










-
Lignocellulose is a complex biopolymer composed primarily of cellulose, hemicellulose, and lignin, displaying significant resistance to hydrolysis[1,2]. Deriving nutritional value from lignocellulose is challenging due to the highly resistant crystalline cellulose regions and the lignin coating that encapsulates the polysaccharide network[3]. Mammals typically lack the enzymes to efficiently break down complex lignocellulosic biomass, relying heavily on microbes residing in the digestive tract to perform this function[4]. Especially in ruminants, the symbiotic microbiota process the efficient conversion of lignocellulose into high-nutritious foods. For decades, studies have shown that lignocellulolytic degradation is primarily conducted by the rumen microbial community sequentially and synergistically, and the rumen microbiota has thus been employed as a model system to discover the enzyme repertoires for lignocellulose depolymerization[5,6]. Despite the extensive research on lignocellulose degradation in the rumen, the specific diversity of the rumen microbiota responsible for each stage of the degradation cascades of various lignocellulosic components remains to be elucidated. Understanding the microbial diversity and substrate specificity is crucial for developing targeted strategies to enhance the efficiency of lignocellulose degradation and harness its full potential as a sustainable resource.
Microorganisms employ diverse enzymatic strategies for lignocellulose degradation. One strategy involves cellulosomes, multienzyme assemblies efficiently degrading lignocellulose[7,8]. These assemblies consist of cell surface-anchored scaffolding proteins with cohesion and dockerin domains, binding multiple carbohydrate-active enzymes (CAZymes, such as glycoside hydrolases)[9]. Another strategy, encoded by specific polysaccharide utilization loci (PULs), exhibits somewhat 'selfish' behavior by transporting depolymerized products of complex polysaccharides into microbial cells, limiting sugar release into the environment and access for other scavenging populations[10,11]. Additionally, non-catalytic module carbohydrate-binding modules (CBMs) with various substrate-binding capabilities play an important role in lignocellulose degradation[12,13]. Despite the identification of numerous lignocellulose-degrading enzymes at the molecular level, the enzymatic strategies employed by ruminal microbiota for the degradation cascades of lignocellulosic components remain limited.
The lignocellulose degradation process involves fiber colonization, amorphous region degradation, specific bacterial population increase, and crystalline region degradation[14]. Different microorganisms sequentially dominate these phases, driven by niche partitioning and microbial interactions[14]. Previous studies have reported that the physicochemical properties of feed can be the primary factors that determine microbial colonization and digestion in the rumen[8,15,16]. In particular, the diversity and composition of microbiota in the rumen differ between forage-fed and grain-fed animals[4], implying that their microbial colonization and digestion in lignocellulosic biomass is different. Despite the recognized influence of high-grain diets on altering microbial communities and reducing lignocellulose degradation, it remains unclear which phase of degradation cascades is mainly affected and how this reduction occurs.
Here, 244 rumen metagenome samples were analyzed from Holstein cows, constructing 5034 microbial metagenome-assembled genomes (MAGs). These MAGs underwent functional comparison, serving as a base for subsequent experiments, including in vivo high-grain diet interventions and in sacco rumen incubation. This comprehensive approach provided insights into lignocellulose degradation from both spatial and temporal perspectives. The integrated datasets allowed (1) identification of the diversity of lignocellulolytic microbiomes (LMs) and their diverse enzymatic strategies for specific lignocellulosic components during the degradation cascades, (2) elucidate the primary lignocellulosic components affected by high-grain diets and identify the key microbial players involved, and (3) clarify which stage of the lignocellulolytic cascades is predominantly influenced by high-grain diets and identify the primary microbial contributors to these effects. Based on a vast array of uncultured microbial genomes, the findings provide a more in-depth understanding of the lignocellulose degradation cascades of rumen microbiota, particularly in relation to diet, offering insights for promoting the efficient conversion of low-quality lignocellulosic biomass into highly nutritious milk in dairy cattle in the future.
-
Twelve lactating Holstein cows with rumen fistulas, weighing 651 ± 54 kg, were housed in tie stalls for a one-month experiment[4]. Before the animal trial, all cows were fed a diet with a forage-to-grain ratio of 6:4 for one week. After this preparation period, six cows were fed on a high-forage diet with a forage-to-grain ratio of 6:4 on a dry matter (DM) basis (CON group), whereas the six other cows were fed a high-grain diet with a forage-to-grain ratio of 4:6 on a DM basis (HG group; Supplementary Table S7). The feeding trial lasted for 21 d, and the animals were fed ad libitum twice a day (07:00 and 19:00). The feed and fecal samples were collected at 07:00 and 19:00 for three consecutive days before the cows were slaughtered. The feed and fecal samples were processed for chemical composition analysis, including dry matter (DM), neutral detergent fiber (NDF), and acid detergent fiber (ADF)[17]. To assess the apparent digestibility of DM, NDF, and ADF, acid insoluble ash (AIA) was used as an internal marker[18]. After the experiment, all cows were slaughtered to collect rumen contents and stored at −80 °C. The rumen contents were further used for DNA extraction.
Ruminal in situ incubations and sample collection
-
Six lactating rumen-fistulated Holstein cows (CON group: n = 3; HG group: n = 3) were selected for in sacco rumen incubation. The Leymus chinensis materials were dried and cut into 2.5 mm pieces. The pieces were weighed and 4 g of them were placed in each heat-sealed nylon bag (bag size: 8 cm × 12 cm; pore size: 300 μm). A total of 96 heat-sealed bags, 16 per cows, were simultaneously placed into each rumen before morning feeding. After 0.5, 2, 4, 6, 8, 12, 24, and 36 h of incubation, two nylon bags were retrieved at each time point from each cow's rumen, washed three times with distilled water to eliminate liquid-borne and loosely attached microbes, and then squeezed to remove excess water. The incubated Leymus chinensis samples in nylon bags were transferred to the laboratory in liquid nitrogen. One replicate was used for subsequent DNA extraction, while the other was subjected to chemical composition analysis, including DM, NDF, and ADF[17]. The Leymus chinensis samples were also stored for chemical composition analysis.
DNA extraction and metagenomic sequencing
-
The DNA from the incubated Leymus chinensis samples (including 0.5, 2, 4, 6, 8, 12, 24, and 36 h of rumen incubation) was extracted using a microbead stirrer (BioSpec Products, Inc., Bartlesville, OK, USA)[19] and the E.Z.N.A.® Stoo1 DNA Kit (Omega Bio-tek, Norcross, GA, USA) according to the manufacturer's protocols. The quality and quantity of the extracted DNA were determined using the Nanodrop ND-1000 (Thermo Scientific, Wilmington, USA), and the integrity of the DNA was evaluated through electrophoresis on 0.8% agarose gels. The high-quality DNA samples were then stored at −80 °C until further processing.
16S rRNA gene sequencing
-
The extracted high-quality DNA was further used for 16S rRNA gene sequencing. The V3 and V4 regions of the gene were amplified using universal primers (341F: 5′-CCTAYGGGRBGCASCAG-3′, 806R: 5′-GGACTACNNGGGTATCTAAT-3′), with a 6 bp barcode unique to each sequence. The Illumina MiSeq platform was used for sequencing, and the barcodes and sequencing primers were removed for data processing. After removing low-quality reads, the remained paired-end reads were merged using FLASH[20] (v.1.2.7). The sequences were further screened to remove chimeras and dereplication by the procedure of 'removeBimeraDenovo', and ASV feature tables were constructed using the DADA2[21] (v.1.18) plug-in in QIIME 2[22] (v.2021.08). The ASVs were taxonomically assigned against the SILVA v.138 database[23] using the naive Bayes classifier[24].
Fiber-adherent rumen metagenomic sequencing
-
The microbial DNA extracted from 18 ruminal incubated Leymus chinensis samples (0.5, 8, and 36 h) was used for metagenomic sequencing. For each sample, the high-quality DNA samples were utilized to generate a metagenomic library with an insert size of 350 bp following the manufacturer's instructions for the TruSeq DNA PCR-Free Library Preparation Kit (Illumina, San Diego, CA, USA). The resulting library was sequenced on the Illumina NovaSeq platform to obtain the sequence data.
