07 0.25 0.29 8.46 OfIAA3 & OfIAA29 0.18 0.45 0.41 15.02 OfIAA5 & OfIAA12 0.22 0.92 0.24 30.57 OfIAA5 & OfIAA28 0.22 1.67 0.13 55.65 OfIAA5 & OfIAA36 0.04 0.22 0.21 7.27 OfIAA5 & OfIAA38 0.18 0.73 0.25 24.38 OfIAA9 & OfIAA10 0.01 0.02 0.69 0.56 OfIAA8 & OfIAA13 0.1 0.67 0.16 22.2 OfIAA8 & OfIAA19 0.07 0.12 0.62 3.89 OfIAA6 & OfIAA18 0.11 0.28 0.41 9.26 OfIAA7 & OfIAA17 0.08 0.24 0.35 8.16 OfIAA6 & OfIAA29 0.27 0.72 0.37 23.97 OfIAA8 & OfIAA38 0.36 2.28 0.16 75.9 OfIAA13 & OfIAA19 0.16 0.72 0.22 24.03 OfIAA13 & OfIAA27 0.2 1.65 0.12 55.07 OfIAA13 & OfIAA33 0.21 1.16 0.19 38.59 OfIAA12 & OfIAA36 0.21 0.76 0.28 25.36 OfIAA13 & OfIAA37 0.25 1.7 0.15 56.59 OfIAA12 & OfIAA38 0.1 0.2 0.48 6.59 OfIAA14 & OfIAA24 0.07 0.19 0.39 6.34 OfIAA15 & OfIAA25 0.09 0.28 0.3 9.44 OfIAA16 & OfIAA20 0.18 0.43 0.43 14.39 OfIAA18 & OfIAA29 0.3 0.97 0.31 32.19 OfIAA21 & OfIAA31 0.07 0.19 0.35 6.36 OfIAA22 & OfIAA27 0.14 0.52 0.27 17.18 OfIAA23 & OfIAA28 0.07 0.75 0.1 24.89 OfIAA23 & OfIAA28 0.07 0.75 0.1 24.89 OfIAA22 & OfIAA37 0.06 0.23 0.27 7.69 OfIAA26 & OfIAA35 0.07 0.24 0.3 8.15 OfIAA27 & OfIAA33 0.07 0.25 0.27 8.41 OfIAA27 & OfIAA37 0.12 0.72 0.17 24.12 OfIAA33 & OfIAA37 0.14 0.86 0.16 28.59 OfIAA36 & OfIAA38 0.17 0.67 0.26 22.23 OfIAA37 & OfIAA39 0.22 1.48 0.15 49.41 Ka, nonsynonymous; Ks, synonymous. Multiple alignments of the amino acid sequences of OfIAA proteins revealed that 35 OfIAA proteins contained all four typical conserved domains I, II, III, and IV. However, OfIAA12, 18, 26, and 32 lacked one of these domains (Fig. 4). Specifically, OfIAA32 lacked domain I, OfIAA18 and 26 lacked domain II, and OfIAA12 lacked domain IV. Nuclear localization signals (NLS) were identified at the end of domain IV in 16 OfIAA proteins. Additionally, gene structure analysis showed that the number of exons ranged from two to five (Fig. 5). Furthermore, five different motifs were identified in the 39 OfIAA proteins, and the majority of OfIAA proteins contained all five motifs, with motif 1 being conserved in all the OfIAA proteins (Fig. 5).
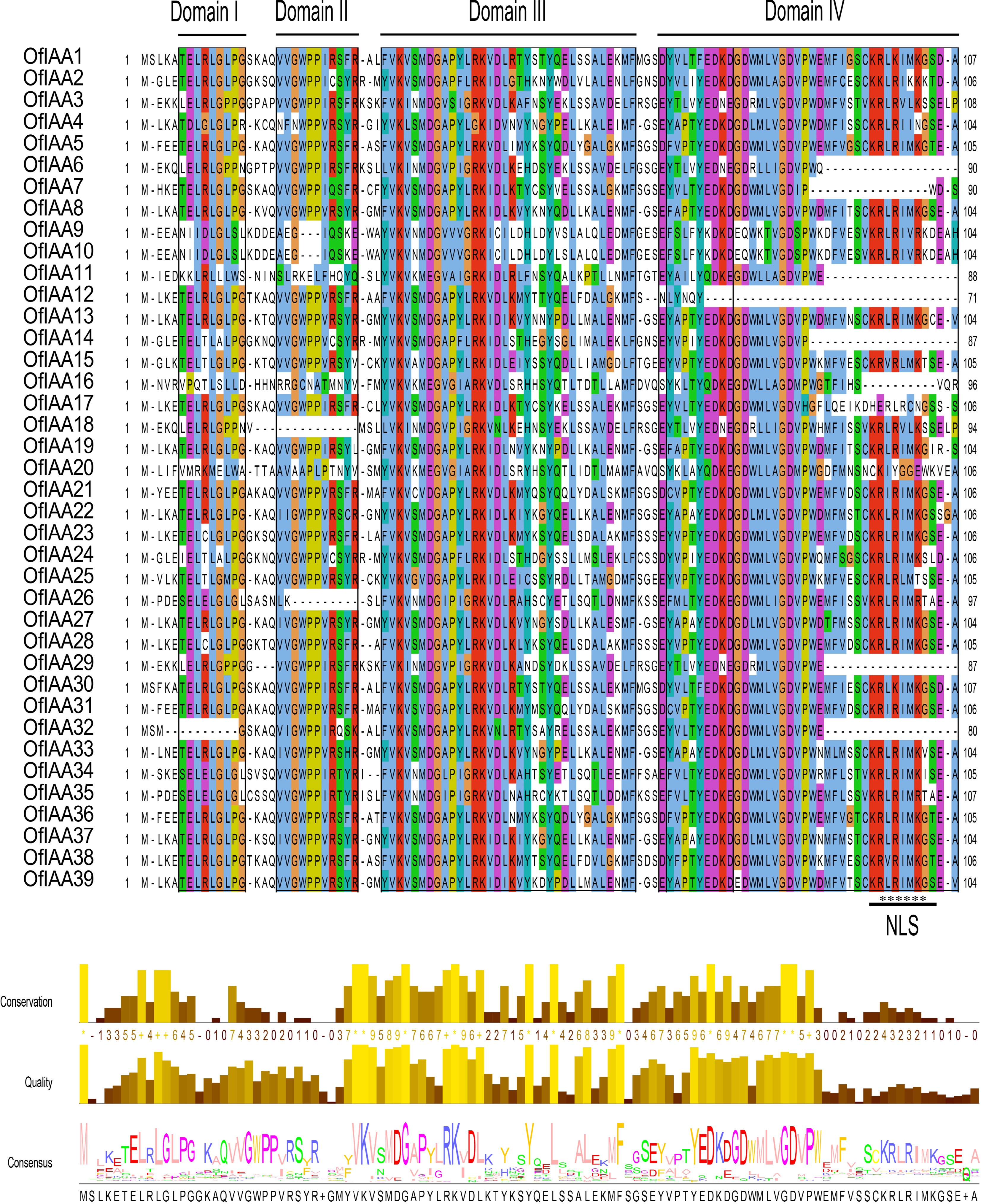
Figure 4.
