









-
The Fabaceae family, the third largest family in angiosperms, contains about 24,480 species (WFO,
https://wfoplantlist.org ), and has been a historically important source of food crops[1−4]. Peas (formerly Pisum sativa L. renamed to Lathyrus oleraceus – Pisum spp. will be used hereafter due to historical references to varietal names and subspecies that may not have been fully synonymized), are a member of the Fabaceae family and is among one of the oldest domesticated food crops with ongoing importance in feeding humans and stock. Peas originated in Western Asia and the Mediterranean basin where early finds from Egypt have been dated to ~4500 BCE and further east in Afghanistan from ~2000 BCE[5], and have since been extensively cultivated worldwide[6,7]. Given that peas are rich in protein, dietary fiber, vitamins, and minerals, have become an important part of people's diets globally[8−10].Domesticated peas are the result of long-term human selection and cultivation, and in comparison to wild peas, domesticated peas have undergone significant changes in morphology, growth habits, and yield[11−13]. From the long period of domestication starting in and around Mesopotamia many diverse lineages of peas have been cultivated and translocated to other parts of the world[14,15]. The subspecies, Pisum sativum subsp. sativum is the lineage from which most cultivars have been selected and is known for possessing large, round, or oval-shaped seeds[16,17]. In contrast, the subspecies, P. sativum subsp. elatius, is a cultivated pea which more closely resembles wild peas and is mainly found in grasslands and desert areas in Europe, Western Asia, and North Africa. Pisum fulvum is native to the Mediterranean basin and the Balkan Peninsula[15,18], and is resistant to pea rust caused by the fungal pathogen Uromyces pisi. Due to its resistance to pea rust, P. fulvum has been cross-bred with cultivated peas in the development of disease-resistant strains[19]. These examples demonstrate the diverse history of the domesticated pea and why further study of the pea pan-plastome could be employed for crop improvement. While studies based on the nuclear genome have been used to explore the domestication history of pea, these approaches do not account for certain factors, such as maternal inheritance. Maternal lineages, which are inherited through plastomes, play a critical role in understanding the full domestication process. A pan-plastome-based approach will no doubt allow us to investigate the maternal genetic contributions and explore evolutionary patterns that nuclear genome studies may overlook. Besides, pan-plastome analysis enables researchers to systematically compare plastome diversity across wild and cultivated species, identifying specific regions of the plastome that contribute to desirable traits. These plastid traits can then be transferred to cultivated crops through introgression breeding or genetic engineering, leading to varieties with improved resistance to disease, environmental stress, and enhanced agricultural performance.
Plastids are organelles present in plant cells and are the sites in which several vital biological processes take place, such as photosynthesis in chloroplasts[20−24]. Because the origin of plastids is the result of an ancient endosymbiotic event, extant plastids retain a genome (albeit much reduced) from the free-living ancestor[25]. With the advancement of high-throughput DNA sequencing technology, over 13,000 plastid genomes or plastomes have been published in public databases by the autumn of 2023[24]. Large-scale comparison of plastomic data at multiple taxonomic levels has shown that plastomic data can provide valuable insights into evolution, interspecies relationships, and population genetic structure. The plastome, in most cases, displays a conserved quadripartite circular genomic architecture with two inverted repeat (IR) regions and two single copy (SC) regions, referred to as the large single-copy (LSC) and small single-copy (SSC) regions. However, some species have lost one copy of the inverted repeated regions, such as those in Erodium (Geraniaceae family)[26,27] and Medicago (Fabaceae)[28,29]. Compared to previous plastomic studies based on a limited number of plastomes, the construction of pan-plastomes attempts to describe all nucleotide variants present in a lineage through intensive sampling and comparisons. Such datasets can provide detailed insights into the maternal history of a species and help to better understand applied aspects such as domestication history or asymmetries in maternal inheritance, which can help guide future breeding programs. Such pan-plastomes have recently been constructed for several agriculturally important species. A recent study focuses on the genus Gossypium[20], using plastome data at the population level to construct a robust map of plastome variation. It explored plastome diversity and population structure relationships within the genus while uncovering genetic variations and potential molecular marker loci in the plastome. Besides, 65 samples were combined to build the pan-plastome of Hemerocallis citrina[30] , and 322 samples for the Prunus mume pan-plastome[31]. Before these recent efforts, similar pan-plastomes were also completed for Beta vulgaris[32], and Nelumbo nucifera[33]. However, despite the agricultural importance of peas, no such pan-plastome has been completed.
In this study, 103 complete pea plastomes were assembled and combined another 42 published plastomes to construct the pan-plastome. Using these data, the following analyses were conducted to better understand the evolution and domestication history of pea: (1) genome structural comparisons, (2) codon usage bias, (3) simple sequence repeat patterns, (4) phylogenetic analysis, and (5) nucleotide variation of plastomes in peas.
-
One hundred and three complete pea plastomes were de novo assembled from public whole-genome sequencing data[34]. For data quality control, FastQC v0.11.5 (
www.bioinformatics.babraham.ac.uk/projects/fastqc/ ) was utilized to assess the quality of the reads and ensure that the data was suitable for assembly. The clean reads were then mapped to a published pea plastome (MW308610) plastome from the GenBank database (www.ncbi.nlm.nih.gov/genbank ) as the reference using BWA v0.7.17[35] and SAMtools v1.9[36] to isolate plastome-specific reads from the resequencing data. Finally, these plastome-specific reads were assembled de novo using SPAdes v3.15.2[37]. The genome annotation was conducted by Geseq online program (https://chlorobox.mpimp-golm.mpg.de/geseq.html ). Finally, the OGDRAW v1.3.1[38] program was utilized to visualize the circular plastome maps with default settings. To better resolve the pan-plastome for peas, 42 complete published pea plastomes were also downloaded from NCBI and combined them with the de novo data (Supplementary Table S1).Codon usage and repeat element patterns
-
To investigate the codon usage in the pan-plastome of pea, we utilized CodonW v.1.4.2 (
http://codonw.sourceforge.net ) to calculate the Relative Synonymous Codon Usage (RSCU) value of the protein-coding genes (PCGs) longer than 300 bp, excluding stop codons. The RSCU is a calculated metric used to evaluate the relative frequency of usage among synonymous codons encoding the same amino acid. An RSCU value above 1 suggests that the codon is utilized more frequently than the average for a synonymous codon. Conversely, a value below 1 indicates a lower-than-average usage frequency. Besides, the Effective Number of Codons (ENC) and the G + C content at the third position of synonymous codons (GC3s) were also calculated in CodonW v.1.4.2. The ENC value and GC3s value were utilized for generating the ENC-GC3s plot, with the expected ENC values (standard curve), are calculated according to formula: ENC = 2 + GC3s + 29 / [GC3s2 + (1 – GC3s)2][39].The MISA program[40] was utilized to detect simple sequence repeats (SSRs), setting the minimum threshold for repeat units at 10 for mono-motifs, 6 for di-motifs, and 5 for tri-, tetra-, penta-, and hexa-motif microsatellites, respectively.