Metagenome assembly and binning
-
The Illumina data from 18 fiber-adherent rumen metagenome samples and 226 published ruminal metagenome samples from dairy cattle was processed[4−6,25−33]. First, quality control was performed on the data using Fastp[34] (v.0.20.1) to trim adapters, then the host (Bos taurus, GCA_002263795.2), food was removed, and human sequences (Homo sapiens, GCA_000001405.28) by using BWA-MEM[35] (v.0.7.17) according to a previous study[4]. The reference genome sets of plants in feed included wheat (Triticum aestivum, GCA_002220415.3), medicago (Medicago truncatula, GCA_000219495.2), rice (Oryza sativa, GCF_000005425.2), maize (Zea mays, GCA_003185045.1 and GCA_000005005.6), and soybean (Glycine max, GCA_000004515.4). MEGAHIT[36] (v.1.2.9) was applied to assemble the high-quality reads from each sample (parameter: --min-contig-len 500 --presets meta-large). The remaining high-quality contigs were binned into genomes by three different approaches, including MaxBin[37] (v.2.2.4), MetaBAT2[35] (v.2.11.1), and CONCOCT[38] (v.0.4.0) with default parameters. The obtained genomes were integrated using the bin refinement module of metaWRAP[39] (v.1.3). Prokaryotic metagenome-assembled genomes (MAGs) were evaluated for completeness and contamination using CheckM[40] (v.1.0.7). Among them, 5,034 rumen microbial MAGs exhibited completeness over 50% and contamination below 10%. The non-redundant 3,808 MAGs were remained, with a dereplication threshold of 99% average nucleotide identity by using dRep[41] (v3.4.0). After filtering for completeness > 80% and contamination < 10%, 1374 MAGs were obtained to predict ORFs by Prokka[42] (v.1.14.6). The estimated genome size of 5,034 MAGs was corrected based on completeness and contamination using the algorithm from Nayfach et al.[43].
Taxonomic and functional annotation
-
All 1374 high-quality genomes were subjected to annotation using GTDB-Tk[44] (v.0.1.6) based on the Genome Taxonomy Database. Subsequently, a maximum-likelihood phylogenomic tree was constructed using PhyloPhlAn[45] (v.1.0) and visualized using iTol[46] (v.4.3.1). The carbohydrate-active enzyme (CAZyme) profiles of each MAG were annotated using dbCAN2[47]. The assignment of dockerin domains of each MAG was predicted based on the hidden Markov model (HMM) using HMMER[48] (v.3.2.1), according to the CAZyme database[49]. Putative lignocellulolytic microbiomes (LMs) as genomes containing any of the CAZyme families capable of lignocellulose degradation were identified, including GH5, GH51, GH48, GH9, GH44, GH74, GH124, GH148, GH45, GH8, GH10, GH2, GH3, GH1, GH116, GH43, GH30, GH98, GH11, GH141, GH39, GH54, GH120, CE1, GH67, CE3, CE5, CE7, GH159, GH4, GH110, GH26, GH113, GH164, CE2, CE4, GH27, GH31, GH36, GH57, CE6, CE12, GH97, CE15, AA1, AA3, AA4, AA6, and AA7. The polysaccharide utilization loci (PUL) of all 1374 MAGs were predicted following PULpy[50] (v.1.0) pipeline. Finally, all 1353 LM-MAGs were employed as a genomic database to assign metagenomic samples from the 12 dairy cattle rumen and 18 fiber-adherent rumen by using CoverM (v.0.6.1;
https://github.com/wwood/CoverM ) (parameter: --min-read-percent-identity 0.95 --min-read-aligned-percent 0.75 --trim-min 0.10 --trim-max 0.90 -m tpm --proper-pairs-only). Subsequently, the transcripts per million (TPM) calculation process was employed to quantify the abundance levels of each genome in these samples.Statistical analysis
-
To compare the feed apparent digestibility of DM, NDF, and ADF between the CON and HG groups, a t-test model was used. For the digestibility of DM, NDF, and ADF in the incubated Leymus chinensis material, a t-test analysis was performed at each time point to compare between the CON and HG groups. To identify the differences between the two groups at the ASV level, Principal Coordinates Analysis (PCoA) based on the Bray-Curtis distance was performed and an ANOSIM test conducted with 9999 permutations using the R vegan package (v.2.6-4). The changes of ASVs between the CON and HG groups among the eight time points during rumen incubation were analyzed using the R packages indicspecies (v.1.7.12) and edgeR (v.3.36.0). Weighted Correlation Network Analysis (WGCNA, v.1.71) was employed to construct co-occurrence modules based on the ASVs with significantly changed abundance, with MEDissThres set to 0.2. Additionally, a Wilcoxon rank-sum test was performed to compare the abundance of MAGs between the CON and HG groups. Relationships between the changes and the number of genes encoding lignocellulolytic CAZymes of Prevotella-affiliated MAGs were based on Spearman.
-
To establish a potent lignocellulolytic consortium in the rumen of dairy cattle, 244 metagenome samples from Holstein cows were used to construct 5034 rumen microbial MAGs, which had completeness of over 50% and contamination below 10% (Supplementary Fig. S1a; Supplementary Tables S1 & S2). The non-redundant 3808 MAGs with a dereplication threshold of 99% average nucleotide identity were observed. Within this subset, 1374 high-quality MAGs were identified with > 80% completeness and < 10% contamination, which had a mean completeness of 89.51% (± 0.15%) and a mean contamination of 3.04% (± 0.06%) (Supplementary Fig. S1b; Supplementary Table S3). For taxonomic profiling, 100%, 99.56%, and 81.88% of MAGs were classified into microbes at the phylum, genus, and species levels, respectively (Supplementary Table S3). The genomic repertoire of the rumen microbiome encompassed 23 phyla, 86 families, and 268 genera in dairy cattle (Supplementary Table S3). The integrated microbial MAGs from dairy cattle rumen are more representative than those previously reported[4].
In the CAZyome analysis, 1353 high-quality MAGs (98.47%) were identified to be involved in the degradation process of lignocellulose, including cellulose, hemicellulose, and lignin (Fig. 1a, Supplementary Table S4). This ability enables cows to efficiently convert complex and recalcitrant plant biomass into valuable nutrients. All 1353 MAGs represented LMs consisting of 23 phyla, with the distribution including Bacteroidota (519), Firmicutes_A (510), Firmicutes (90), Spirochaetota (58), Firmicutes_C (48), Proteobacteria (42), Methanobacteriota (19), Actinobacteriota (18), Fibrobacterota (10), Elusimicrobiota (9), Cyanobacteria (8), and Verrucomicrobiota (8). These LMs contained 35,043 genes encoding lignocellulolytic CAZymes, covering 49 distinct families (Supplementary Table S4). The number of genes encoding lignocellulolytic CAZymes in LMs was strongly positively correlated with their genome size (Mantel test, R = 0.661, p < 0.001; Fig. 1b), suggesting that the lignocellulose degrading ability is closely related to other microbial functions. Certain LM-MAGs, particularly those belonging to the Verrucomicrobiota, Bacteroidota, and Fibrobacterota phyla exhibited the most extensive and diverse repertoire of CAZymes for the lignocellulose degradation (Fig. 1a, c). Members of the Fibrobacterota phylum demonstrated a more varied repertoire of CAZyme families for lignocellulose degradation compared to those in the Actinobacteriota phylum. Additionally, Firmicutes_A exhibited a less diverse repertoire of CAZymes for lignocellulose degradation compared to the Bacteroidota phylum. Furthermore, the Proteobacteria phylum had the lowest number of genes and families encoding lignocellulolytic CAZymes. These observations imply that the diversity and redundancy of lignocellulolytic CAZymes within microbial consortia may contribute to variations in the breakdown and utilization of plant fibers.