Multiple sequence alignment of the OfIAA proteins. The conserved domains (I, II, III, and IV) of the OfIAA gene family are underlined. Nuclear localization signals (NLS) are indicated with black asterisks. Bits indicate amino acid conservation at each position.

Figure 5.
Motif and gene structure analysis of OfIAAs.
Cis-elements analysis of the promoter
-
The 2,000 bp upstream promoter regions of 39 OfIAA genes were identified through CRE analysis (Fig. 6). A total of 11 types of CREs were identified, with the majority being associated with hormone responses, such as auxin, ABA (abscisic acid), GA (gibberellin), MeJA (methyl jasmonate), and SA (salicylic acid). In addition, a significant number of OfIAA promoters contained ABA-responsive elements (31 OfIAA genes), MeJA-responsive elements (26 OfIAA genes), and GA-responsive elements (23 OfIAA genes). Furthermore, the promoters of OfIAA genes related to defense and stress responsiveness, and low-temperature responsiveness were also identified.
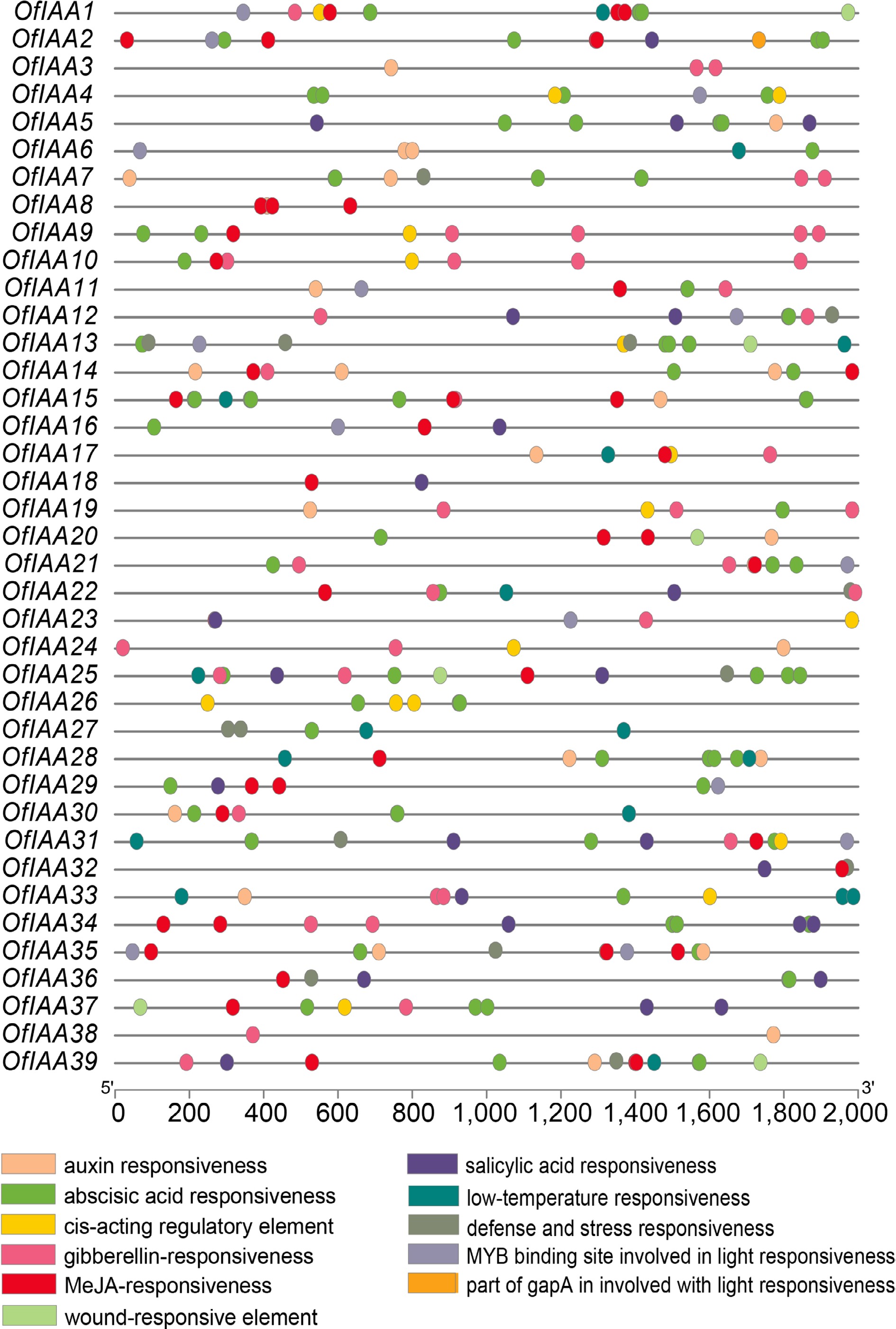
Figure 6.
