









-
Morus L., commonly known as mulberry trees, is a genus of flowering plants in the family Moraceae. These deciduous trees are widely distributed and/or naturalized in all temperate areas across the globe, also in the mountains of Indonesia and South America[1]. Mulberry trees typically have broad, lobed leaves and produce small, sweet, multiple-fruit aggregates that resemble berries, with the fruits varying in color depending on the species, ranging from white to deep purple-black[2]. As the sole food consumed by the silkworm (Bombyx mori), mulberry represents a vital component of sericulture, which originated more than 5,000 years ago and strongly influenced world history and the expansion of a global economy[3−5]. Mulberry holds potential beyond its use of foliage to feed the silkworm, particularly in the horticultural industry, food industry, and human health applications worldwide[6,7]. Various parts of mulberry trees, including leaves, barks, and fruits, have been utilized in traditional medicine for antioxidant, anti-inflammatory, and antimicrobial properties[8,9]. Beyond medicine, mulberry trees offer lush foliage, colorful seasonal changes, delicious fruit, wildlife attraction, adaptability to diverse environments, graceful form, and rich historical significance, making them a desirable addition to any native landscape[2,10].
Since Linnaeus first established the genus Morus with the description of seven species[11], over 150 Morus species names have been cited in the Index Kewensis to date[12]. However, 17 of these species are currently accepted by the vast majority of botanical authorities[1]. Interspecies hybridization occurs frequently in Morus[4], a phenomenon reported many times in previous studies. Hybridization has been detected between white mulberry (M. alba) and red mulberry (M. rubra), and asymmetrical introgression of these two species directly led to the decline of the local populations of red mulberry[13,14]. Hybridizations have also been documented between M. alba and M. australis[15], M. alba and M. serrata[16], as well as between M. boninensis and M. australis[17]. Frequent hybridization, combined with a wide geographical distribution, a long domestication history, and considerable morphological plasticity, make the taxonomy of Morus challenging[5,18]. With the increasing amounts of genomic data and many newly developed bioinformatic pipelines, more and more low or single-copy nuclear genes and plastomes are available for species identification and phylogeny of different plant lineages[19−22]. Our scientific understanding of Morus has benefited from these innovations, with the phylogeny and taxonomy of Morus successfully resolved[5] based on nuclear and plastid data.
Mitochondria play a vital role in the growth, development, programmed cell death, and male sterility of angiosperms[23,24]. Unlike most plastomes with stable size and gene content, the plant mitochondrial genome (mitogenome) is extremely variable in its size, collinearity, number of repeats, and general structure[25]. The size of the plant mitogenome ranges from 66 kb[26] to nearly 12 Mb[27] across different species, and the mitogenomes exhibit circular, linear, and even complex branching and reticular structures[28,29]. Most changes in the size and structure of mtDNA are thought to be caused by repeat-mediated duplication and recombination[30], and frequent gene transfer between organelle genomes and between organelle and nuclear genomes aggravate these changes[31,32]. Consequently, the assembly and conformational determination of the mitogenome can be more complex and challenging than that of plastomes.
Innovations with the genomics era has brought new opportunities for mitogenome research and promoted the rapid development of mitochondrial genetics and genomics. Different pipelines have been developed to assemble the mitogenome, such as GetOrganelle[33], GSAT[34], and PMAT[35], which facilitated the generation of reliable plant mitogenome assembly graphs. A series of studies have indicated that mitogenomes can also be used for population genetics, molecular ecology, plant phylogeny, and adaptive evolution[36−39]. To date, plastomes from 4,019 genera and 16,435 species have been published based on the Chloroplast Genome Information Resource website (
https://ngdc.cncb.ac.cn/cgir/statistic ), but only 673 plant mitogenomes are reported according to the result of Wang et al.[40]. For the genus Morus, a total of 104 plastomes from 13 species have been assembled. Still, data on the mitogenomes are lacking with only three incomplete mitogenomes involving two species (M. alba and M. notabilis) released in GenBank as of June 2024. This limitation significantly hinders research on the evolution of Morus mitogenomes.The current study sequenced and assembled the complete mitogenomes of three Morus species: M. alba, M. mongolica, and M. notabilis. Using Illumina short reads and PacBio HiFi long reads, the dynamic transformation of mitogenome conformation was investigated across these species. Their genomic characteristics, including gene content, intracellular gene transfer, and organellar phylogeny were also compared. These findings enhance our understanding of the evolution of organelle genomes in Morus species and offer insights into their phylogeny, germplasm resource identification, genetic diversity conservation, and environmental adaptation mechanisms.