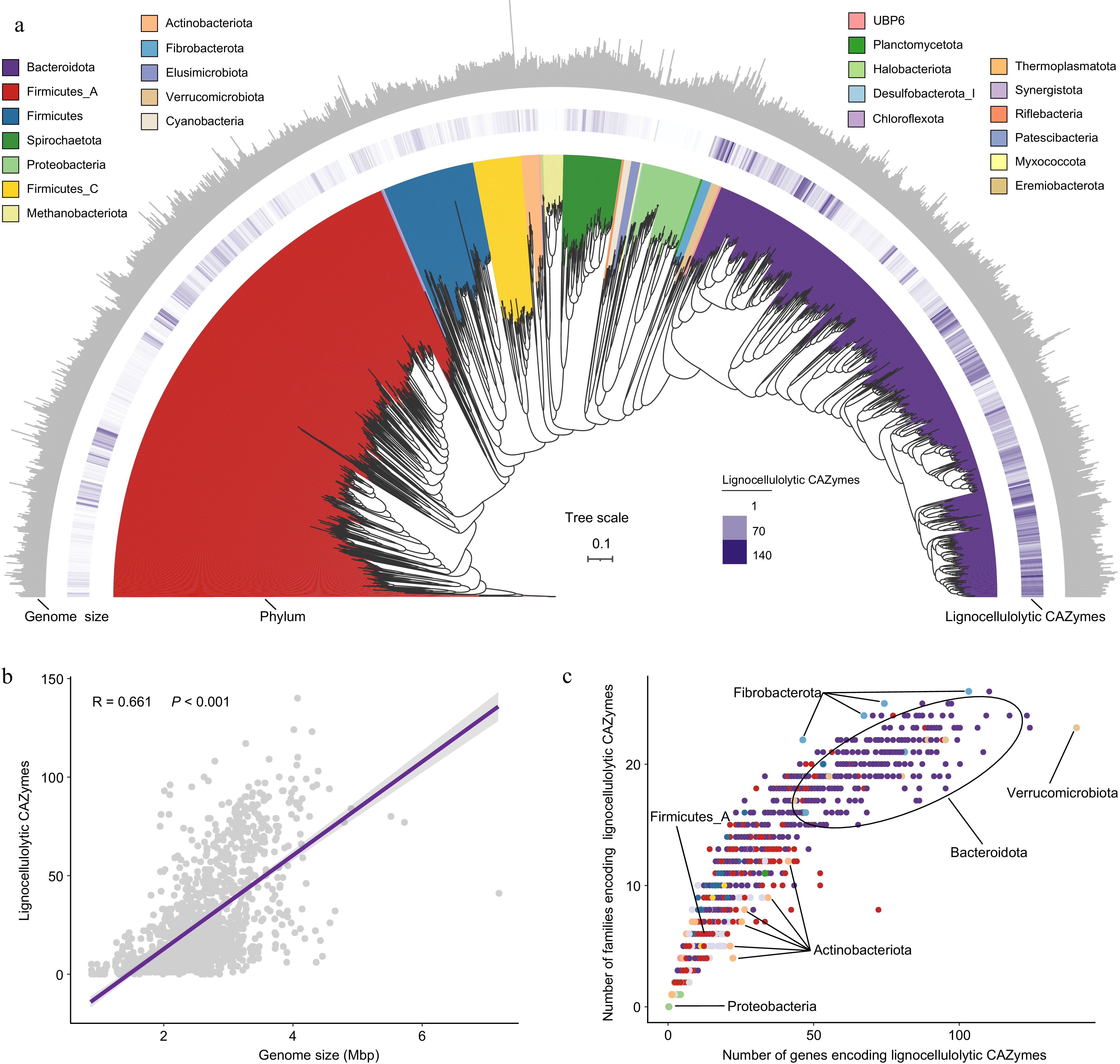
Figure 1.
The CAZyme profiles in lignocellulolytic microbiomes (LMs) from dairy cattle rumen. (a) Phylogenetic tree of 1353 microbial metagenome-assembled genomes (MAGs) coding lignocellulolytic CAZymes. The maximum-likelihood tree is constructed using PhyloPhlAn. Branches are shaded with color to highlight phylum-level affiliations. The inside layer of the heat map represents number of genes encoding lignocellulolytic CAZymes of each LM-MAG. The outside layer of the bar graph represents the genome size of each LM-MAG. (b) The correlation between number of genes encoding lignocellulolytic CAZymes and genome size of LM-MAGs. (c) Number of degradative CAZymes in distinct families in each LM-MAG. Genomes are colored by phylum.
Deciphering lignocellulolytic consortia and their enzymatic strategies involved in degradation cascades of various lignocellulosic components
-
Lignocellulose is a complex and recalcitrant structure composed primarily of cellulose, hemicellulose, and lignin, which provide plants with rigidity and durability. For cellulose, GH5 was identified as the most abundant cellulolytic enzyme in rumen LMs, particularly within the Bacteroidota phylum (Fig. 2a). Members of Fibrobacter and Ruminococcus (e.g., Ruminococcus flavefaciens) had the largest number of CAZyme genes coding endo- and exoglucanases (primarily GH5 and GH9; Fig. 2b). Therefore, these microbial populations play a significant role in cellulose degradation by primarily attacking cellulose fibrils at the amorphous regions, followed by cutting at the crystalline regions from both the reducing and non-reducing ends. Ruminococcus MAGs were found to encode GH48, enabling them to target both amorphous and crystalline cellulose (Fig. 2b). Compared with Ruminococcus spp., Fibrobacter spp. exhibited a more diverse array of CAZyme families to attack amorphous regions of cellulose by encoding endoglucanases GH45 and GH8 (Fig. 2b). In addition, the enzymes involved in lignocellulose degradation often feature a CBM attached to the catalytic domain[12]. Notably, Ruminococcus spp. (25) and Fibrobacter spp. (23) displayed the highest counts of CBM-containing enzymes, many of which had N-terminal signal peptides (Supplementary Table S5). This highlights the abundance of secreted or periplasmic multi-domain enzymes in these populations, which are highly effective in lignocellulose degradation. Ruminococcus spp., in particular, had adapted to produce complex proteins with multiple catalytic domains, often accompanied by one or two CBMs to target amorphous cellulose and hemicellulose. This was particularly evident in numerous PULs (CBM4 + GH9; Fig. 3). In contrast, certain proteins identified in Fibrobacter spp. contained three tandemly arranged CBMs and one GH domain (CBM11 + CBM11 + CBM11 + GH51) to adhere cellulosic biomass (Fig. 3). The most significant disparity between Ruminococcus and Fibrobacter was that Ruminococcus had a multitude of dockerin and cohesion domains, whereas Fibrobacter lacked such domains, implying that Ruminococcus possesses the ability to produce cellulosomes for fiber digestion[51] (Supplementary Table S3). Additionally, no genes encoding lytic polysaccharide monooxygenases (LPMOs) targeting cellulosic crystalline substrates were found in the LMs of dairy cattle rumen. For β-glucosidases, members of the Bacteroidales order were the major agents of hydrolyzing glucose dimers into glucose (e.g., GH2 and GH3; Supplementary Table S4).