Phylogenetic analysis
-
The 145 complete pea plastomes were aligned using MAFFT v 7.487[41]. Single nucleotide variants (SNVs)-sites were used to derive an SNV only dataset from the entire-plastome alignment[42]. A total of 959 SNVs were analyzed using IQ-TREE v2.1[43] with a TVMe + ASC + R2 substitution model, determined by ModelTest-NG[44] based on BIC, and clade support was assessed with 1,000 bootstrap replicates. Vavilovia formosa (MK604478) was chosen as an outgroup. The principal coordinates analysis (PCA) was conducted in TASSEL 5.0[45].
Haplotype and genetic diversity analyses
-
DnaSP v6[46] was utilized to identify different haplotypes among the plastomes, with gaps and missing data excluded. Haplotype networks were constructed in Popart v1.7[47] using the median-joining algorithm. Haplotype diversity (Hd) for each group was calculated by DnaSP v6[46], and the evolutionary distances based on the Tamura-Nei distance model were computed based on the population differentiation index (FST) between different groups with the plastomic SNVs.
-
In this study, the pan-plastome structure of peas was elucidated (Fig. 1). The length of these plastomes ranged from 120,826 to 122,547 bp. And the overall GC content varied from 34.74% to 34.87%. In contrast to typical plastomes characterized by a tetrad structure, the plastomes of peas contained a single IR copy. The average GC content among all pea plastomes was 34.8%, with the highest amount being 34.84% and the lowest 34.74%, with minimal variation among the pea plastomes.
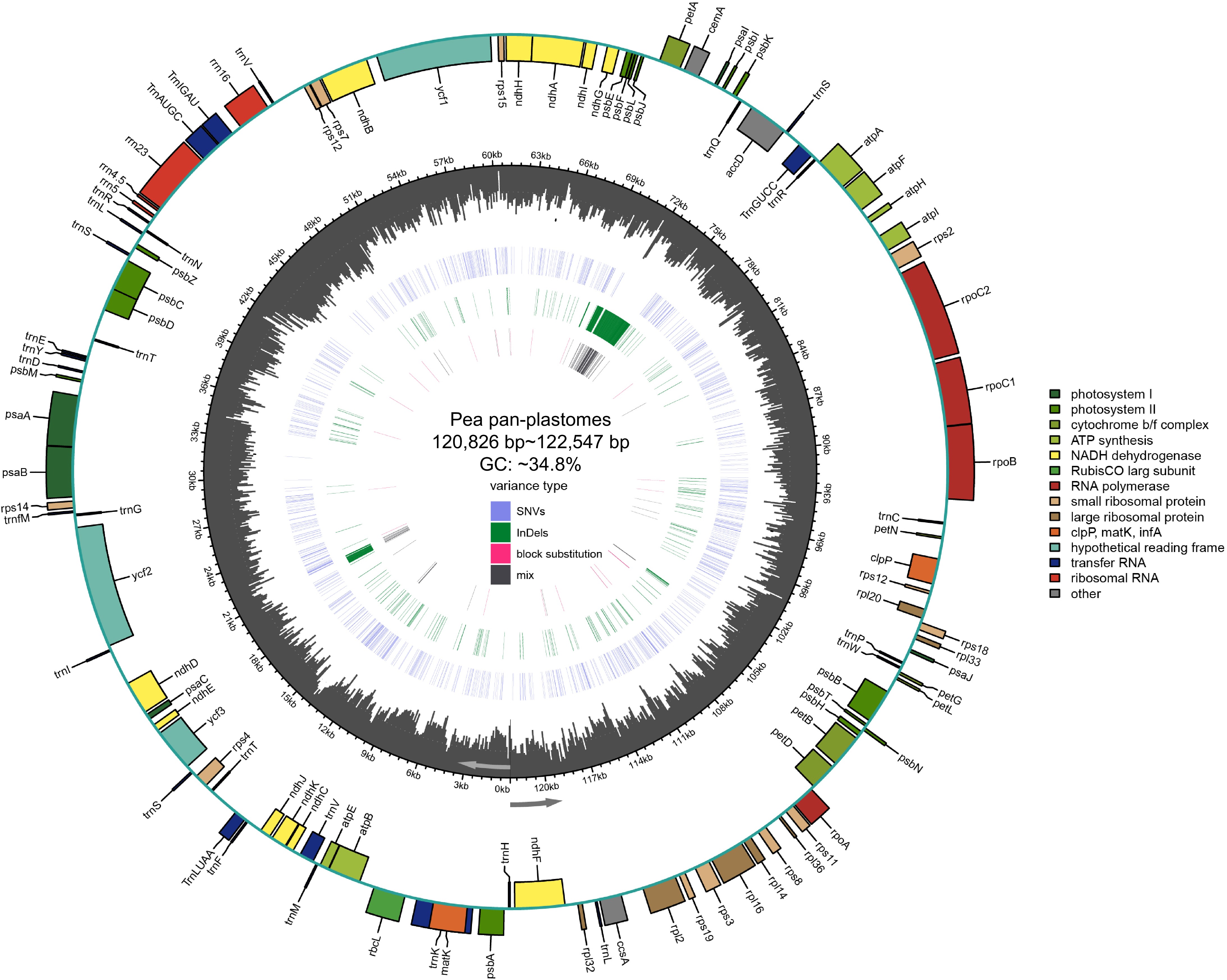
Figure 1.
Pea pan-plastome annotation map. Indicated by arrows, genes listed inside and outside the circle are transcribed clockwise and counterclockwise, respectively. Genes are color-coded by their functional classification. The GC content is displayed as black bars in the second inner circle. SNVs, InDels, block substitutions and mixed variants are represented with purple, green, red, and black lines, respectively. Single nucleotide variants (SNVs), block substitutions (BS, two or more consecutive nucleotide variants), nucleotide insertions or deletions (InDels), and mixed sites (which comprise two or more of the preceding three variants at a particular site) are the four categories into which variants are divided.
A total of 110 unique genes were annotated (Supplementary Table S2), of which 76 genes were PCGs, 30 were transfer RNA (tRNA) genes and four were ribosomal RNA (rRNA) genes. Genes containing a single intron, include nine protein-coding genes (rpl16, rpl2, ndhB, ndhA, petB, petD, rpoC1, clpP, atpF) and six tRNA genes (trnK-UUU, trnV-UAC, trnL-UAA, trnA-UGC, trnI-GAU, trnG-UCC). Additionally, two protein-coding genes ycf3 and rps12 were found to contain two introns.