-
Fresh leaves from one individual of Morus mongolica were collected from the Baligou scenic area, Henan, China (113°34′25.12″ E, 35°36′26.66″ N), immediately frozen in liquid nitrogen and then stored at −80 °C. Dr. Luxian Liu undertook the identification of M. mongolica, and a voucher specimen was deposited at the Herbarium of Henan University with the accession number LVV20240608. No specific permissions were required for sample collection which are neither privately owned nor protected and the field study did not involve endangered or protected species. Genomic DNA was extracted using a QIAGEN Genomic Kit (Qiagen, CA, USA). Illumina paired-end (PE) reads were generated on the Illumina NovaSeq 6000 (Illumina Inc., San Diego, CA, USA) and long-read sequencing was conducted using the PacBio Revio sequencer (Pacific Biosciences of California, Inc., USA). All of the above sequencing protocols were performed by the Genome Center of NextOmics (Wuhan, China). To further explore the mitogenome evolution of Morus, HiFi data and Illumina data of two M. alba cultivars and one M. notabilis were downloaded from the National Genomics Data Center (Supplementary Table S1).
Assembly and annotation of organelle genomes
-
For the plastome assembly, GetOrganelle v1.7.5.1[33] was used to assemble the Illumina paired-end reads with default parameters directly. For the mitogenome assembly, the 'autoMito' module in PMAT v1.5.1[35] software was utilized to conduct de novo assembly of the PacBio HiFi data derived from Morus species. Next, 24 conserved plant mitochondrial protein-coding genes (PCGs) from the 15 representative species were used to identify the draft mitogenome by the BLASTn v2.2.30+[41] based on the assembled contigs. Then, the mitogenome graph was simplified using Bandage v 0.8.1[42] and the plastid or nuclear contigs removed. The Illumina paired-end reads were mapped to the mitogenome using CLC Genomics Workbench 12 (CLC Inc. Aarhus, Denmark) to improve assembly accuracy.
The plastomes of Morus were annotated in Geneious R11[43], and putative starts stops, and intron positions of genes were corrected by comparison with homologous genes of the M. alba plastome (GenBank accession number: OQ644502). The mitogenomes were annotated with the IPMGA webserver (
www.1kmpg.cn/mga ), and all annotations were further manually checked and corrected for start and stop codons of PCGs using Geneious R11. The tRNA genes were identified using tRNAscan-SE v1.21[44]. Finally, the plastome and mitogenome maps were generated using the OrganellarGenomeDRAW tool (OGDRAW v1.3.1)[45] followed by manual modification.Repeat-mediated homologous recombination prediction
-
In the initial assembly, there were multiple repeats present in the mitogenomes, which resulted in different mitogenome conformations for Morus species. Therefore, the following strategy for each repeat was used to validate the potential conformations of the mitogenome. First the sequences of the repetitive elements and their flanking regions of 1 kb from the genome assembled by PMAT were extracted to ensure that long reads can completely cross the repetition. Subsequently, the long reads were mapped to the above extracted sequences using BWA v0.7.17[46], and the alignment file was converted to BAM format using SAMtools v1.13[47]. Finally, each long read was carefully checked using Tablet[48] and the number of long reads that completely crossed the repeat region counted. The conformation with the largest number of long reads was defined as the dominant conformation. For the branch nodes, a 2 kb region on each side of the node was extracted as the reference sequence and the BAM file obtained was visualized using the above method by IGV v2.17.4[49]. The depth at the node was calculated and the path with higher depth selected as the dominant conformation.