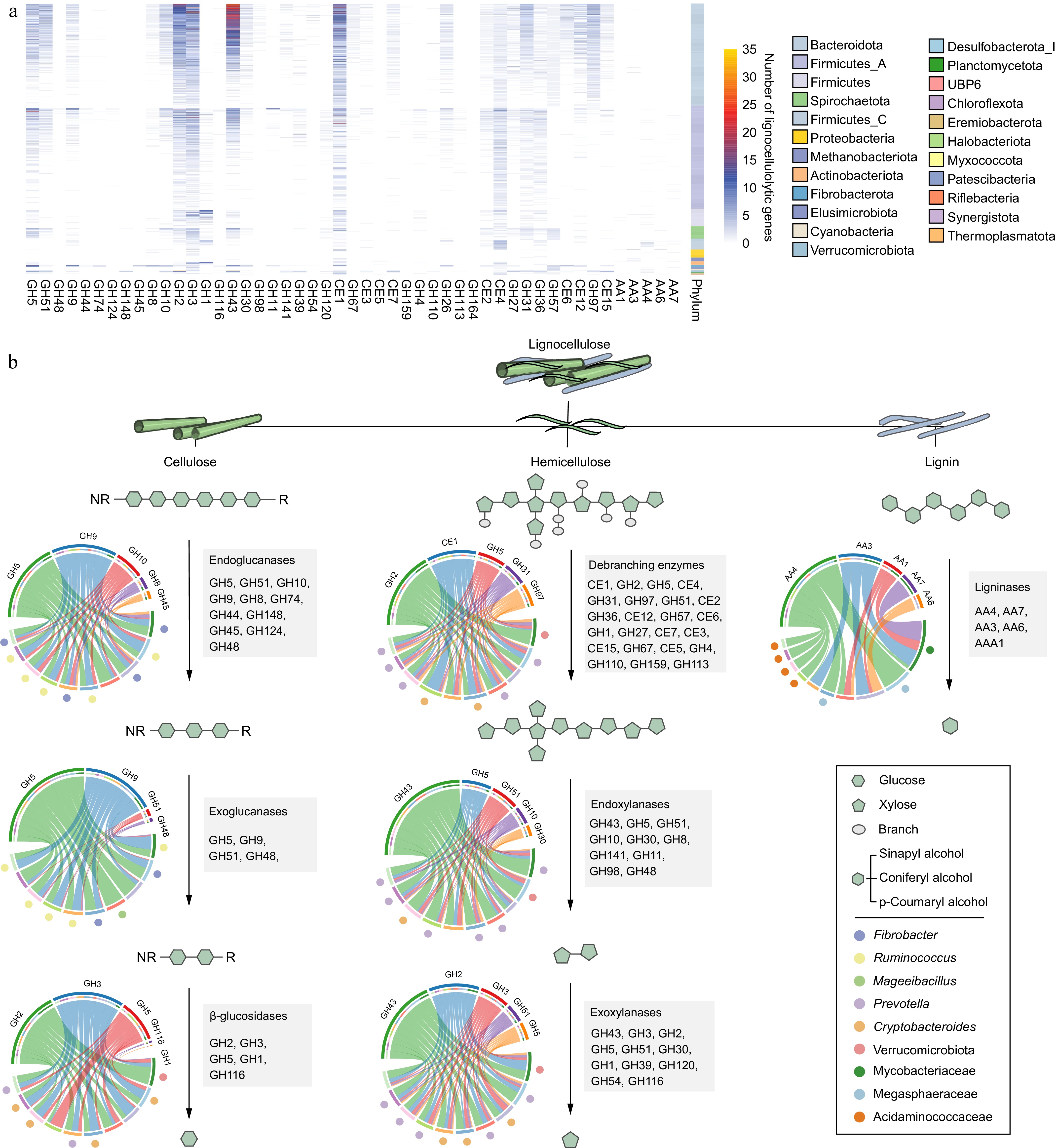
Figure 2.
The distribution and diversity of lignocellulolytic microbiomes (LMs) from dairy cattle rumen. (a) Distribution of lignocellulolytic CAZymes (GH, glycoside hydrolases; CE, carbohydrate esterases; AA, Auxiliary Activities) in LM-MAGs. Genomes are colored by phylum. (b) Cooperative model of cellulases, hemicellulases, and ligninases in lignocellulose degradation in the LM-MAGs. Chord Diagram represent the top families of lignocellulolytic CAZymes contributed by ruminal LM-MAGs. The detailed information regarding LMs involved in the degradation process of lignocellulose, including cellulose, hemicellulose, and lignin, can be found in Table S4.
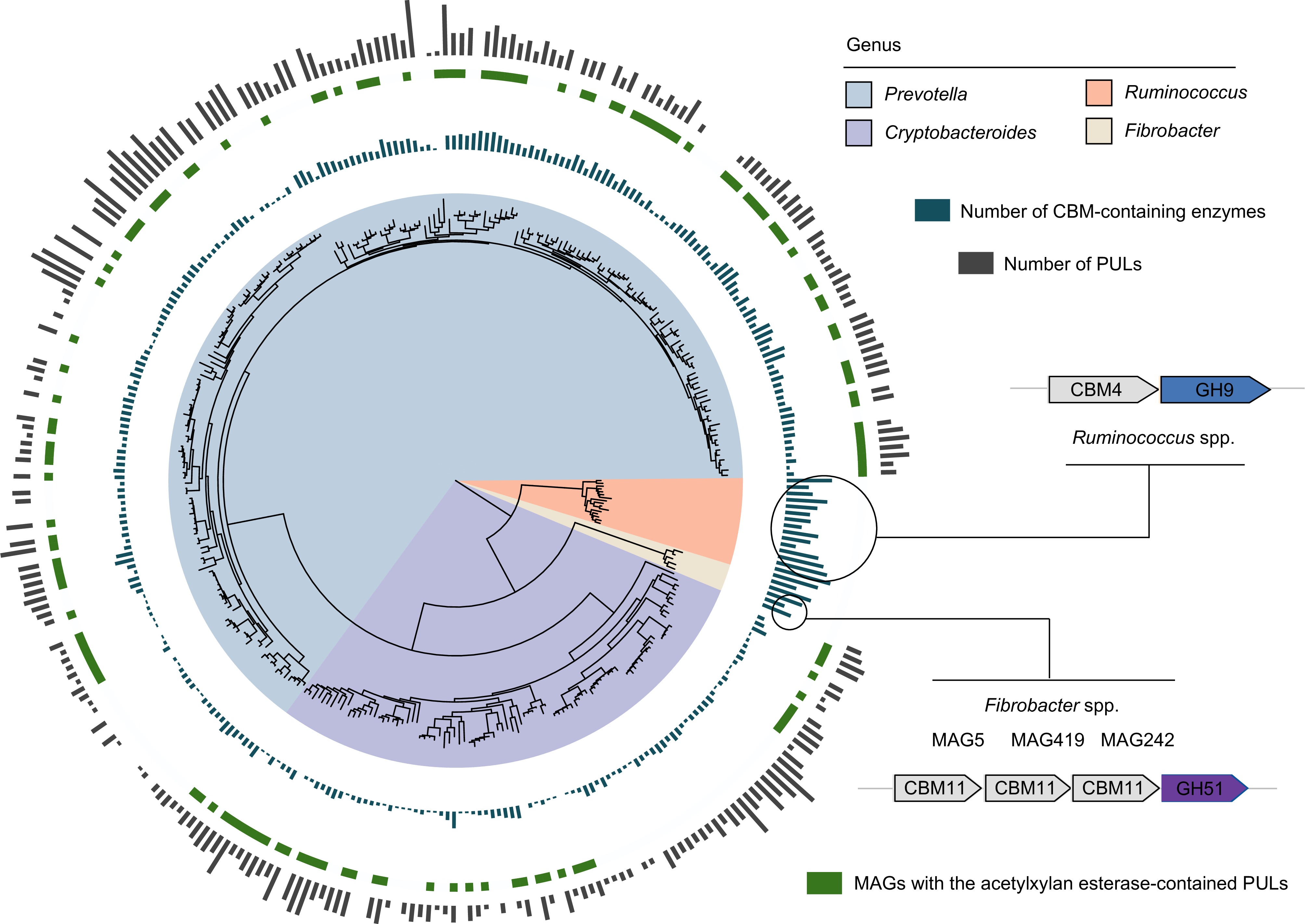
Figure 3.
Different lignocellulose degradation strategies used by taxa present in the dairy cattle rumen. Phylogenetic tree of lignocellulolytic microbiomes (LMs) belonged to genera Prevotella, Cryptobacteroides, Ruminococcus, and Fibrobacter. The maximum-likelihood tree is constructed using PhyloPhlAn. The background of branches is shaded with color to highlight these four taxa. The inside layer of the bar graph represents number of CBM-containing enzymes. The green color represents the specific MAGs with the acetylxylan esterase-contained PULs. The outside layer of the bar graph represents the number of polysaccharide utilization loci (PULs) encoded by the targeted MAGs.
For hemicellulose degradation, the predominant CAZyme family identified within LMs was GH43, with a notable presence in the Bacteroidota phylum (Fig. 2a). Within the Bacteroidota population, the distribution of lignocellulolytic CAZymes exhibited a similar pattern (Fig. 2a), suggesting a considerable degree of conservation in their enzymatic systems. Members of the Bacteroidota phylum, primarily Prevotella spp. and Cryptobacteroides spp., were found to encode enzymes such as endoxylanase, β-xylosidase, and de-branching activities (primarily GH43, GH2, GH3, and CE1; Fig. 2b). Furthermore, it was observed that Prevotella spp. and Cryptobacteroides spp. encoded a substantial number of PULs to handle the chemical and structural complexity of hemicelluloses such as xylose, arabinose, galactose, mannose, and ferulic acid (Supplementary Table S6). It is noteworthy that most of the Prevotella and Cryptobacteroides MAGs contained acetylxylan esterase-containing PULs (Fig. 3), facilitating the breakdown of the xylan backbone. These results indicate that Prevotella spp. and Cryptobacteroides spp. have developed PULs to address the challenges posed by complex and diverse hemicelluloses, which are subsequently transported into the cells for their utilization.