Codon usage and simple sequence repeats (SSRs) patterns in peas
-
The codon usage frequency in pea plastome genes is shown in Fig. 2a. The analysis of codon usage in the pea plastome indicated significant biases for specific codons across various amino acids. Here a nearly average usage in some amino acids was observed, such as Alanine (Ala) and Valine (Val). For most amino acids, the usage of different synonymous codons was not evenly distributed. Regarding stop codons, a nearly even usage was found, with 37.0% for TAA, 32.2% for TAG and 30.8% for TGA.
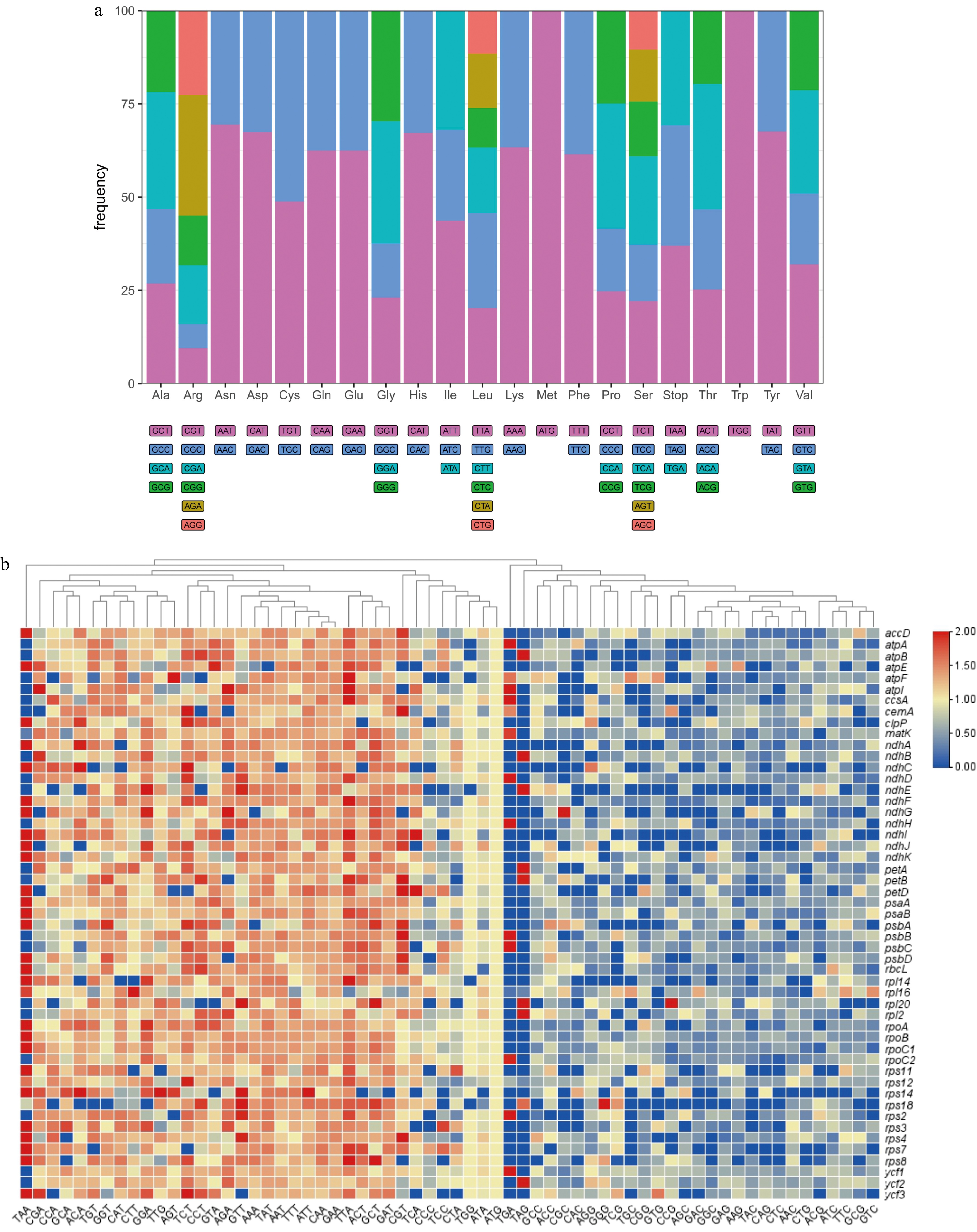
Figure 2.
(a) The overall codon usage frequency in 51 CDSs (length > 300 bp) from the pea pan-plastome. (b) The heatmap of RSCU values in 51 CDSs (length > 300 bp) from the pea pan-plastome. The x-axis represents different codons and the y-axis represents different CDSs. The tree at the top was constructed based on a Neighbor-Joining algorithm.
The RSCU heatmap (Fig. 2b) showed different RSCU values for all codons in plastomic CDSs. In general, a usage bias for A/T in the third position of codons was found among CDSs in the pea pan-plastome. The RSCU values among these CDSs ranged from 0 to 4.8. The highest RSCU value (4.8) was found with the CGT codon in the cemA gene, where six synonymous codons exist for Arg but only CGT (4.8) and AGG (1.2) were used in this gene. This explained in large part the extreme RSCU value for CGT, resulting in an extreme codon usage bias in this amino acid.
In the ENC-GC3s plot (Fig. 3), 31 PCGs were shown below the standard curve, while 20 PCGs were above. Besides, around 12 PCGs were near the curve, which meant these PCGs were under the average natural selection and mutation pressure. This plot displayed that the codon usage preferences in pea pan-plastomes were mostly influenced by natural selection. Five genes were shown an extreme influence with natural selection for its extreme ΔENC (ENCexpected – ENC) higher than 5, regarding as petB (ΔENC = 5.18), psbA (ΔENC = 8.96), rpl16 (ΔENC = 5.62), rps14 (ΔENC = 14.29), rps18 (ΔENC = 6.46) (Supplementary Table S3).