Analysis of RSCU and repeated sequences
-
The PCGs of the mitogenome were extracted using PhyloSuite v1.2.3[50], and the relative synonymous codon usage (RSCU) in protein coding sequences of Morus was investigated using the program CodonW v 1.4.2 (
http://codonw.sourceforge.net/ ).Forward, reverse, palindromic, and complementary repeats were detected using the online REPuter software (
https://bibiserv.cebitec.uni-bielefeld.de/reputer )[51] with maximum computed repeats, minimal repeat size, and hamming distance set to 5,000, 30, and 3 respectively. The tandem repeats with longer than 6 bp repeat units were identified using Tandem Repeats Finder v.4.09 (http://tandem.bu.edu/trf/trf.submit.options.html )[52] with default parameters. Simple sequence repeats (SSRs) were detected using MISA (https://webblast.ipk-gatersleben.de/misa/ )[53], and a threshold of ten, five, four, three, three, and three repeat units for mono-, di-, tri-, tetra-, penta-, and hexanucleotide SSRs, were applied respectively.Identification of mitochondrial plastid DNA (MTPT)
-
To identify plastid-derived DNA fragments in the mitogenome, the plastome and mitogenome sequences were compared for each Morus species using BLASTn[41] with the following parameters: -evalue 1e-5, -word_size 9, -gapopen 5, -gapextend 2, -reward 2, -penalty -3, and -dust no. The BLASTn results were visualized using the Circos package[54], and the identified MTPTs were also annotated using the IPMGA webserver.
Phylogenetic inference of Rosales
-
In phylogeny inference, 40 Rosales accessions including four Morus accessions analyzed in this study, and another 36 Rosales species downloaded from GenBank were selected to reconstruct phylogenetic trees with two Cucurbitales species selected as the outgroups (Supplementary Table S2). To minimize the impact of missing data or insufficiently informative loci on phylogenetic inference, the minimum number of species that share PCGs was set to 35. Maximum likelihood (ML) and Bayesian inference (BI) were performed to estimate the phylogeny based on all shared PCGs. PARTITIONFINDER v2.1.1[55] was used to determine the optimal nucleotide substitution models. ML analysis was performed using RAxML v8.1.11[56] with 1000 rapid bootstrap iterations and the default settings for other parameters. BI analysis was carried out using MrBayes v3.2.3[57], and the posterior probability was estimated using four chains running 5,000,000 generations sampling every 1000 generations. Convergence of the MCMC chains was assumed when the average standard deviation of split frequencies reached 0.01 or less, and the first 25% of sampled trees were considered burn‐in trees.
-
The mitogenomes of Morus species showed a complex assembly graph mediated by different numbers of long repeats (LRs) or node branches based on long-reads data (Fig. 1). One pair of LRs existed in each of the mitogenome of M. notabilis and M. alba 'Zhongsang5801' (M. alba-ZS5801; Fig. 1a, c), three pairs of LRs occurred in M. alba 'Zhenzhubai' (M. alba-ZZB; Fig. 1d), and LRs-mediated multiple conformations were not detected in M. mongolica (Fig. 1b). These repeats were resolved by artificially simulating four possible paths and making judgments based on the mapping results of long reads, and the dominant conformation of the mitogenome for Morus species was obtained by using this strategy. For M. notabilis, the repeat sequence (LR1) with 9,951 bp length mediated genomic recombination and formed one master circular structure or two small circular structures with equal probability (Fig. 2a), and here the master circle representing the complete mitogenome was used for subsequent analysis. In the mitogenome of M. mongolica, the obtained complex structure was disassembled into a linear molecule and a circular molecule after calculating the depth at the node (Fig. 2b). The two cultivars of M. alba showed two completely different mitogenomic conformations, the genomic recombination mediated by one pair of LRs (LR1) resulted in the mitogenome disassembled into a linear molecule and a circular molecule in the cultivar 'Zhongsang5801' (Fig. 2c), and three pairs of LRs (LR1, LR2, and LR3) occurred in the mitogenome of the cultivar 'Zhenzhubai' which divided the complex structure into three circular molecules and one linear molecule (Fig. 2d). Finally, the paired-end reads and long reads were mapped to the mitogenomes, and the statistics of the sequencing depth showed that a gap-free genome assembly of high quality was obtained (Supplementary Fig. S1).