Despite the presence of diverse CAZyme repertoires, only a few enzymes involved in lignin breakdown were identified in the rumen microbiome of dairy cattle (Fig. 2a). Specifically, only 135 MAGs (9.82%) encoded AA1, AA3, AA4, AA6, or AA7 enzymes involved in lignin modification and degradation, primarily belonging to the classes Clostridia (47 MAGs), Negativicutes (26 MAGs), and Methanobacteria (17 MAGs; Supplementary Table S4). The AA family with the highest number of genes was vanillyl-alcohol oxidase AA4 (consisting of 89 genes) used for lignin degradation (Fig. 2a). In contrast, only eight genes coding for laccase AA1 were identified for lignin modification (Fig. 2a). These findings suggest that the rumen microbiota in dairy cattle has a limited capacity to degrade lignin from plant biomass.
Novel consortium involved in degradation cascades of various lignocellulosic components
-
Among the 1353 LM-MAGs identified, some members of the Fibrobacteraceae family, such as Hallerella spp., were isolated from pig intestines in 2020[52] and had not been previously reported in ruminants. Genome annotation against the CAZy database revealed that four Hallerella-affiliated MAGs possessed a large number of cellulases belonging to the families GH5 and GH9, but only a few of these cellulases were found to be fused with CBM domains (Supplementary Tables S4 & S5). This suggests that Hallerella spp. may have the potential to utilize cellulose as a carbohydrate substrate[52]. Additionally, Mageeibacillus spp., previously isolated from the human vagina, contained the highest number of endo- and exoglucanases belonging to the GH5 family, as well as dockerin (Fig. 2b; Supplementary Table S3). Sodaliphilus is a recently described genus, with its type species S. pleomorphus, initially isolated from pig feces[52]. It is noteworthy that it was identified that 32 MAGs could be annotated to Sodaliphilus (including S. pleomorphus) in the dairy cattle rumen, which had the largest number of dockerins and was enriched in hemicellulose and cellulose-degrading enzymes (Supplementary Table S3). However, Sodaliphilus did not contain any cohesion domains, suggesting that the numerous non-cellulosomal dockerins may have other functions[7]. Analysis of PULs revealed that each of the Sodaliphilus-affiliated MAGs contained seven PULs, with S. pleomorphus having more than ten PULs (Supplementary Table S6). Therefore, we hypothesize that Sodaliphilus is a potential lignocellulose degrader in the rumen. Overall, the identification of potential lignocellulolytic consortia may open new opportunities for enhancing the degradation of plant fibers in dairy cattle production.
Selective reduction of hemicellulose degradation in the rumen by high-grain diets
-
The 1353 LM-MAGs were further employed as a genomic database to assign metagenomic samples from the CON and HG groups in dairy cattle rumen to explore the substantial alterations in microbial communities and enzyme abundances during the degradation processes of cellulose, hemicellulose, and lignin affected by high-grain diets (Fig. 4a; Supplementary Table S7). In the depolymerization phase of celluloses, genomes containing endoglucanases, particularly from the genera Ruminococcus and Hallerella exhibited a significant increase in abundance under high-grain diet conditions (Wilcoxon rank-sum test, p < 0.05; Fig. 4b & Supplementary Table S8). Meanwhile, the grain-based diet increased the abundance of endo-β-1,4-glucanase GH124 (Fig. 4b). Therefore, Ruminococcus spp. and Hallerella spp. emerged as the primary agents targeting cellulose fibrils at the amorphous regions during high-grain diet feeding conditions. In the subsequent cellulose degradation process, a noteworthy shift transpired in the majority of Prevotella-affiliated genomes encoding exoglucanases and β-glucosidases under high-grain diet feeding conditions, notably marked by the decreased abundance in Prevotella ruminicola (Supplementary Table S8).
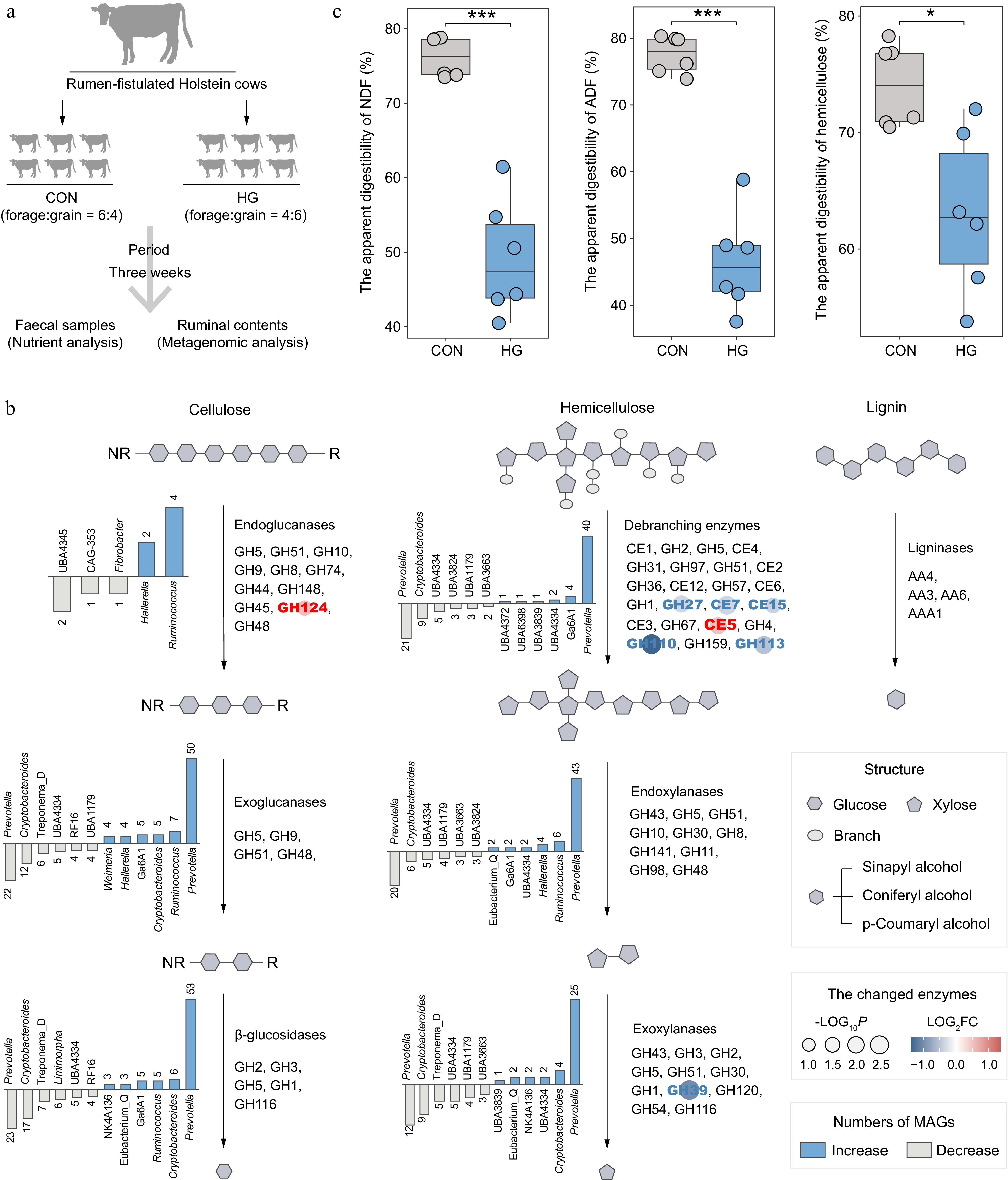
Figure 4.
Alterations in the lignocellulose degradation potential of rumen microbiota between the CON and HG groups in dairy cattle. (a) Experimental scheme of high-grain diet intervention. (b) The number and top taxonomic populations of the significantly increased and decreased abundances of lignocellulolytic microbiome (LM-MAGs) during the degradation of various lignocellulosic components in the rumen in the HG group, compared with the CON group (Wilcoxon rank-sum test, p < 0.05). The bar chart illustrates the number of genomes classified into specific genera with significantly different abundances, containing at least half of the CAZyme families encoding the same type of enzyme. (c) Comparison of the apparent digestibility in neutral detergent fiber (NDF), acid detergent fiber (ADF), and hemicellulose between the CON and HG groups, respectively. Significance is based on the relative index of each cohort according to the t-test. * p < 0.05, ** p < 0.01, *** p < 0.001.