Cis-elements distribution of OfIAA genes.
Expression patterns of OfIAAs in different tissues
-
To gain further insight into the expression patterns of the OfIAA genes in different tissues, transcriptome sequencing was performed. The expression profiles of the 39 OfIAA genes were examined in the six different tissues, including root, annual stem, perennial stem, young leaf, mature leaf, and flower (Fig. 7). The expression analysis revealed that the majority of the OfIAA genes exhibited broad expression patterns in all six tissues, except for OfIAA4, 9, and 10. Of particular interest was the predominant expression of OfIAA6 in the root, while OfIAA7, 16, 20, 28, and 30 exhibited high expression levels in the annual stem. Additionally, OfIAA29 and 32 had high expression levels in the perennial stem, and OfIAA14 was overrepresented in the young leaf. Furthermore, the transcriptome analysis revealed that OfIAA24 and 25 were predominantly expressed in the flower (Fig. 7).
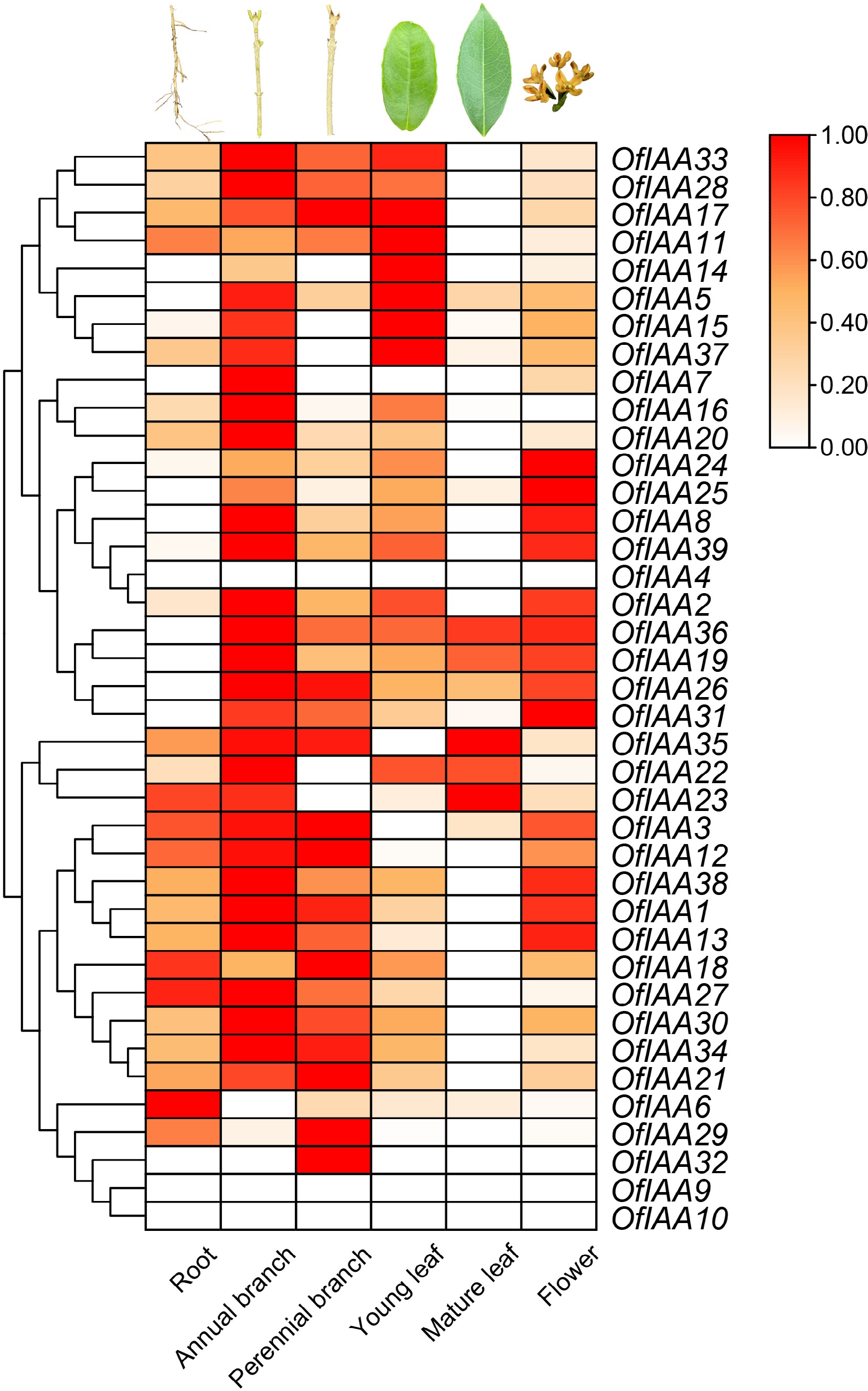
Figure 7.
Expression analysis of OfIAAs in different tissues.