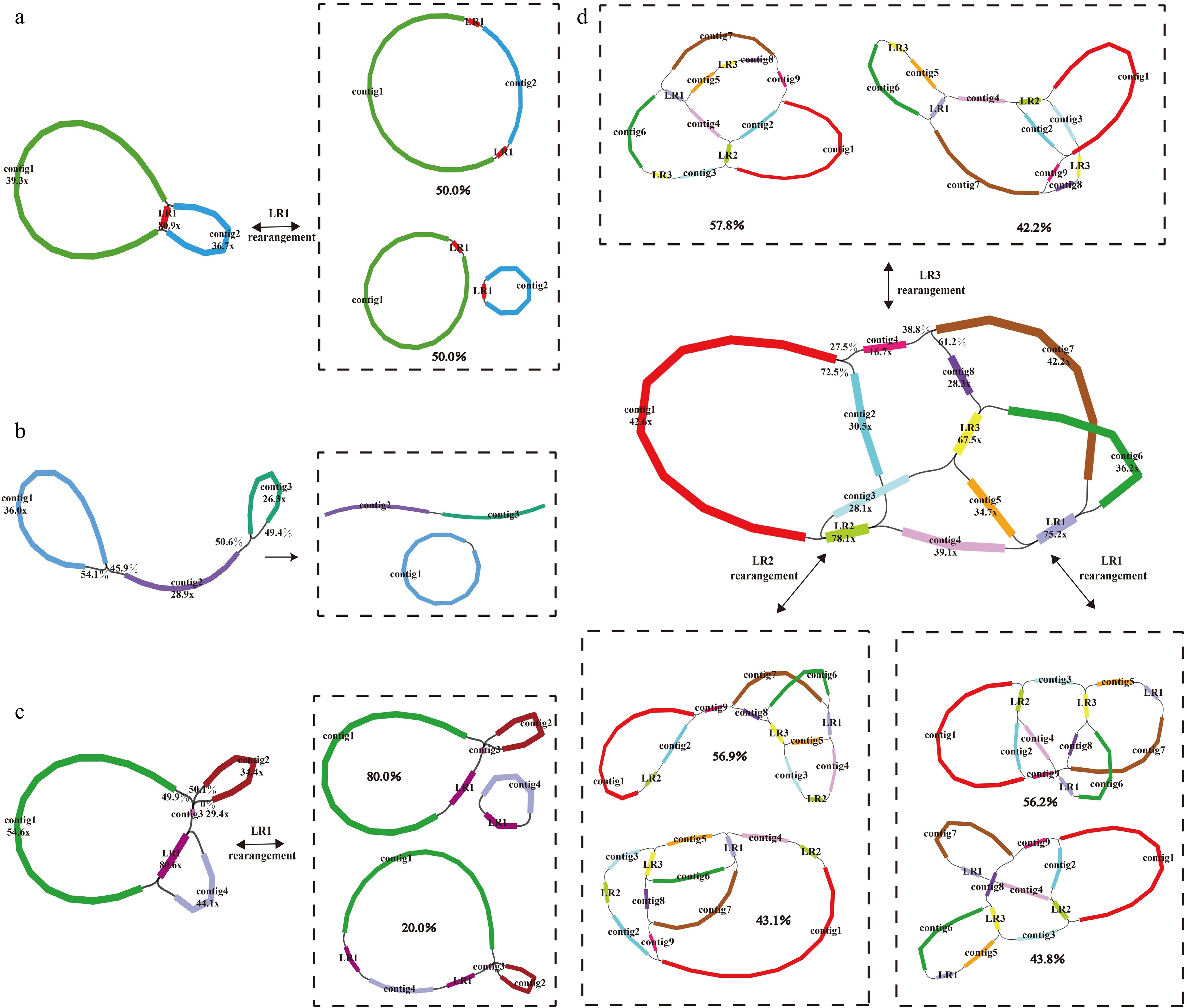
Figure 1.