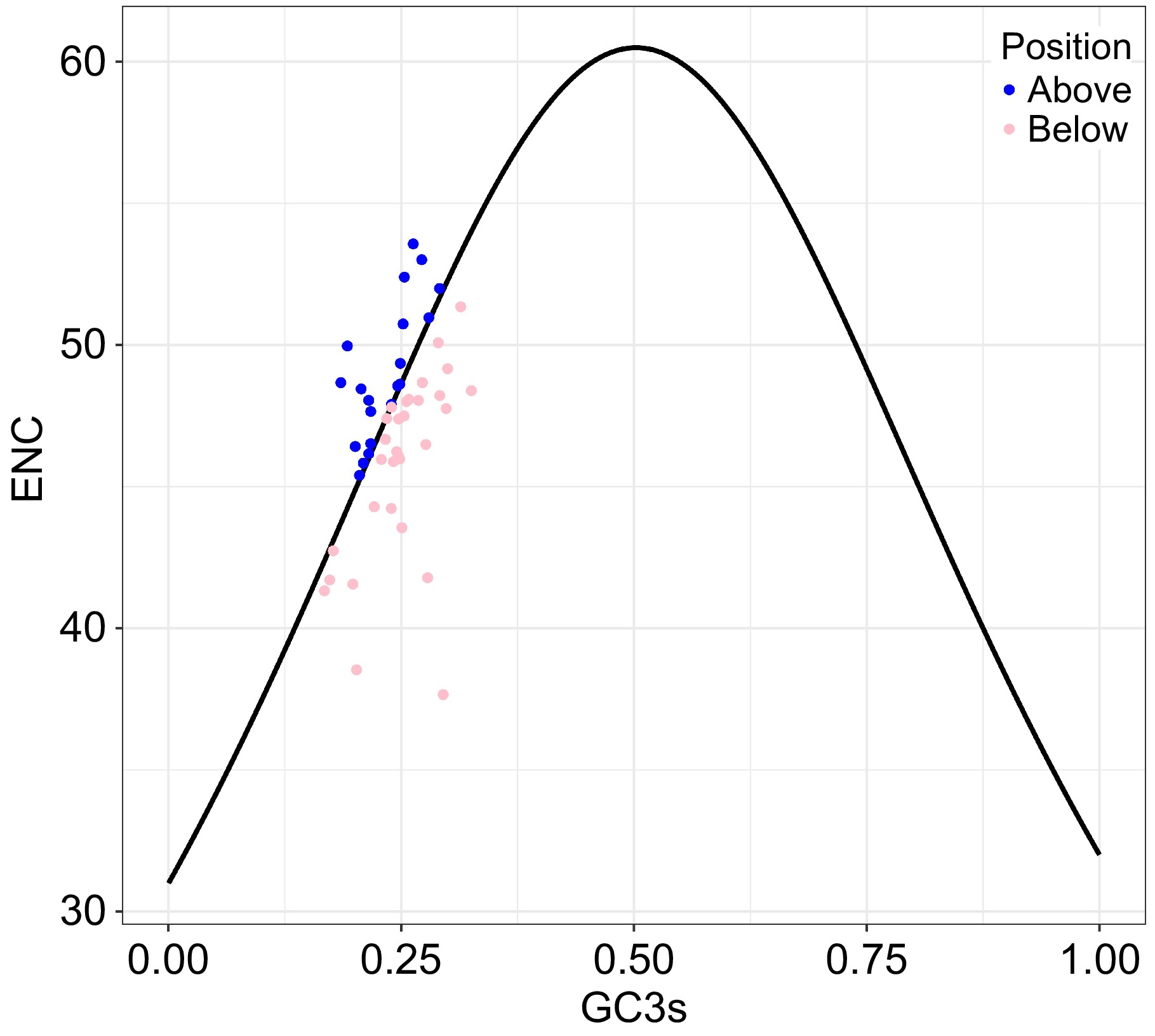
Figure 3.
The ENC-GC3s plot for pea pan-plastome, with GC3s as the x-axis and ENC as the y-axis. The expected ENC values (standard curve) are calculated according to formula: ENC = 2 + GC3s + 29 / [GC3s2 + (1 − GC3s)2].
For SSR detection (Fig. 4), mononucleotide, dinucleotide, and trinucleotide repeats were identified in the pea pan-plastome including A/T, AT/TA, and AAT/ATT. The majority of these SSRs were mononucleotides (A/T), accounting for over 90% of all identified repeats. Additionally, we observed that A/T and AT/TA repeats were present in all pea accessions, whereas only about half of the plastomes contained AAT/ATT repeats. It was also found that the number of A/T repeats exhibited the greatest diversity, while the number of AAT/ATT repeats showed convergence in all plastomes that possessed this repeat.
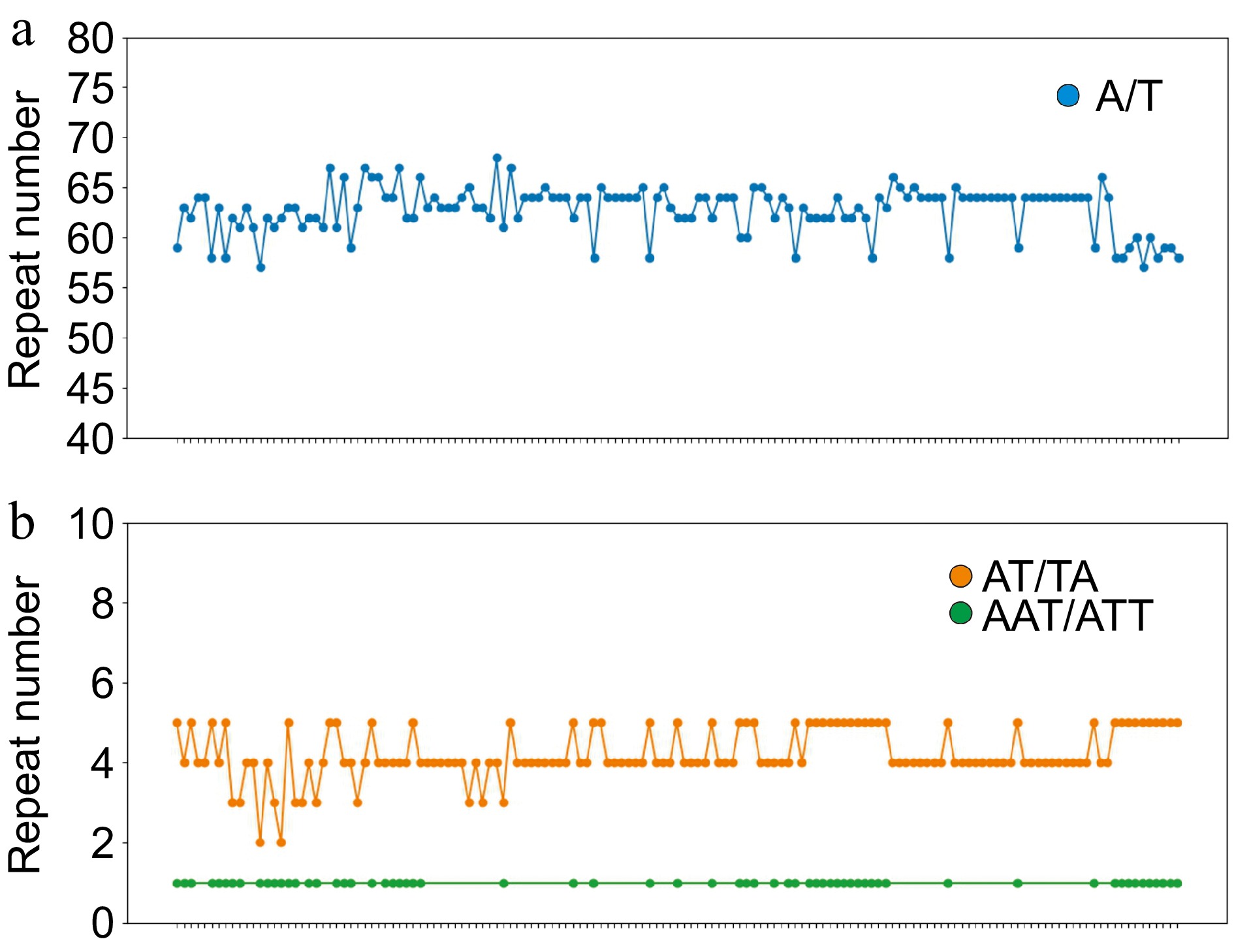
Figure 4.