Analysis of OfIAAs expression in flower opening processes
-
In O. fragrans, the scent release is significantly influenced by the flower opening process, with the transformation from the S1 to S2 stages playing a particularly crucial role. To investigate the potential involvement of OfIAA genes in this process, a comprehensive analysis of their expression profiles was conducted via RNA-seq in flower buds at the S1 and S2 stages (Table 2). Through analysis, a total of 18 DEGs (differentially expressed genes) were identified from the S1 to S2 stage (Table 2). Among these DEGs, 16 genes were upregulated, including OfIAA2, 8, 11, 12, 13, 14, 15, 20, 25, 27, 28, 33, 36, 37, 38, and 39, while two genes, namely OfIAA6 and 29, were downregulated. Subsequently, the expression patterns of all DEGs were validated in flower buds at the S1 and S2 stage using qRT‒PCR (Fig. 8). The qRT-PCR results were consistent with the expression trends observed in the transcriptomic data. These results suggest that these differentially expressed OfIAA genes may have a potential role in regulating the flowering process.
Table 2. Identification of DEGs in the flower opening processes of O. fragrans.
Gene Expression FoldChange Padj value S1 S2 OfIAA2 0.00 22.82 7.00 6.69 × 10−7 OfIAA6 1,121.83 435.01 −1.37 3.97 × 10−29 OfIAA8 28.68 789.65 4.79 1.93 × 10−99 OfIAA11 5.05 24.49 2.26 1.20 × 10−3 OfIAA12 13.62 145.78 3.42 2.75 × 10−21 OfIAA13 18.45 60.19 1.70 8.14 × 10−5 OfIAA14 0.32 7.61 4.45 9.80 × 10−3 OfIAA19 92.29 490.40 2.41 1.30 × 10−41 OfIAA20 0.30 6.22 4.15 4.90 × 10−2 OfIAA25 25.79 131.80 2.36 1.76 × 10−15 OfIAA27 4.82 23.02 2.25 5.20 × 10−3 OfIAA28 3.73 173.53 5.60 5.41 × 10−28 OfIAA29 1,136.52 235.71 −2.27 4.41 × 10−60 OfIAA33 4.68 33.32 2.86 9.99 × 10−5 OfIAA36 44.20 92.87 1.06 7.90 × 10−4 OfIAA37 101.89 292.73 1.52 1.54 × 10−15 OfIAA38 110.89 292.85 1.40 1.45 × 10−13 OfIAA39 24.51 134.72 2.44 2.23 × 10−11 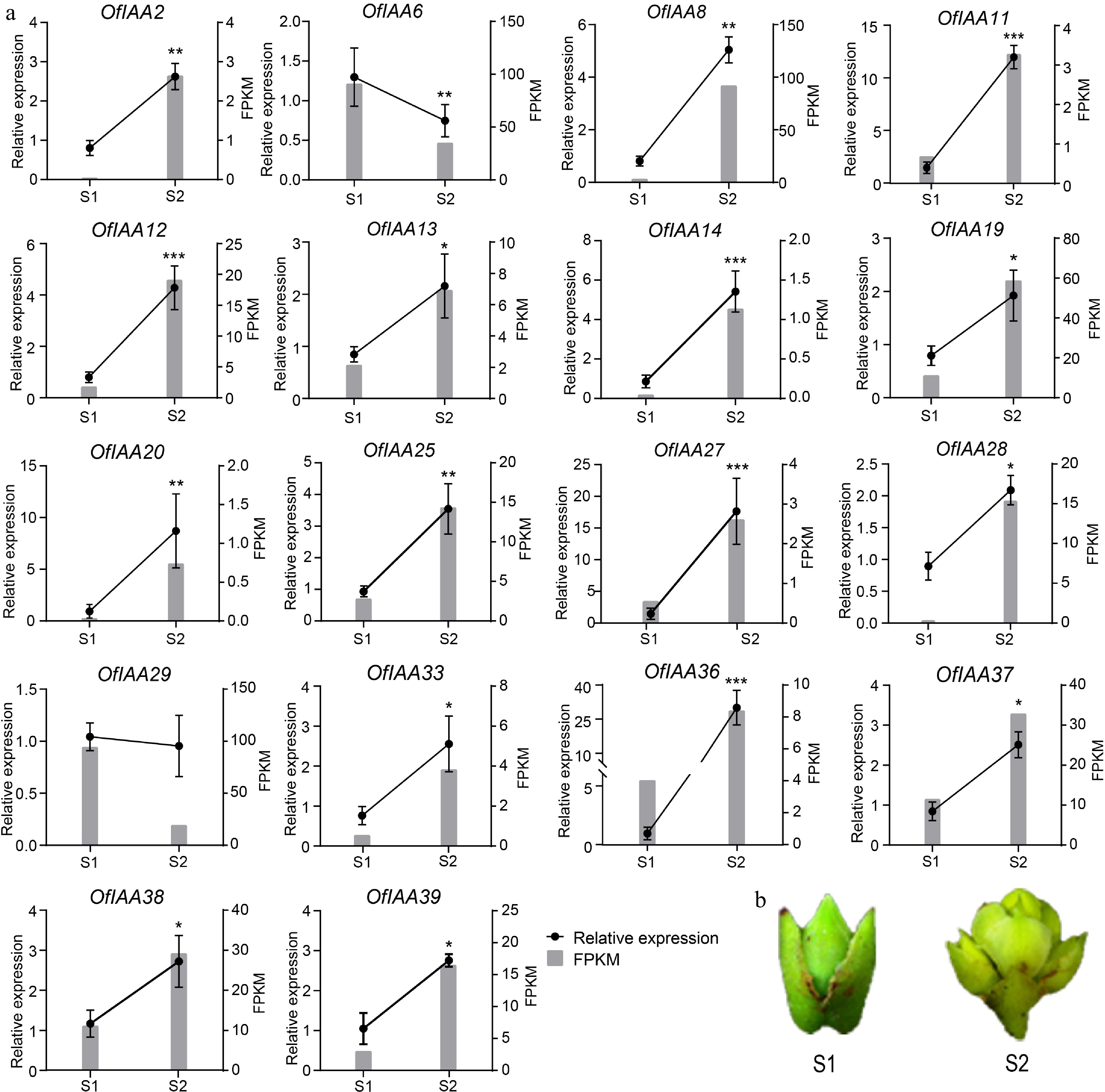
Figure 8.