Mitogenome assembly graph and possible connections (black lines) mediated by repeats for (a) M. notabilis, (b) M. mongolica, (c) M. alba-ZS5801, and (d) M. alba-ZZB. Each colored segment is labeled with its name and coverage. The boxes represent the different mitogenome conformations mediated by each pair of repeats and the probability of being supported.
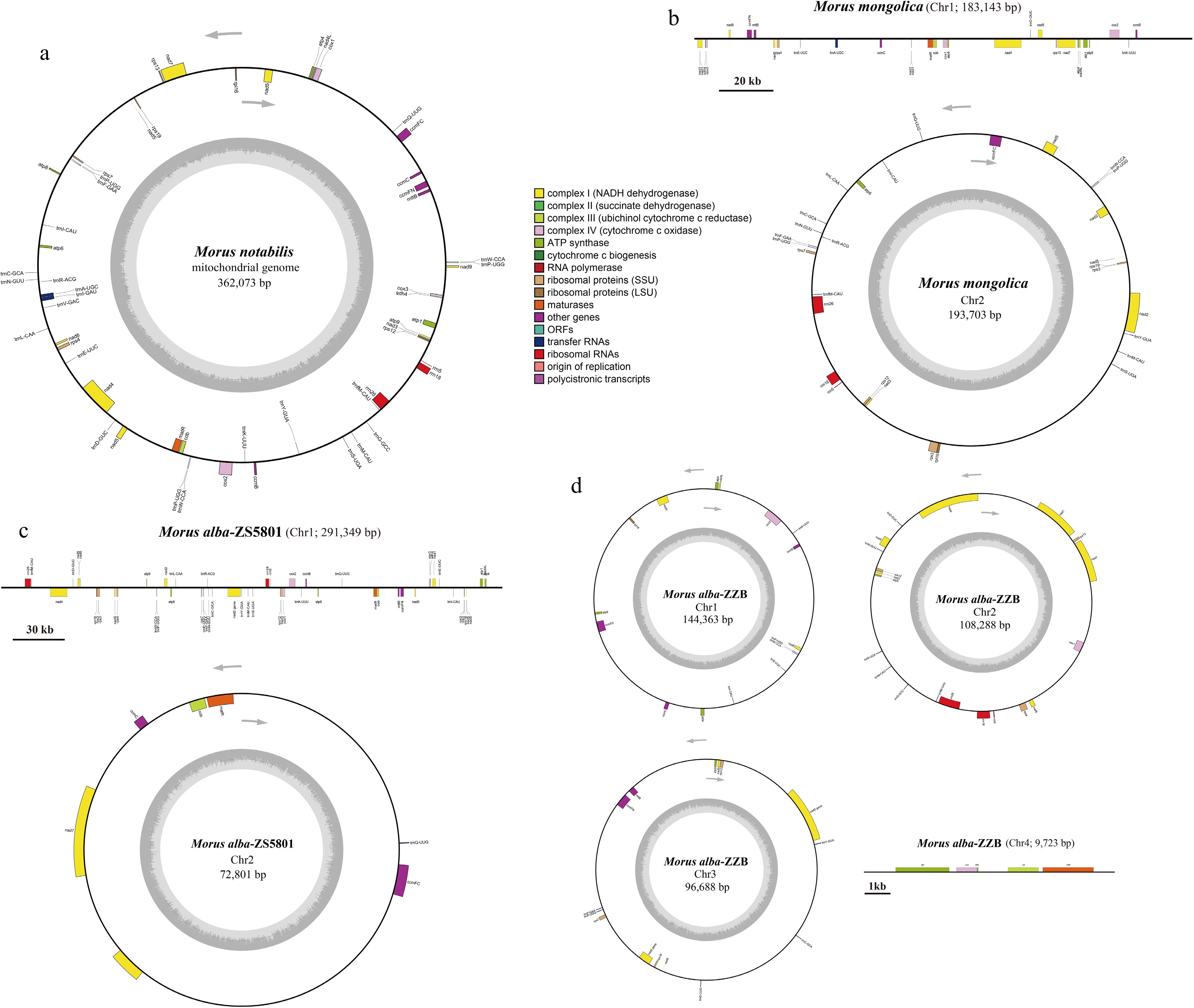
Figure 2.