Simple sequence repeats (SSRs) in the pea pan-plastome. The x-axis represents different samples of pea and the y-axis represents the number of repeats in this sample. (a) The number of A/T repeats in the peapan-plastome. (b) The number of AT/TA and AAT/ATT repeats of pea pan-plastomes.
Phylogenetic analysis
-
To better understand the phylogenetic relationships and evolutionary history of peas, a phylogenetic tree was reconstructed using maximum likelihood for 145 pea accessions utilizing the whole plastome sequences (Fig. 5a). The 145 pea accessions were grouped into seven clades with high confidence. These groups were named the 'PF group', 'PSeI-a group', 'PSeI-b group', 'PA group', 'PSeII group', 'PSeIII group', and the 'PS group'. The naming convention for these groups relates to the majority species names for accessions in each group, where P. fulvum makes up the 'PF group', P. sativum subsp. elatius in the 'PSeI-a group', 'PSeI-b group', 'PSeII group', and 'PSeIII group', P. abyssinicum in the 'PA group', and P. sativum in the 'PS group'. From this phylogenetic tree, it was observed that the 'PSeI-a group' and the 'PSeI-b group' had a close phylogenetic relationship and nearly all accessions in these two groups (except DCG0709 accession was P. sativum) were identified as P. sativum subsp. elatius. In addition to the P. sativum subsp. elatius found in PSeI, seven accessions from the PS group were identified as P. sativum subsp. elatius.
The PCA results (Fig. 5b) also confirmed that domesticated varieties P. abyssinicum were closer to cultivated varieties PSeI and PSeII, while PSeIII was more closely clustered with cultivated varieties of P. sativum. A previous study has indicated that P. sativum subsp. sativum and P. abyssinicum were independently domesticated from different P. sativum subsp. elatius populations[34].
The complete plastome sequences were utilized for haplotype analysis using TCS and median-joining network methods (Fig. 5c). A total of 76 haplotypes were identified in the analysis. The TCS network resolved a similar pattern as the other analyses in that six genetic clusters were resolved with genetic clusters PS and PSeIII being very closely related. The genetic cluster containing P. fulvum exhibits greater genetic distance from other genetic clusters. The genetic clusters containing P. abyssinicum (PA) and P. sativum (PS) had lower levels of intracluster differentiation. In the TCS network, Hap30 and Hap31 formed distinct clusters from other haplotypes, such as Hap27, which may account for the genetic difference between the 'PSeI-a group' and 'PSeI-b group'. The network analysis results were consistent with the findings of the phylogenetic tree and principal component analyses results in this study.
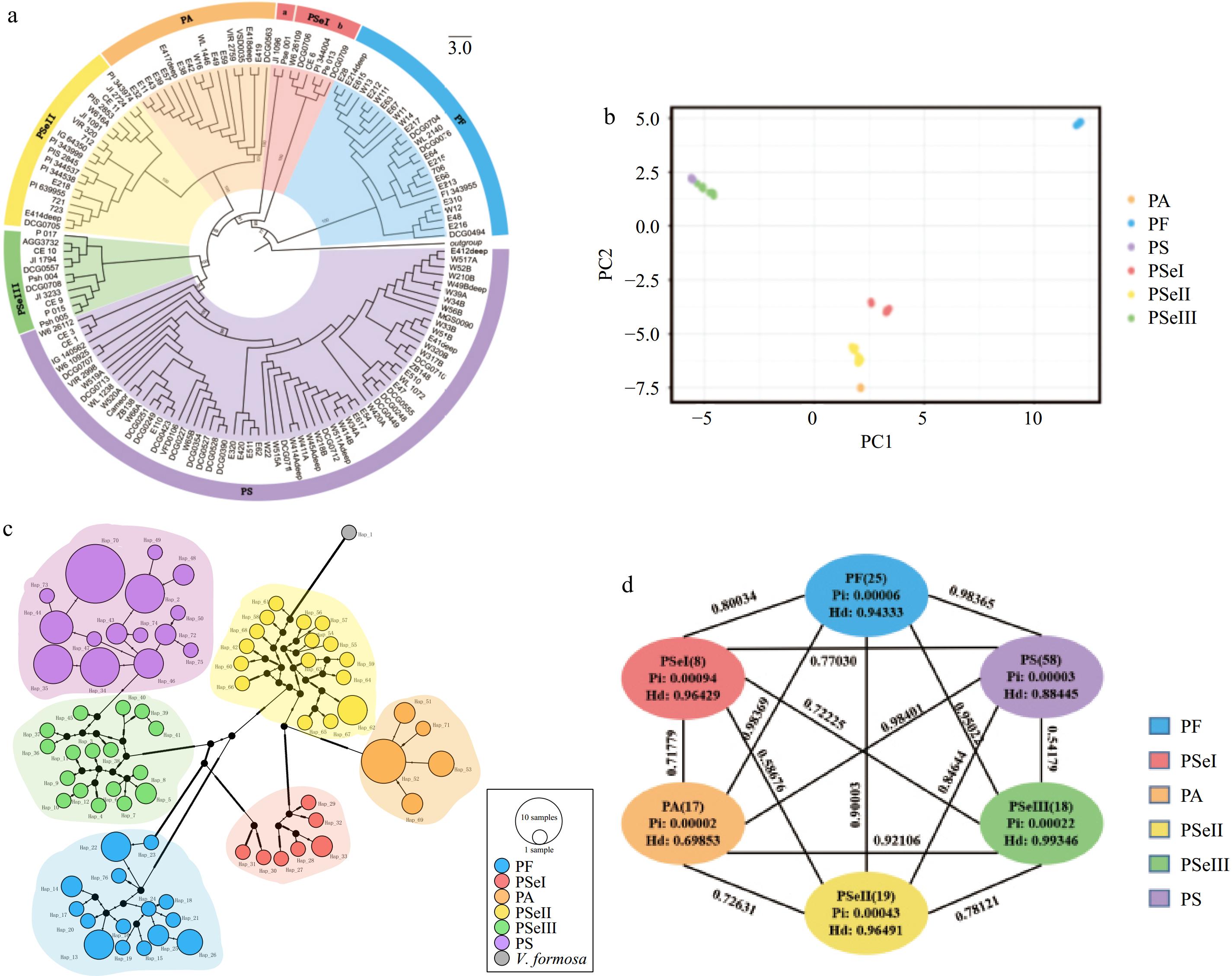
Figure 5.