Expression analysis of the differently expressed OfIAA genes during flower opening processes in O. fragrans. (a) FPKM and qRT‒PCR analysis of the differentially expressed OfIAA genes at stage S1 and S2. Error bars represent the standard error of three replicates. (b) Phenotypes of flower buds at S1 and S2 stage. Significance was assessed by Duncan's multiple-range test (DMRT) at p < 0.001 (***), p < 0.01 (**) and p < 0.05 (*).
Analysis of OfIAAs expression under different abiotic stresses
-
Salinity and drought stress have a profound impact on the growth, development, and natural distribution of O. fragrans. To investigate the potential role of OfIAA genes in the responding to salt and drought stress, we analyzed their expression patterns under these conditions. The transcriptomic results revealed that the expression of the majority of OfIAA genes were significantly altered in response to salt and drought stress. However, the expression of OfIAA9, 10, 20, 24, and 27 was nearly absent after treatments (Fig. 9a). Based on the DEG thresholds, three DEGs (OfIAA18, 22, and 23) showed downregulation after salt stress, while 11 DEGs were identified under drought stress, including ten downregulated genes (OfIAA13, 15, 18, 21, 22, 23, 28, 29, 31, and 36) and one upregulated gene (OfIAA5) (Table 3). The spatiotemporal expression of all the DEGs was further verified during 24-h after salt and drought treatments, respectively. In particular, OfIAA18, 22, and 23 showed a decreasing trend within 12 h after salt treatment, but their expression levels increased after 12 h (Fig. 9a). In the context of the drought treatment, OfIAA5 and 28 exhibited a decreasing trend within 12 h, followed by an increase at 24 h (Fig. 9c). Furthermore, OfIAA15, 18, 21, 22, 23, 29, 31, and 36 exhibited a general decline over the 24-h period following the drought treatment (Fig. 9b). OfIAA13 showed an increasing trend from 0 to 12 h, but was downregulated at 24 h after the drought treatment (Fig. 9c). These results indicate that the differentially expressed OfIAA genes are involved in the response to salt and drought stress in O. fragrans.
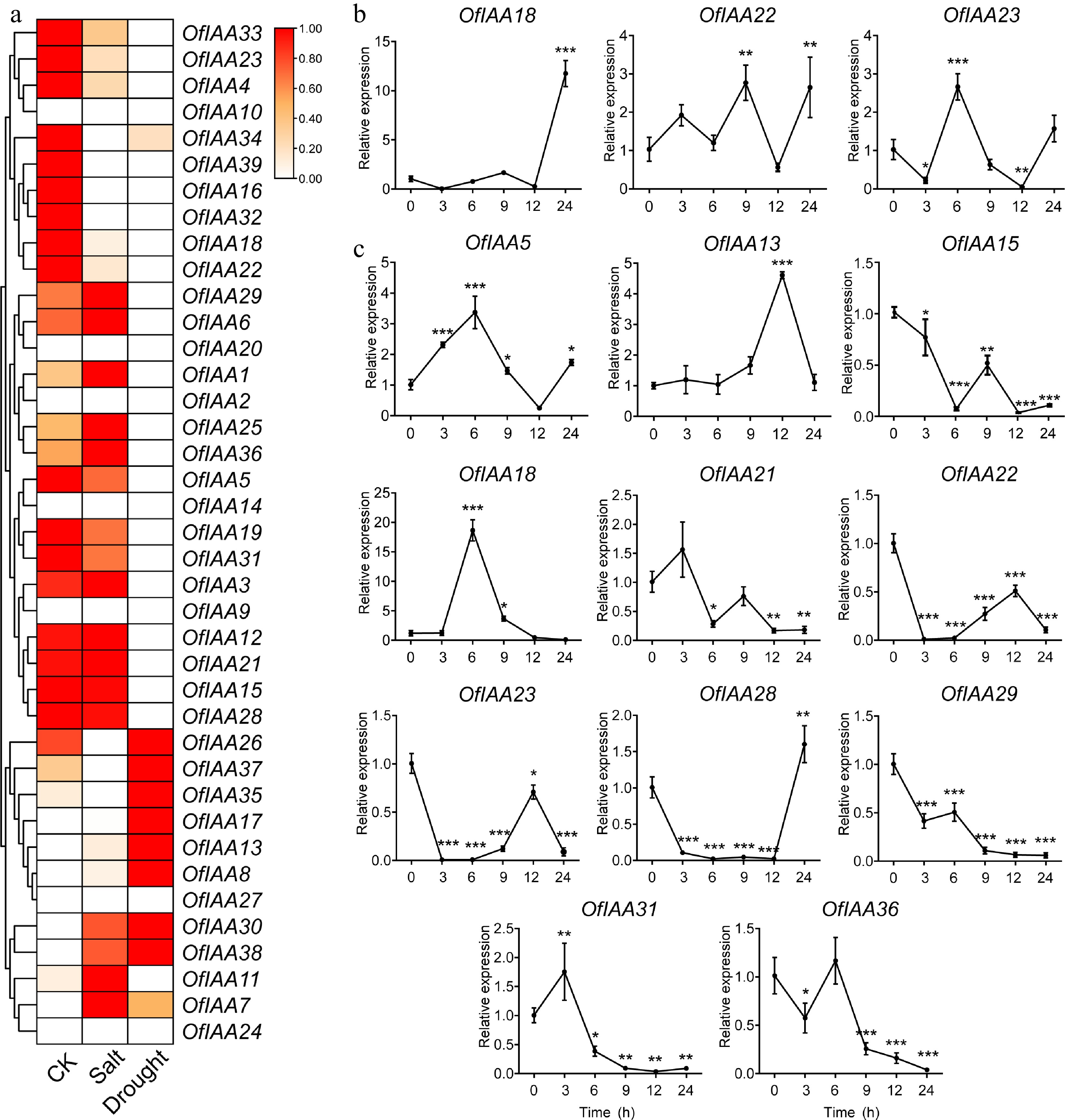
Figure 9.