The mitogenome map of Morus species. (a) M. notabilis, (b) M. mongolica, (c) M. alba-ZS5801, and (d) M. alba-ZZB. Genes belonging to different functional groups are color-coded.
The complete mitogenomes of the four Morus accessions ranged from 359,062 bp (M. alba-ZZB) to 376,846 bp (M. mongolica), revealing a large interspecific variation among different Morus species and cultivars (Fig. 2). The GC content for the four mitogenomes varied slightly from 45.4% (M. alba-ZS5801) to 45.7% (M. notabilis). The four Morus mitogenomes encoded a different set of genes with the number ranging from 49 to 55, including 31−32 PCGs, 15−20 tRNA genes and three rRNA genes (Supplemental Fig. S2, Table 1). Among these genes, 30 PCGs, 14 tRNA genes, and all three rRNA genes (rrn5, rrn18, and rrn26) were shared by all four mitogenomes. Seven PCGs including ccmFC, cox2, nad1, nad2, nad4, nad5, nad7, and two tRNAs (trnA-UGC, trnI-GAU) contained more than one intron (Table 1). For PCGs, rps3 and rps13 were lost in M. alba-ZZB and M. alba-ZS5801 respectively, three PCGs including atp9, nad3, and rps12 had two copies in M. alba-ZZB and two genes cob and matR had also two copies in M. alba-ZS5801. For tRNA genes, three, five, and four tRNA genes were missing in M. mongolica, M. alba-ZZB, and M. alba-ZS5801 respectively, only trnQ-UUG had two copies in M. alba-ZS5801 and two tRNA genes (trnP-UGG, trnW-CCA) had one more copy in M. notabilis compared with the other three genomes.
Table 1. Summary of genes contained in the four Morus mitogenomes.
Group of genes M. notabilis M. mongolica M. alba-ZS5801 M. alba-ZZB Core genes ATP synthase atp1, atp4, atp6, atp8, atp9 atp1, atp4, atp6, atp8, atp9 atp1, atp4, atp6, atp8, atp9 atp1, atp4, atp6, atp8, atp9 (2) Cytochrome c biogenesis ccmB, ccmC, ccmFC*, ccmFN ccmB, ccmC, ccmFC*, ccmFN ccmB, ccmC, ccmFC*, ccmFN ccmB, ccmC, ccmFC*, ccmFN Ubichinol cytochrome c reductase cob cob cob (2) cob Cytochrome c oxidase cox1, cox2*, cox3 cox1, cox2*, cox3 cox1, cox2*, cox3 