(a) An ML tree resolved from 145 pea plastomes. (b) PCA analysis showing the first two components. (c) Haplotype network of pea plastomes. The size of each circle is proportional to the number of accessions with the same haplotype. (d) Genetic diversity and differentiation of six clades of peas. Pairwise FST between the corresponding genetic clusters is represented by the numbers above the lines joining two bubbles.
Among the six genetic clusters, the highest haplotype diversity (Hd) was observed in PSeIII (Hd = 0.99, π = 0.22 × 10−3), followed by PSeII (Hd = 0.96, π = 0.43 × 10−3), PSeI (Hd = 0.96, π = 0.94 × 10−3), PF (Hd = 0.94, π = 0.6 × 10−4), PS (Hd = 0.88, π = 0.3 × 10−4), and PA (Hd = 0.70, π = 0.2 × 10−4). Genetic differentiation was evaluated between each genetic cluster by calculating FST values. As shown in Fig. 5d, except for the relatively lower population differentiation between PS and PSeIII (FST = 0.54), and between PSeI and PSeII (FST = 0.59), the FST values between the remaining clades ranged from 0.7 to 0.9. The highest population differentiation was observed between PF and PA (FST = 0.98). The FST values between PSeI and different genetic clusters were relatively low, including PSeI and PF (FST = 0.80), PSeI and PS (FST = 0.77), PSeI and PSeIII (FST = 0.72), PSeI and PSeII (FST = 0.59), and PSeI and PA (FST = 0.72).
Nucleotide variation in the pea pan-plastome
-
To further determine the nucleotide variations in the pea pan-plastome, 145 plastomes were aligned and nucleotide differences analyzed across the dataset. A total of 1,579 variations were identified from the dataset (Table 1), including 965 SNVs, 24 Block Substitutions, 426 InDels, and 160 mixed variations of these three types. Among the SNVs, transitions were more frequent than transversions, with 710 transitions and 247 transversions. In transitions, T to G and A to C had 148 and 139 occurrences, respectively, while in transversions, G to A and C to T had 91 and 77 occurrences, respectively.
Table 1. Nucleotide variation in the pan-plastome of peas.
Variant Total SNV Substitution InDel Mix
(InDel, SNV)Mix
(InDel, SUB)Total 1,576 965 24 426 156 4 CDS 734 445 6 176 103 4 Intron 147 110 8 29 0 0 tRNA 20 15 1 4 0 0 rRNA 11 3 0 6 2 0 IGS 663 392 9 211 51 0 When analyzing variants by their position to a gene (Fig. 6), there were 731 variations in CDSs, accounting for 46.3% of the total variations, including 443 SNVs (60.6%), six block substitutions (0.83%), 175 InDels (23.94%), and four mixed variations (14.64%). There were 104 variants in introns, accounting for 6.59% of the total variations, including 78 SNVs (75%), seven block substitutions (6.73%), and 19 InDels (18.27%). IGS (Intergenic spacers) contained 660 variations, accounting for 41.8% of the total variations, including 394 SNVs (59.7%), nine block substitutions (1.36%), 207 InDels (31.36%), and 50 mixed variations (7.58%). The tRNA regions contained 63 variants, accounting for 3.99% of the total variations, including 47 SNVs (74.6%) and 14 InDels (22.2%). The highest number of variants were detected in the IGS regions, while the lowest were found in introns. Among CDSs, accD (183) had the highest number of variations. In introns, rpL16 (18) and ndhA (16) had the most variants. In the IGS regions, ndhD-trnI-CAU (73), and trnL-UAA-trnT-UGU (44) possessed the greatest number of variants.
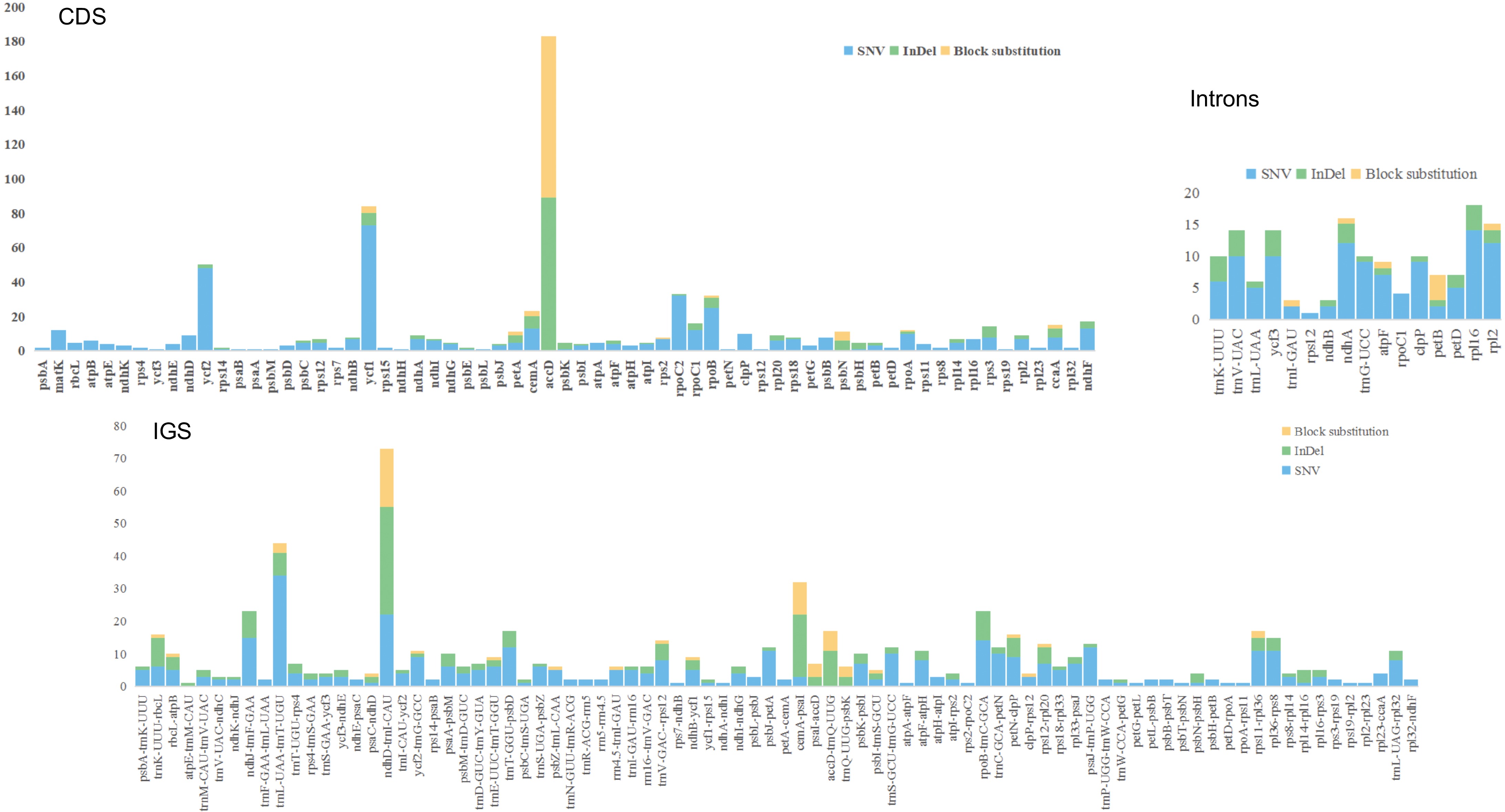
Figure 6.