Expression profiles of the differentially expressed OfIAA genes after (a) salt and (b) drought treatments by qRT‒PCR in O. fragrans. Error bars represent the standard error for three replicates. Significance was assessed by Duncan's multiple-range test (DMRT) at p < 0.001 (***), p < 0.01 (**) and p < 0.05 (*).
Table 3. Identification of DEGs under salt and drought treatments in O. fragrans.
Stress Gene Expression FoldChange Padj value CK Treatment Salt OfIAA18 1.24 0.28 −2.18 2.2 × 10−3 OfIAA22 4.047 0.67 −2.59 3.6 × 10−5 OfIAA23 6.47 1.92 −1.75 1.5 × 10−4 Drought OfIAA5 35.73 12.14 −1.54 1.6 × 10−17 OfIAA13 0.38 6.59 4.15 8.1 × 10−12 OfIAA15 2.87 0.41 −2.80 2.3 × 10−4 OfIAA18 1.24 0.23 −2.41 6.4 × 10−3 OfIAA21 79.82 29.78 −1.41 3.3 × 10−26 OfIAA22 4.05 0.20 −4.30 4.1 × 10−9 OfIAA23 6.47 1.06 −2.59 2.9 × 10−7 OfIAA28 2.07 0.71 −1.53 3.7 × 10−2 OfIAA29 13.73 6.75 −1.01 4.9 × 10−5 OfIAA31 60.42 15.61 −1.94 1.9 × 10−26 OfIAA36 52.94 16.89 −1.63 3.1 × 10−17 -
Auxin, an essential plant hormone, plays a crucial role in plant growth, development, and physiological processes. In the context of auxin signaling, the Auxin/IAA gene serves as a key regulator of downstream responses. Identification of the Aux/IAA gene family provides a valuable foundation for further research into auxin signaling and regulatory mechanisms. With the advent of molecular biology techniques and genome sequencing, comprehensive analyses of the Auxin/IAA gene family have been conducted in various species, such as Arabidopsis[8], rice[25], maize (Zea mays)[26], and Populus[9]. However, limited information is available on the Aux/fIAA gene family in O. fragrans. Therefore, it is imperative to conduct a comprehensive investigation of this gene family in O. fragrans to gain a better understanding of its potential functions in the flower opening processes and responses to abiotic stress.
In this study, a genome-wide identification and comprehensive analysis of the OfIAA gene family in O. fragrans was performed. A total of 39 OfIAA genes were identified in O. fragrans, which is more than the number found in Arabidopsis (29) and Populus (35) (Supplemental Table S1). The OfIAA gene family exhibited significant variation in protein length, MW, and pI consistent with findings in Arabidopsis and rice[8,25] (Supplemental Table S1). Upon analysis of the chromosomal distribution, we observed a non-uniform distribution of the OfIAA genes across the 19 chromosomes (Fig. 1). Notably, OfIAAs located on the same chromosome had similar gene structures and gene motifs. A typical Aux/IAA protein consists of four conserved domains[27]. The present analysis revealed that 35 OfAux/IAA proteins had all four domains, while the remaining proteins lacked one domain (Fig. 2). For instance, OfIAA32 lacked domain I, indicating that it cannot repress via interaction with the TPL (TOPLESS) corepressor, thereby losing its repressor function in auxin signaling[28]. Similarly, the absence of domain II in OfIAA18 and 26 result in reduced protein stability and an inability to participate in the TIR1 (transport inhibitor response 1) degradation pathway, making them resistant to degradation at elevated auxin levels[29]. Moreover, the absence of domain IV in OfIAA12 indicates that it cannot form homo- and heterodimers with ARF proteins[30]. Gene duplications, particularly segmental duplications, play a key role in the expansion of gene families[31]. A total of 34 pairs of segmental duplications were found among the OfIAA genes, with no tandem duplications observed (Fig. 3, Table 1). It was hypothesized that segmental duplications may have contributed to the expansion of the Aux/IAA gene family in O. fragrans. Phylogenetic analysis classified the 39 OfIAA proteins into two main groups, consistent with Arabidopsis and rice[32]. Notably, OfIAA genes within the same clade displayed conserved intron-exon structures (Fig. 5), suggesting potential functional similarities. Five types of motifs in the OfAux/IAA gene family were also identified (Fig. 5), which is consistent with Hordeum vulgare[33] and pepper[34]. In addition, the promoter regions of the OfIAA genes contained numerous hormone-responsive and stress-responsive elements (Fig. 6). These elements are involved in regulating the expression of the OfIAA genes in response to hormone signals and stress conditions in O. fragrans.