In the depolymerization process of hemicelluloses, it is notable that the high-grain diet significantly decreased the abundance of various debranching enzymes, including acetylxylan esterases (CE7 and CE15), α-galactosidases (GH27 and GH110), and mannosidase (GH113) (Fig. 4b). Furthermore, a subsequent decline in the abundance of exoxylanases GH39 was observed after feeding the high-grain diets (Fig. 4b), leading to the inhibition of debranching activity in hemicellulose and subsequent degradation of xylan chains. This reduction was primarily attributed to a substantial decrease in the abundance of Prevotella-affiliated genomes (Fig. 4b). However, enzymes involved in lignin modification and degradation processes exhibited no significant changes after high-grain diet feeding.
As expected, nutrient content analysis of fecal samples revealed a significant reduction in the apparent digestibility of neutral detergent fiber (NDF) and acid detergent fiber (ADF) in response to the high-grain diet (t-test, p < 0.001; Fig. 4c). In addition, the high-grain diet significantly decreased the apparent digestibility of hemicellulose (p = 0.01). Therefore, the high-grain diet reduces the degradation of lignocellulose, primarily manifested in its impact on the degradation of hemicellulose.
The critical stages affected by high-grain diets in the lignocellulolytic cascades
-
To gain a deeper understanding of the specific stage of lignocellulose degradation affected by high-grain diets, resulting in an overall decrease in digestibility, the study was extended to encompass a more extensive temporal dimension. Leymus chinensis was subjected to incubation within the rumens of the six fistulated cows, comprising three forage-fed and three grain-fed cows, over 36 h (Fig. 5a). The samples were taken at nine different time points (0, 0.5, 2, 4, 6, 8, 12, 24, and 36 h) for nutrient analysis, 16S rRNA gene sequencing, and metagenomics (Fig. 5a). Nutrient analysis revealed a decrease in the digestibility of NDF, cellulose, and hemicellulose of leymus chinensis during in situ rumen later-stage incubation, especially at 36 h (t-test, p < 0.05; Fig. 5b). It is noteworthy that high-grain diets significantly impacted hemicellulose degradation during the initial phase of rumen incubation, resulting in reduced digestibility at 0.5 h and 2 h (t-test, p < 0.05; Fig. 5b). However, the digestibility of cellulose during the early stage of rumen incubation was unaffected by high-grain diets (t-test, p < 0.05; Fig. 5b). Therefore, the effect of high-grain diets on the degradation of hemicellulose during the initial stage of rumen incubation emerges as a crucial factor contributing to the overall reduction in lignocellulose degradation.
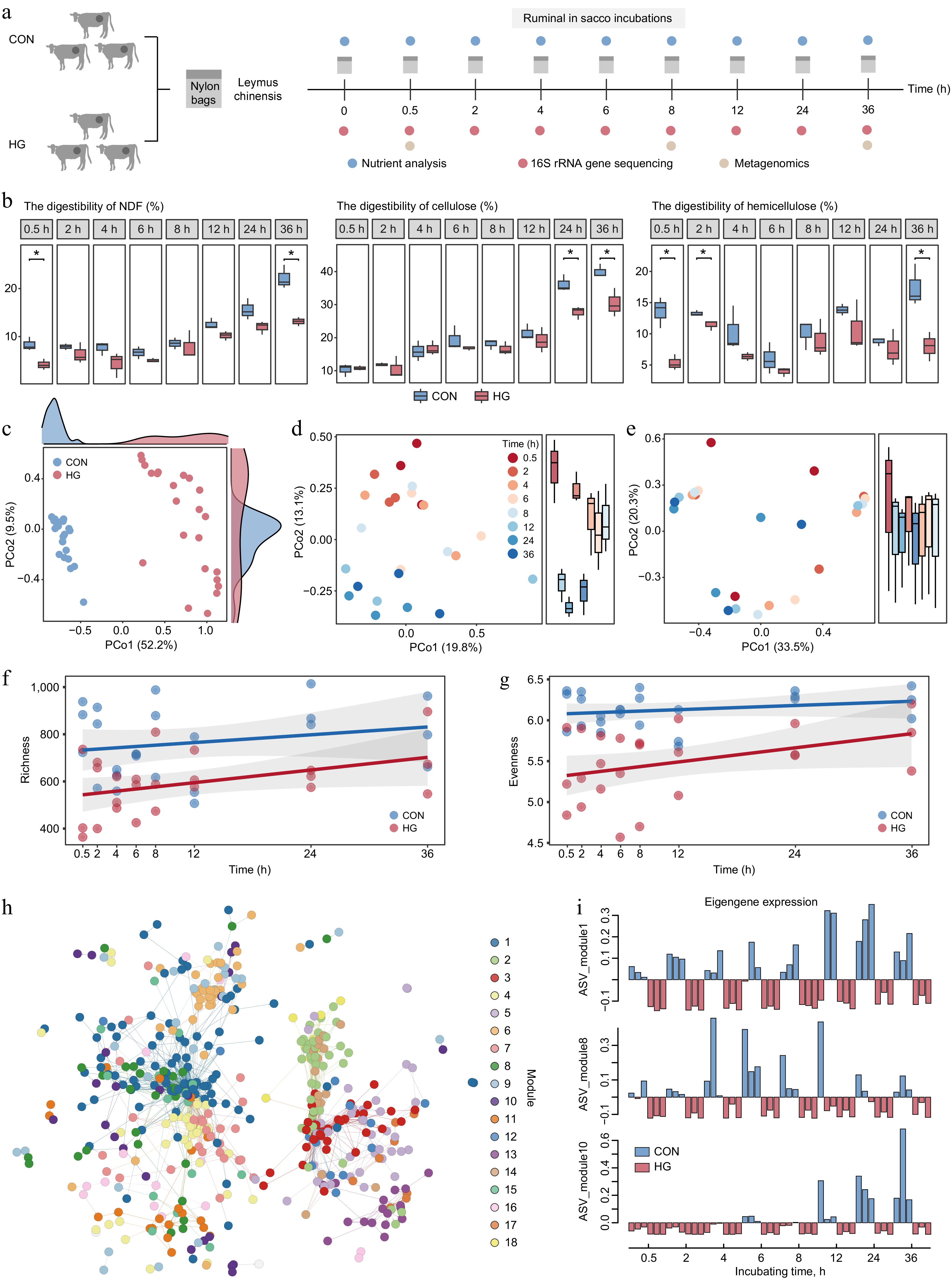
Figure 5.
Key stages of reduction in lignocellulose degradation with high-grain diet intervention. (a) Experimental scheme of rumen in situ incubations. (b) Comparison of digestibility in neutral detergent fiber (NDF), cellulose, and hemicellulose of incubated Leymus chinensis materials at each time point between the CON and HG groups according to the t-test, respectively. * p < 0.05, ** p < 0.01, *** p < 0.001. (c) Principal Coordinate Analysis (PCoA) plot generated from Bray–Curtis dissimilarity matrices using Leymus chinensis-adherent ASV abundances during rumen incubation in both the CON and HG groups. (d) Principal Coordinate Analysis (PCoA) plot generated from Bray–Curtis dissimilarity matrices using Leymus chinensis-adherent ASV abundances at 0.5, 2, 4, 6, 8, 12, 24, and 36 h during rumen incubation in the CON group. (e) Principal Coordinate Analysis (PCoA) plot generated from Bray–Curtis dissimilarity matrices using Leymus chinensis-adherent ASV abundances at 0.5, 2, 4, 6, 8, 12, 24, and 36 h during rumen incubation in the HG group. Temporal shift of richness ((f), Observed number of ASVs) and evenness ((g), Shannon diversity) indexes of ASVs between the CON and HG groups, respectively. (h) The co-occurrence network visualizing significant correlations of diet-sensitive 786 ASVs among eight time points during the rumen incubation (R > 0.7). (i) Eigengene expression of module 1, module 8, and module 10 in the CON and HG groups. CON, high-forage diet; HG, high-grain diet; ASV, amplicon sequence variants.