Variant locations within the pea pan-plastome categorized by genic position (Introns, CDS, and IGS).
Finally, examples of some genes with typical variants were provided to better illustrate the sequence differences between clades (Fig. 7). For example, the present analysis revealed that the ycf1 gene exhibited a high number of variant loci, which included unique single nucleotide variants (SNVs) specific to the P. abyssinicum clade. Additionally, a unique InDel variant belonging to P. abyssinicum was identified. Similar unique SNVs and InDels were also found in other genes, such as matK and rpoC2, distinguishing the P. fulvum clade from others. These unique SNVs and InDels could serve as DNA barcodes to distinguish different maternal lineages of peas.
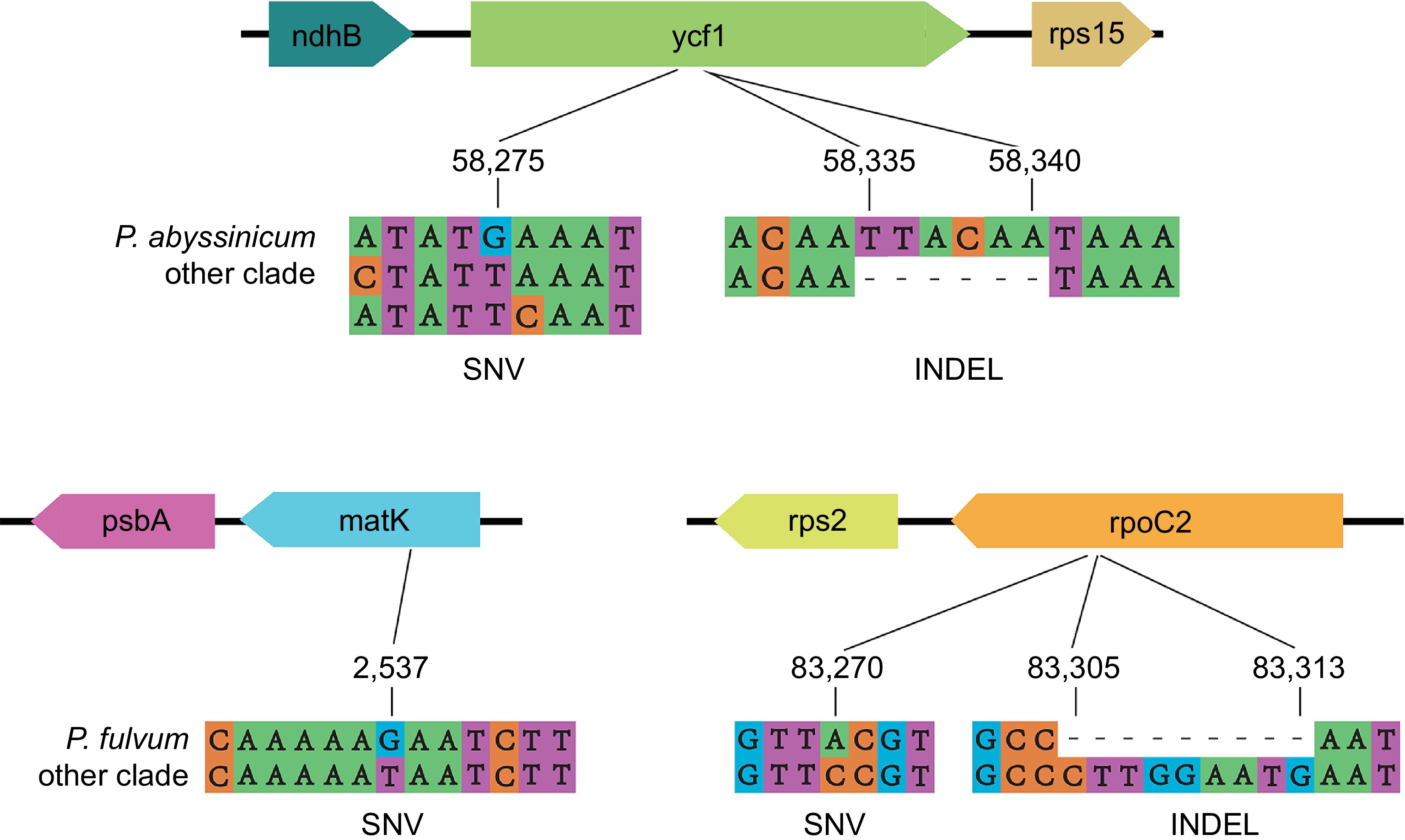
Figure 7.
Examples of variant sites.
-
The present research combined 145 pea plastomes to construct a pan-plastome of peas. Compared to single plastomic studies, pan-plastome analyses across a species or genus provide a higher-resolution understanding of phylogenetic relationships and domestication history. Most plastomes in plants possess a quadripartite circular structure with two inverted repeat (IR) regions and two single copy regions (LSC and SSC)[20−24]. However, the complete loss of one of the IR regions in the pea plastome was observed which is well-known among the inverted repeat-lacking clade (IRLC) species in Fabaceae. The loss of IRs has been documented in detail from other genera such as Erodium (Geraniaceae family)[26,27] and Medicago (Fabaceae family)[28,29]. This phenomenon although not commonly observed, constitutes a significant event in the evolutionary trajectories of certain plant lineages[26]. Such large-scale changes in plastome architecture are likely driven in part by a combination of selective pressures and genetic drift[48]. In the pea pan-plastome, it was also found that, compared to some plants with IR regions, the length of the plastomes was much shorter, and the overall GC content was lower. This phenomenon was due to the loss of one IR with high GC content.
Repetitive sequences are an important part of the evolution of plastomes and can be used to reconstruct genealogical relationships. Mononucleotide SSRs are consistently abundant in plastomes, with many studies identifying them as the most common type of SSR[49−52]. Among these, while C/G-type SSRs may dominate in certain species[53,54], A/T types are more frequently observed in land plants. The present research was consistent with these previous conclusions, showing an A/T proportion exceeding 90% (Fig. 4). Due to their high rates of mutation, SSRs are widely used to study phylogenetic relationships and genetic variation[55,56]. Additionally, like other plants, pea plastome genes have a high frequency of A/Ts in the third codon position. This preference is related to the higher AT content common among most plant plastomes and Fabaceae plastomes in particular with their single IRs[57,58]. The AT-rich regions are often associated with easier unwinding of DNA during transcription and potentially more efficient and accurate translation processes[59]. The preference for A/T in third codon positions may also be influenced by tRNA availability, as the abundance of specific tRNAs that recognize these codons can enhance the efficiency of protein synthesis[60,61]. However, not all organisms exhibit this preference for A/T-ending codons. For instance, many bacteria have GC-rich genomes and thus show a preference for G/C-ending codons[62−64]. This variation in codon usage bias reflects the differences in genomic composition and the evolutionary pressures unique to different lineages.