The analysis of transcriptomic expression of the OfIAA genes in different tissues revealed that the OfIAA genes are widely expressed in stem tissue. Moreover, certain OfIAA genes show unique preferential expression patterns (Fig. 7). Among the OfIAA genes, 16 OfIAA genes (OfIAA2, 6, 7, 14, 16, 17, 20, 21, 24, 25, 28, 30, 32, 33, 34, and 35) showed the highest expression in different tissues of O. fragrans (Fig. 7), indicating the different functions of the OfIAA gene family in these tissues. Aux/IAA gene play important roles in regulating plant growth and development, particularly in the flowering processes. For example, in rose (Rosa hybrida), repressing the specific ethylene-repressed RhIAA14 gene causes restricted cell expansion, leading to reduced flower size and limited petal expansion[18]. Similarly, petal abscission is delayed when RhIAA4-1 is silenced[35], while downregulation of RhIAA16 promotes petal abscission[19]. In O. fragrans, the functions of OfIAAs in flower opening processes remain unexplored. The present study identified 18 differentially expressed OfIAA genes (16 upregulated and two downregulated) from the S1 stage to the S2 stage during flower opening (Fig. 8). These DEGs suggest a possible involvement in the regulation of flower opening in O. fragrans. Furthermore, the Aux/IAA gene family has been proven to be involved in the abiotic stress response and improved abiotic stress tolerance. Overexpression of TaIAA15-1A in bread wheat (Triticum aestivum) improves drought tolerance by regulating the expression of genes involved in auxin, ABA, phenolamide, and antioxidant signaling pathways[36]. In grapevine (Vitis vinifera), VvIAA18 significantly enhances salt tolerance by upregulating downstream genes, as demonstrated in transgenic tobacco plants and Escherichia[37]. Three DEGs (OfIAA18, 22, and 23) were identified after salt treatment (Fig. 9), indicating that salt stress inhibits auxin signaling. Drought treatment significantly induced 11 DEGs, suggesting their potential role in regulating drought tolerance in O. fragrans. The responses of OfIAA genes to both salt and drought suggest their involvement in abiotic stress adaptation in O. fragrans. These results highlight the multiple roles of OfIAA genes in regulating flower opening and abiotic stress response in O. fragrans.
-
A comprehensive genome-wide analysis was conducted to identify 39 OfIAA genes in O. fragrans. The analysis of gene characterization revealed the evolutionary conservation of the OfIAA gene family. Additionally, synteny analysis suggested that segmental duplication had a significant role in the evolution of the OfIAA gene family. Through transcriptomic and qRT‒PCR analyses, it was found that almost all the OfIAAs were expressed in the stem tissue. Furthermore, 18 DEGs were identified, which contributed to the regulation of the flower opening in O. fragrans. Moreover, three and 11 DEGs were obtained after salt and drought treatments, respectively. The expression patterns of differentially expressed OfIAA genes suggested their essential role in responding to abiotic stress tolerance.
-
The potted material of O. fragrans 'Yanhonggui' is maintained in the Osmanthus Germplasm Resource Garden of Zhejiang Agriculture and Forestry University in Hangzhou, China. All the materials were kept in a greenhouse under natural conditions. To investigate the tissue-specific expression patterns, samples were collected from different plant tissues, including the root, annual stem, perennial stem, young leaf, mature leaf, and flower. During flower opening, flower buds were collected at two different developmental stages. The S1 stage involved the buds in the globular form with the inner bracts visibly covering the inflorescence. The S2 stage involved the buds when the inflorescence had burst through the bracts, and the florets were closely crowded. According to Dong et al.[38], uniform branch cuttings of O. fragrans 'Yanhonggui', each 20 cm long, were used for salt and drought treatment. All the cuttings were subjected to 200 mmol/L D-mannitol and 200 mmol/L NaCl, respectively, in a growth chamber under controlled conditions (temperature of 23 °C, 14/10-h light/dark cycle, and relative humidity of 60%). Three biological replicates were performed for each treatment. The third or fourth fully expanded leaf from the top was collected from each treatment at 0, 3, 6, 9, 12, and 24 h. All samples were immediately frozen in liquid nitrogen and stored at −80°C.
Genome-wide identification of the AUX/IAA gene family
-
The nucleotide and protein sequences of O. fragrans were downloaded from the O. fragrans genome database in NCBI (
www.ncbi.nlm.nih.gov/genome/?term=Osmanthus+fragrans ). The Hidden Markov Model (HMM) for the Aux/IAA gene family (PF02309) was downloaded from the pfam database (www.ebi.ac.uk/interpro ) and utilized with the hmmbuild tool (www.hmmer.org ) to identify potential Aux/IAA genes in the genome of O. fragrans. The amino acid sequences of OfIAA proteins were analyzed using the online tool Expasy (www.expasy.org ) to calculate the length of amino acid, molecular weight (MW), and isoelectric point (pI). Additionally, the Protein Parameter Calculator in TBtools (https://github.com/CJ-Chen/TBtools ) was employed to determine the instability index, aliphatic index, and grand average of hydropathicity for the OfIAA proteins. The subcellular localization prediction of OfIAA proteins was performed using WoLF PSORT (https://wolfpsort.hgc.jp/ ).Phylogenetic tree construction, gene structure, and motif analysis
-
The AUX/IAA protein sequences of Arabidopsis thaliana and rice (Oryza sativa) were obtained from the NCBI (National Center for Biotechnology Information) database (Supplemental Tables S2 & S3). Subsequently, the Aux/IAA protein sequences of O. fragrans, Arabidopsis, and rice were aligned using the MEGA11 software[39], and a phylogenetic tree was generated by the neighbor-joining method with a bootstrap value of 1,000. In addition, the exon-intron distribution of the OfAux/IAA genes were analyzed using Gene Structure Display Sever 2.0 (
http://gsds.cbi.pku.edu.cn/ ). The motifs of the OfAux/IAA proteins were predicted by the Multiple Expectation Maximization for Motif Elicitation (MEME) online program (https://meme-suite.org/meme/tools/meme ).Promoter region analysis of OfAux/IAA genes
-
To investigate cis-regulatory elements (CREs) in promoter sequences of OfAux/IAA genes, 2,000 bp genomic regions upstream of the translational start codons were analyzed using the Plantcare program (
http://bioinformatics.psb.ugent.be/webtools/plantcare/html/ ). The putative CREs in the promoter sequences of each OfAux/IAA gene were visualized in TBtools software[40].Chromosomal location, sequence, and synteny analysis
-
The chromosome information of OfAux/IAA genes was analyzed by TBtools software[40], including chromosome length, numbers, and the start and end sites of genes. The domains of the predicted OfAux/IAA proteins were visualized using DNAMAN version 7 software. The pattern of gene duplication of OfAux/IAAs was assessed by MCScanX v1.0 software. The DnaSP v5.0 soft was used to analyze the synonymous (Ks) and nonsynonymous (Ka) substitution ratios of the gene pairs. The divergence time was calculated using the formula T = Ks/2r, where Ks represents the synonymous substitutions per site and r represents the rate of divergence of nuclear genes in plants. For dicotyledonous plants, the rate of divergence (r) was considered to be 1.5 × 10−8 synonymous substitutions per site per year. Synteny analyses of OfAux/IAA genes, OfAux/IAAs and AtAux/IAAs, and OfAux/IAAs and OsAux/IAAs were conducted using the Quick MCScanX Wrapper program in TBtools.