To understand the effect of high-grain diets on microbial colonization trajectories, fiber-adherent 16S rRNA gene sequencing data was analyzed at the amplicon sequence variant (ASV) level. The ordination analysis revealed a distinct distribution between the CON and HG groups (p = 0.0001, ANOSIM 9999 permutations; Fig. 5c). Considering the variable 'timepoint' showed that microbial taxa in the CON group exhibited a clear separation over time, indicating a regular colonization pattern following the degradation process (Fig. 5d). In contrast, the high-grain diet disrupted the colonization pattern of microorganisms, resulting in a mixing of microbiota at different time points (Fig. 5e). I was speculated that the high-grain diet affects the initial stage of the degradation period, resulting in no separation of microbial colonization in different degradation periods. Moreover, the diversity curve showed that alpha diversity (richness and evenness) in the HG group was lower than that in the CON group from 0.5 h during the rumen incubation (Fig. 5f, g).
Indicator species and edge analysis were conducted to identify 786 ASVs with significantly changed abundance during rumen incubation under grain introduction, mainly belonging to Prevotella (120 ASVs), Rikenellaceae RC9 gut group (72 ASVs), Unclassified F082 (59 ASVs), Christensenellaceae R-7 group (57 ASVs), and Ruminococcus (39 ASVs) (indicator species: p < 0.05 and edgeR: FDR < 0.05; Supplementary Table S9). These diet-sensitive 786 ASVs were further clustered into 18 dominant co-occurrence modules using weighted correlation network analysis (WGCNA) among eight time points during the rumen incubation (Fig. 5h). Specifically, the ASVs from modules 1, 8, and 10 significantly decreased in abundance at different time points under the high-grain diet conditions (Fig. 5i). The ASVs from module 1 belonged tothe Rikenellaceae RC9 gut group and showed a notable decline in abundance during the later stages (12−36 h) of rumen incubation under high-grain diet conditions (Supplementary Table S9). The ASVs from module 10 belonged to Treponema and Fibrobacter also demonstrated a substantial reduction in abundance during the later stages (12−36 h) of rumen incubation under high-grain diet conditions (Supplementary Table S9). Furthermore, ASVs from module 8, predominantly attributed to Prevotella (70.97%), exhibited a significant decrease in abundance during the early stages (0.5−8 h) of rumen incubation under high-grain diet conditions (Supplementary Table S9). This implies that Prevotella spp. may play a pivotal role as a key executor in the reduction of early-stage hemicellulose degradation.
The 1353 LM-MAGs were further utilized as a genomic database to analyze the metagenomic data from fiber-adherent samples and found that 740 LM-MAGs colonized leymus chinensis during rumen incubation (TPM > 0), with the majority (about 19.6%) belonging into Prevotella (Supplementary Table S10). Notably, a decrease in the abundance of Prevotella-affiliated MAGs were observed in the HG-fed cow rumen, which was most significant from 0.5 to 8 h of rumen incubation compared to the CON group (Supplementary Fig. S2). This phenomenon reiterated the initial 16S rRNA gene observations. In detail, MAG168, MAG160, MAG421, MAG1102, MAG646, MAG1207, and MAG763 mainly colonized from 0.5 to 8 h of rumen incubation in the CON group, while their colonization abundance was much lower in the HG group. Additionally, Prevotella ruminicola (MAG71, MAG1076, and MAG906) and Prevotella sp900100635 (MAG170 and MAG413) mainly colonized at 8 h of rumen incubation in the CON group. These Prevotella spp. showed a strong potential for hemicellulose degradation, as inferred from the enrichment family GH43 (Supplementary Table S4). The present genome-centric analysis further supports the hypothesis that the high-grain diet affects the degradation of amorphous regions in lignocellulose during the initial stage of rumen incubation. This effect was largely dependent on the Prevotella-dominated reduction in hemicellulose degradation.
-
Despite the impressive ability of rumen microbiota in dairy cattle to convert low-quality lignocellulose into nutrient-rich milk, approximately 50% of plant biomass is still resistant to degradation[53]. Previous studies have explored carbohydrate-degrading enzyme libraries within the rumen microbiomes of dairy cattle[4,6]. However, the limited number of dairy cattle has hindered our understanding of the microbial mechanisms involved in the degradation cascades of lignocellulose. In this study, all publicly available rumen metagenomes of dairy cattle were compiled and supplemented them with our own data, creating a comprehensive dataset to elucidate 1353 high-quality microbial genomes involved in lignocellulose degradation in the rumen. The present study provided a systematic description of these 1353 high-quality LM-MAGs spanning 23 phyla, representing a crucial foundation for a comprehensive understanding of rumen lignocellulose degradation efficiency in dairy cattle and creating opportunities for further improvement.
Given the diversity and redundancy of lignocellulolytic CAZymes, it was observed that different microbial consortia employed distinct enzymatic strategies in the degradation cascades of specific lignocellulosic components, including cellulose, hemicellulose, and lignin. Cellulose, the main component of lignocellulose, is degraded through the synergistic action of three classes of enzymes, including endoglucanases, exoglucanases, and β-glucosidases[54−56]. Members of Fibrobacter and Ruminococcus (e.g., Ruminococcus flavefaciens) were predicted as powerful degraders to process endo- and exoglucanases to target amorphous and crystalline cellulose[57]. An interesting phenomenon was the striking enrichment of non-catalytic domain CBMs in genera Fibrobacter and Ruminococcus. It was found that Fibrobacter spp. contained tandemly arranged CBM families anchored to cellulosic biomass. The absence of CBMs leads to a significant reduction in the binding affinity and enzymatic activity of enzyme proteins towards crystalline cellulose[58]. In contrast, the substrate adhesion effects of multiple CBMs can greatly promote the enzymatic activities of the catalytic domains of CAZymes[13,59]. Therefore, the presence of multiple CBMs-harboring GH domains in Fibrobacter spp. in cow rumen is more advantageous for cellulases to attach to the hydrophobic surface of the crystalline substrate, thereby enhancing the degradation processivity. Compared to cellulose-degrading taxa, the main players in hemicellulose degradation, Prevotella spp. and Cryptobacteroides spp., had relatively fewer CBM domains but encoded numerous PULs to degrade the main and side chains of hemicellulose[14]. The enrichment of acetylxylan esterase-contained PULs contributes largely to hemicellulose degradation. A significant proportion of the xylose residues in hemicellulose are estimated to be substituted with acetyl groups at the O-2 or O-3 position, ranging from approximately 22% to 50%[60]. The acetylxylan esterase encoded by Prevotella spp. and Cryptobacteroides spp. can remove these acetyl groups of hemicellulose to improve the degradation efficiency of other enzymes. Therefore, the different bacterial consortia employed diverse enzymatic strategies for degradation cascades of various lignocellulosic components.
Previous studies have shown that feeding high-grain diets impacts the ability of ruminal microbes to degrade fiber[4,61]. The present findings further suggest that a high-grain diet specifically disrupts the degradation cascade of hemicellulose. This was evident in the significant decrease of acetylxylan esterases (CE7 and CE15), α-galactosidases (GH27 and GH110), mannosidases (GH113), and exoxylanases (GH39) under high-grain diet conditions, which leads to a decrease in debranching activity in hemicellulose and subsequent degradation of xylan chains. Notably, this process was primarily driven by the reduction of Prevotella spp. The decreased apparent digestibility of hemicellulose under high-grain diet conditions supported this phenomenon. Through further extending the present study to multiple time points, it was discovered that a high-grain diet decreased the early-stage degradation of hemicellulose by Prevotella spp., leading to the reduced degradation of the amorphous regions of lignocellulose. This reduction in turn reduces the accessibility of microbial degrading enzymes to the ordered crystalline cellulose and results in ineffective degradation of lignocellulose[12]. The present results showed that feeding high-grain diets disrupted the ordered colonization of microorganisms on Leymus chinensis, resulting in the non-separation of microbiota at different colonization time points. Therefore, the Prevotella-dominated reduction in hemicellulose degradation during the initial stage of rumen incubation may be the primary reason for the reduced digestibility of lignocellulose under high-grain diet feeding.