cox1, cox2*, cox3 Maturases matR matR matR (2) matR Transport membrane protein mttB mttB mttB mttB NADH dehydrogenase nad1*, nad2****, nad3, nad4***, nad4L, nad5**, nad6, nad7****, nad9 nad1*, nad2***, nad3, nad4***, nad4L, nad5**, nad6, nad7****, nad9 nad1*, nad2***, nad3, nad4***, nad4L, nad5**, nad6, nad7****, nad9 nad1*, nad2***, nad3 (2), nad4***, nad4L, nad5**, nad6, nad7****, nad9 Variable genes Large subunit of ribosome rpl16 rpl16 rpl16 rpl16 Small subunit of ribosome rps3*, rps4, rps7, rps12, rps13, rps19 rps3, rps4, rps7, rps12, rps13, rps19 rps3, rps4, rps7, rps12, rps19 rps4, rps7, rps12 (2), rps13, rps19 Succinate dehydrogenase sdh4 sdh4 sdh4 sdh4 rRNA genes Ribosomal RNAs rrn5, rrn18, rrn26 rrn5, rrn18, rrn26 rrn5, rrn18, rrn26 rrn5, rrn18, rrn26 tRNA genes Transfer RNAs trnA-UGC*, trnC-GCA, trnD-GUC, trnE-UUC, trnF-GAA, trnfM-CAU, trnG-GCC, trnI-CAU, trnI-GAU*, trnK-UUU, trnL-CAA, trnM-CAU, trnN-GUU, trnP-UGG (3), trnQ-UUG, trnR-ACG, trnS-UGA, trnV-GAC, trnW-CCA (2), trnY-GUA trnA-UGC*, trnC-GCA, trnD-GUC, trnE-UUC, trnF-GAA, trnfM-CAU, trnI-CAU, trnK-UUU, trnL-CAA, trnM-CAU, trnN-GUU, trnP-UGG (2), trnQ-UUG, trnR-ACG, trnS-UGA, trnW-CCA, trnY-GUA trnC-GCA, trnD-GUC, trnE-UUC, trnF-GAA, trnfM-CAU, trnI-CAU, trnK-UUU, trnL-CAA, trnM-CAU, trnN-GUU, trnP-UGG (2), trnQ-UUG (2), trnR-ACG, trnS-UGA, trnW-CCA, trnY-GUA trnC-GCA, trnD-GUC, trnE-UUC, trnF-GAA, trnfM-CAU, trnG-GCC, trnI-CAU, trnK-UUU, trnM-CAU, trnN-GUU, trnP-UGG (2), trnQ-UUG, trnS-UGA, trnW-CCA, trnY-GUA The number of asterisks indicate the number of introns that the genes contained. The number in the parentheses indicate the number of copies of the genes. Repeat sequences analysis
-
The dispersed repeats within each Morus mitogenome were identified as two major types, including 105 (M. notabilis)−141 (M. mongolica) forward and 103 (M. notabilis)−125 (M. alba-ZZB) palindromic repeats and one complementary repeat was found in three mitogenomes except M. notabilis (Fig. 3a). No reverse repeat