This study also comprehensively examined the variant loci of the pea pan-plastome. Among these variant sites, some could potentially serve as DNA barcode sites for specific lineages of peas, such as ycf1, rpoC2, and matK. Both ycf1 and matK have been widely used as DNA barcodes in many species[65−68], as they are hypervariable. Researchers now have a much deeper understanding of the crucial role plastomes have played in plant evolution[69−71]. By generating a comprehensive map of variant sites, future researchers can now more effectively trace differences in plastotypes to physiological and metabolic traits for use in breeding elite cultivars.
Evolutionary history
-
The development of a pan-plastome for peas provides new insights into the maternal domestication history of this important food crop. Based on the phylogenetic analysis in this study, we observed a clear differentiation between wild and cultivated peas, with P. fulvum being the earliest diverging lineage, and was consistent with former research[34]. The ML tree (Fig. 5a) indicated that cultivated peas had undergone at least two independent domestications, namely from the PA and PS groups, which is consistent with former research[34]. However, as the present study added several accessions over the previous study and plastomic data was utilized, several differences were also found[34], such as the resolution of the two groups, referred to as PSeI-a group and PSeI-b group which branched between the PA group and PF group. Previous research based on nuclear data[34] only and with fewer accessions showed that the PA group and PF group were closely related in phylogeny, with no PSeI group appearing between them. One possible explanation is that the PSeI-a and PSeI-b lineages represents the capture and retention of a plastome from a now-extinct lineage while backcrossing to modern cultivars has obscured this signal in the nuclear genomic datasets. However, procedural explanations such as incorrectly identified accessions might have also resulted in such patterns. In either case, the presence of these plastomes in the cultivated pea gene pool should be explored for possible associations with traits such as disease resistance and hybrid incompatibility. This finding underscores the complexity of the domestication process and highlights the role of hybridization and selection in shaping the genetic landscape of cultivated peas. As such, future studies integrating data from the nuclear genome, mitogenome, and plastome will undoubtedly provide deeper insights into the phylogeny and domestication of peas. This pan-plastome research, encompassing a variety of cultivated taxa, will also support the development of elite varieties in the future.
-
This study newly assembled 103 complete pea plastomes. These plastomes were combined with 42 published pea plastomes to construct the first pan-plastome of peas. The length of pea plastomes ranged from 120,826 to 122,547 bp, with the GC content varying from 34.74% to 34.87%. The codon usage pattern in the pea pan-plastome displayed a strong bias for A/T in the third codon position. Besides, the codon usage of petB, psbA, rpl16, rps14, and rps18 were shown extremely influenced by natural selection. Three types of SSRs were detected in the pea pan-plastome, including A/T, AT/TA, and AAT/ATT. From phylogenetic analysis, seven well-supported clades were resolved from the pea pan-plastome. The genes ycf1, rpoC2, and matK were found to be suitable for DNA barcoding due to their hypervariability. The pea pan-plastome provides a valuable supportive resource in future breeding and selection research considering the central role chloroplasts play in plant metabolism as well as the association of plastotype to important agronomic traits such as disease resistance and interspecific compatibility.
This study was funded by the Guangdong Pearl River Talent Program (Grant No. 2021QN02N792) and the Shenzhen Fundamental Research Program (Grant No. JCYJ20220818103212025). This work was also funded by the Science Technology and Innovation Commission of Shenzhen Municipality (Grant No. RCYX20200714114538196) and the Innovation Program of Chinese Academy of Agricultural Sciences. We are also particularly grateful for the services of the High-Performance Computing Cluster in the Agricultural Genomics Institute at Shenzhen, Chinese Academy of Agricultural Sciences.
-
The authors confirm contribution to the paper as follows: study conception and design: Wang J; data collection: Kan J; analysis and interpretation of results: Kan J, Wang J; draft manuscript preparation: Kan J, Wang J, Nie L; project organization and supervision: Tiwari R, Wang M, Tembrock L. All authors reviewed the results and approved the final version of the manuscript.
-
The annotation files of newly assembled pea plastomes were uploaded to the Figshare website (https://figshare.com/, doi: 10.6084/m9.figshare.26390824).
-
The authors declare that they have no conflict of interest.
-
# Authors contributed equally: Junhu Kan, Liyun Nie, Meixia Wang
- Supplementary Table S1 List of 145 pea plastomes accessions.
- Supplementary Table S2 Annotated genes in pea pan-plastomes.
- Supplementary Table S3 Dataset of ENC-GC3s plot.
- Copyright: © 2024 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Kan J, Nie L, Wang M, Tiwari R, Tembrock LR, et al. 2024. The Mendelian pea pan-plastome: insights into genomic structure, evolutionary history, and genetic diversity of an essential food crop. Genomics Communications 1: e004 doi: 10.48130/gcomm-0024-0004 

The Mendelian pea pan-plastome: insights into genomic structure, evolutionary history, and genetic diversity of an essential food crop
- Received: 12 August 2024
- Revised: 29 October 2024
- Accepted: 29 October 2024
- Published online: 27 November 2024
Abstract: The Mendelian pea (Pisum sativa), a member of the Fabaceae family, is widely cultivated worldwide as an important food resource. While extensive genetic studies have been conducted on pea, a comprehensive pan-plastome assembly has not yet been achieved. The present study combined 103 newly assembled pea plastomes with 42 previously published plastomes to construct the first pea pan-plastome. The lengths of plastomes varied from 120,826 to 122,547 bp, with an average GC content of 34.8%. Protein-coding genes in the pan-plastome exhibited a strong bias towards A/T in the third codon position, with a notably high frequency of the amino acid arginine (RSCU value = 4.8) among plastome-encoded proteins. Additionally, the codon usage of petB, psbA, rpl16, rps14, and rps18 showed extreme influence from natural selection. Moreover, the genes ycf1, rpoC2, and matK were identified as hypervariable regions, suggesting their potential utility as DNA barcoding loci to distinguish maternal lineages for breeding and other agronomic purpose. The phylogenetic results indicated that cultivated peas had undergone at least two independent domestications, originating from the PA and PS groups. Compared to former research based on nuclear data, the PSeI-a group and PSeI-b group were newly found branched between the PA group and PF group.
-
Key words:
- Pisum sativa /
- Pan-plastome /
- Population structure /
- DNA barcoding