RNA isolation and RNA-sequencing
-
A total of 0.5 g of material was used for total RNA extraction using a FastPure Universal Plant Total RNA Isolation Kit (Vazyme, Nangjing, China) according to the manufacturer's instructions. For library construction and RNA sequencing, 3.0 μg of RNA from each sample was utilized. The RNA sequencing method employed was previously described by Ye et al.[37]. Three biological replicates were performed for RNA sequencing. Fragments Per Kilobase of exon model per Million mapped fragments (FPKM) was employed to calculate the expression, the differentially expressed genes (DEGs) were determined by the threshold of |log2(ratio treatment/CK)| ≥ 1, along with Padj: (p.adjust) < 0.001. The RNA sequencing data were accessed at the SRA (Sequence Read Archive) database (
www.ncbi.nlm.nih.gov/sra ) under Bioproject number PRJNA961323. TBtools software was used to generate the heatmap to visualize the expression of OfIAA genes.cDNA synthesis and quantitative real-time PCR (qRT‒PCR)
-
cDNA synthesis was carried out using ToloScript All-in-one RT EasyMix for qPCR (TOLOBIO, Shanghai, China) according to the manufacturer's recommendations. qRT‒PCR analysis was performed using the LightCycler480II System (Roche, Basel, Switzerland) as described by Yang et al.[41]. Three biological repetitions were performed. The relative expression level of genes was calculated by the 2−ΔΔCᴛ method. The sequence-specific primers are listed in Supplemental Table S4.
Statistical analysis
-
Data treatment and analysis were performed by Excel 2022 (Microsoft, Redmond, USA). IBM SPSS 20 software (IBM, Armonk, USA) was utilized to determine significant differences. Significance was assessed by Duncan's multiple-range test (DMRT) at p < 0.001 (***), p < 0.01 (**) and p < 0.05 (*).
-
The authors confirm contribution to the paper as follows: study design and supervision: Zhao H, Dong B; participating in the entire thesis writing and data analysis: Cao S, Dong B; assisting in the bioinformatics analysis of the gene family: Ye Y, Wang Y; participated in the experiment process: Zheng Z, Zhong S, Xiao Z; providing guidance and manuscript revision: Fang Q, Deng J. All authors reviewed the results and approved the final version of the manuscript.
-
The plant materials are preserved in the Osmanthus Germplasm Resource Garden of Zhejiang Agriculture and Forestry University (Hangzhou, China). The raw reads files have been accessed on NCBI Sequence Read Archive (SRA) under the BioProject number of PRJNA961323.
This work was funded by the National Natural Science Foundation of China (Grant No. 31902057 and 32401643), the Key research and development program of Zhejiang Province (Grant No. 2021C02071), and the Zhejiang Provincial Natural Science Foundation of China (Grant No. LQ19C160012).
-
The authors declare that they have no conflict of interest.
-
# Authors contributed equally: Shanshan Cao, Yong Ye
- Supplemental Table S1 The characteristics of OfIAA proteins in Osmanthus fragrans.
- Supplemental Table S2 Information on IAA genes in Arabidopsis thaliana and Oryza sativa.
- Supplemental Table S3 The amino acid sequences of AtIAA, OsIAA, and OfIAA proteins.
- Supplemental Table S4 The sequence of primers.
- Copyright: © 2024 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Cao S, Ye Y, Zheng Z, Zhong S, Wang Y, et al. 2024. Aux/IAA gene family identification and analysis reveals roles in flower opening and abiotic stress response in Osmanthus fragrans. Ornamental Plant Research 4: e027 doi: 10.48130/opr-0024-0025 

Aux/IAA gene family identification and analysis reveals roles in flower opening and abiotic stress response in Osmanthus fragrans
- Received: 30 June 2024
- Revised: 13 August 2024
- Accepted: 19 August 2024
- Published online: 08 October 2024
Abstract: The Aux/IAA (auxin/indole-3-acetic acid) gene family plays a crucial role in regulating various aspects of plant growth, development, and abiotic tolerance in the auxin transduction pathway. However, limited information is available about the Aux/IAA family in Osmanthus fragrans. This study aims to comprehensively analyze the Aux/IAA gene family on a genome-wide scale. A total of 39 OfIAA genes containing four conserved domains were identified. These genes were unevenly distributed across 19 chromosomes and grouped into six clades based on phylogenetic analysis, showing conserved gene structure and motif composition. The expansion of OfIAA genes in the O. fragrans genome was partially due to segmental duplication events. Analysis of cis-regulatory elements (CREs) in the promoters of the OfIAA genes revealed the presence of many CREs related to different hormones and abiotic stresses. Through transcriptome and expression pattern analysis, we found that the majority of OfIAA genes were expressed in the stem tissue. Moreover, during the flower opening process, 18 OfIAA genes exhibited differential expression, while three and 11 OfIAA genes, respectively, showed altered expression patterns after salt and drought treatments. These differentially expressed genes are likely involved in the regulation of flower opening and abiotic stress response. This study provides new insights into the potential roles of OfIAAs and contributes to a better understanding of the regulatory mechanisms of flower opening and abiotic stress tolerance in O. fragrans.
-
Key words:
- OfIAA /
- Expression pattern /
- Flower opening /
- Abiotic stress /
- Osmanthus fragrans