In addition, it was found that some lignocellulose-degrading bacteria were significantly enriched under high-grain conditions, and importantly, they belong to microbial taxa that have not been described before in ruminants. For example, Hallerella spp., the nearest phylogenetic neighbor of Fibrobacter spp.[52], were found to possess a large amount of endoglucanases and exoglucanases. Hallerella-affiliated MAGs were found to only attach to fiber in cows fed high-grain diets, indicating their important role in lignocellulose degradation in such environments. Despite both belonging to the Fibrobacteraceae family, Fibrobacter spp. and Hallerella spp. had different adaptabilities in various environments. In detail, Fibrobacter was more suitable for high-fiber environments, while Hallerella exhibited fiber degradation ability in low-fiber environments. Additionally, members of Sodaliphilus were predicted to possess the capability of lignocellulose degradation through extracellular enzymes or polysaccharide utilization loci (PULs), and their abundance exhibited a significant increase under high-grain diet conditions. Previous studies have shown that high-grain diets can lead to a decrease in the abundance of lignocellulose-degrading microorganisms[4,62]. Therefore, these newly discovered potential lignocellulose-degrading bacteria in the rumen of dairy cattle represent important resources for development. These bacteria could help mitigate the reduced lignocellulose degradation capacity associated with high-grain feeding. Although further research is needed to confirm the actual lignocellulose degradation capabilities of these microorganisms, our findings provide valuable support for future efforts aimed at enhancing the degradation efficiency of lignocellulose in the rumen of dairy cattle fed high-grain diets.
In addition to bacteria, several studies have explored the genomes of eukaryotic organisms, such as fungi and ciliates, in the rumen[63,64]. These studies have revealed that these organisms encode a variety of carbohydrate-related genes and enzymes, showing their significant capabilities for degrading plant fiber. The present study primarily focuses on prokaryotic genomic research related to the microbial degradation of rumen carbohydrates. Future research should clarify how high-grain diets influence different lignocellulosic components and the various stages of lignocellulolytic cascades through specific protozoa and fungi. This will contribute to a more comprehensive understanding of the role of rumen microorganisms in carbohydrate degradation.
-
The present study utilized a genome-centric approach to identify 1353 high-quality MAGs involved in lignocellulose degradation in the cow rumen. By analyzing their enzymatic strategies for different substrate types, insights into the complexity and specialization of ruminal microbial populations in degrading various lignocellulosic components were gained, with a particular emphasis on cellulose and hemicellulose degradation, while highlighting their limited role in lignin degradation. Through spatial and temporal studies involving diet interventions and rumen in situ incubation, it was discovered that a high-grain diet primarily interfered with the degradation of amorphous regions of lignocellulose and significantly reduced hemicellulose degradation by Prevotella-dominated communities. These findings underscore the intricate interplay among diet, microbial consortia, and enzymatic strategies in the rumen, highlighting the potential for manipulating the rumen microbiota to improve the efficiency of lignocellulose degradation in dairy cattle.
-
All procedures were reviewed and preapproved by the the Nanjing Agricultural University Institutional Animal Care and Use Committee, identification number: SYXK-2017–0027, approval date: 2019-3-12. The research followed the 'Replacement, Reduction, and Refinement' principles to minimize harm to animals. This article provides details on the housing conditions, care, and pain management for the animals, ensuring that the impact on the animals is minimized during the experiment.
-
The authors confirm contribution to the paper as follows: study conception and design: Mao S, Zhu W; samples collection and experiments conduction: Lin L, Yang H, Zhang Jiyou, Lai Z, Qi W, Ma H, Zhang Jiawei; published rumen metagenome collection: Xie F, Lin L; bioinformatic analyses, data visualization and interpretation, draft manuscript preparation: Lin L; manuscript revision: Mao S. All authors read, edited, and approved the final manuscript.
-
Raw sequence reads for all 18 incubated leymus chinensis samples are available under European Nucleotide Archive (ENA) project PRJNA955930. All MAGs produced and utilized in this study have been deposited in Figshare (
https://figshare.com/s/dcaad3555e23f2551029 ). The data sources for an additional 226 published ruminal metagenome samples from dairy cattle are provided in Supplementary Table S1. Please note that although there were 48 samples available under PRJNA214227 as reported by Wolff et al.[27], we exclusively utilized 16 rumen samples from dairy cattle. For other projects, we also exclusively utilized rumen samples from dairy cattle. The current project was supported by the high-performance computing platform of Bioinformatics Center, Nanjing Agricultural University. This research was funded by the National Key R&D Program of China (2022YFD1301001).
-
The authors declare that they have no conflict of interest. Shengyong Mao is the Editorial Board member of Animal Advances who was blinded from reviewing or making decisions on the manuscript. The article was subject to the journal's standard procedures, with peer-review handled independently of this Editorial Board member and the research groups.
- Supplementary Table S1 226 published ruminal metagenome samples from dairy cattle.
- Supplementary Table S2 Sample source of 5034 MAGs from dairy cattle rumen.
- Supplementary Table S3 Genomic statistics for 1374 non-redundant high-quality MAGs (Completeness > 80% and Contamination < 10%).
- Supplementary Table S4 1353 high-quality MAGs involved in the degradation process of lignocellulose, including cellulose, hemicellulose, and lignin.
- Supplementary Table S5 Genome-wide prediction of CBM-containing CAZymes in lignocellulolytic MAGs in the rumen of dairy cattle.
- Supplementary Table S6 Genome-wide prediction of total polysaccharide utilization loci (PULs) in lignocellulolytic MAGs in the rumen of dairy cattle.
- Supplementary Table S7 Ingredients and nutritional compositions of the high-forage (CON) and high-grain (HG) diets.
- Supplementary Table S8 The significant changes of microbial genomes and key enzymes involved in the degradation cascade of various lignocellulosic components caused by high-grain diets.
- Supplementary Table S9 Significantly different abundance of 786 microbial ASVs colonized leymus chinensis between the high-forage (CON) and high-grain (HG) diets during the rumen incubation.
- Supplementary Table S10 The abundance of 740 LM-MAGs colonized leymus chinensis during rumen incubation
- Supplementary Fig. S1 A: Pipeline for data processing and integration. B: Distribution of quality metrics across the high-quality MAGs (n = 1374), showing the minimum value, first quartile, median, third quartile and maximum value.
- Supplementary Fig. S2 Heatmap of the abundance and distribution of Prevotella-affiliated LM-MAGs at 0.5, 8, 36 h during the rumen incubation between the CON and HG groups, and a Z-score was used for correction. CON, high-forage diet; HG, high-grain diet.
- Copyright: © 2024 by the author(s). Published by Maximum Academic Press on behalf of Nanjing Agricultural University. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Lin L, Ma H, Zhang J, Yang H, Zhang J, et al. 2024. Lignocellulolytic microbiomes orchestrating degradation cascades in the rumen of dairy cattle and their diet-influenced key degradation phases. Animal Advances 1: e002 doi: 10.48130/animadv-0024-0002 

Lignocellulolytic microbiomes orchestrating degradation cascades in the rumen of dairy cattle and their diet-influenced key degradation phases
- Received: 20 August 2024
- Revised: 14 September 2024
- Accepted: 18 September 2024
- Published online: 21 October 2024
Abstract: Dairy cattle (Bos taurus) can convert lignocellulosic biomass into milk efficiently via their rumen symbiotic microbiota. However, the mechanisms by which the rumen microbiota of cows mediate the degradation cascades of lignocellulose and the specific stages primarily affected by dietary interventions remain unclear. Herein, 244 rumen metagenome samples from Holstein cows were used, identifying 1353 high-quality microbial metagenome-assembled genomes (MAGs) responsible for the degradation cascades of lignocellulose. It was revealed that Fibrobacter spp. and Ruminococcus spp. exhibited numerous endo-/exo-glucanases with accessory non-catalytic multi-carbohydrate binding modules for highly efficient cellulolytic abilities. Prevotella spp. and Cryptobacteroides spp. developed diverse polysaccharide utilization loci (PULs) to tackle the main and side chains of hemicellulose, particularly acetylxylan esterase-contained PULs. Notably, novel and potential lignocellulolytic microbiomes were identified in the rumen of dairy cattle, such as Hallerella spp., Sodaliphilus spp., and Mageeibacillus spp. Through in vivo diet intervention and in sacco rumen incubation, it was discovered that high-grain diets primarily affected Prevotella spp., leading to a reduction in the initial degradation of amorphous regions in lignocellulose. Therefore, the present findings systematically illustrate the orchestrated enzymatic strategies of the cow rumen microbiota for the degradation cascades of lignocellulose, contributing to the dietary regulation of dairy cattle.
-
Key words:
- Lignocellulolytic microbiomes /
- Degradation cascades /
- Enzymatic strategies /
- Rumen /
- Dairy cattle