sequences were observed in any of the four mitogenomes. These repeats ranged from 30 to 22,006 bp (eight were longer than 1 kb) (Supplementary Table S3), with most distributed in non-coding regions and some found in PCGs such as ccmFC, nad2, nad4, nad5, nad7, cob, cox2, matR, and atp9. Additionally, 13−16 tandem repeats with repeat lengths ranging from 12−39 bp were detected in the four genomes and most of these tandem repeats had a length of 15−25 bp (Fig. 3a; Supplementary Table S4). Moreover, 91 (M. notabilis)-109 (M. alba-ZS5801) microsatellites were detected across the four Morus mitogenomes, comprising of 34−48 mono-, 10−15 di-, 11−16 tri-, 29−31 tetra-, 1−2 penta-, and 1−2 hexanucleotide repeat motifs (Fig. 3b).
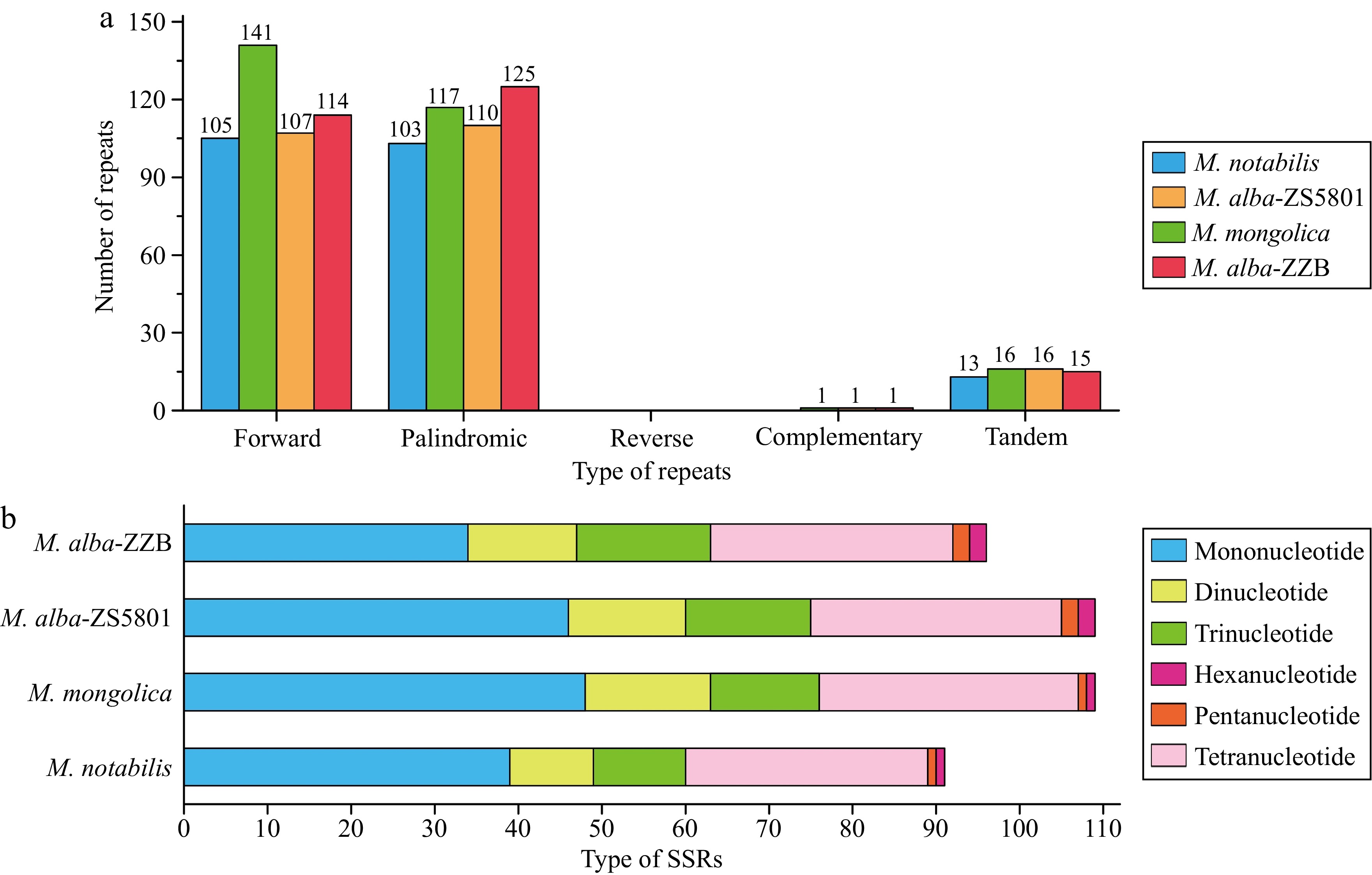
Figure 3.
Repeat sequences in the Morus mitogenomes. (a) Type and number of dispersed repeats and tandem repeats, (b) type and number of simple sequence repeats (SSRs).
Codon bias analysis of mitogenome
-
Codon usage was analyzed in the 32 PCGs of the M. notabilis and M. mongolica mitogenomes, 33 PCGs of M. alba-ZS5801, and 34 PCGs of M. alba-ZZB. A total of 9,619, 9,629, 10,552, and 9,617 codons were identified and a high similarity in codon usage and amino acid frequencies was observed in all four mitogenomes (Fig. 4; Supplementary Table S5). Most of the PCGs used AUG as the start codon, while GUG and AUA were found to be the start codon for rpl16 and nad1 respectively in all four accessions. Three stop codons including UAA, UAG, and UGA were identified in the PCGs, but UAA with RSCU > 1 was the most preferred codon. For the codons of encoding amino acids, there were 31 (M. notabilis), 30 (M. mongolica), 33 (M. alba-ZS5801), and 31 (M. alba-ZZB) codons with RSCU > 1, indicating a higher frequency of these codons compared to other synonymous codons. Among them, the GCU codon for Ala had the highest RSCU value (1.58−1.64), as well as the CAG codon for Gln had the smallest one with an RSCU value ranging from 0.44 to 0.47.
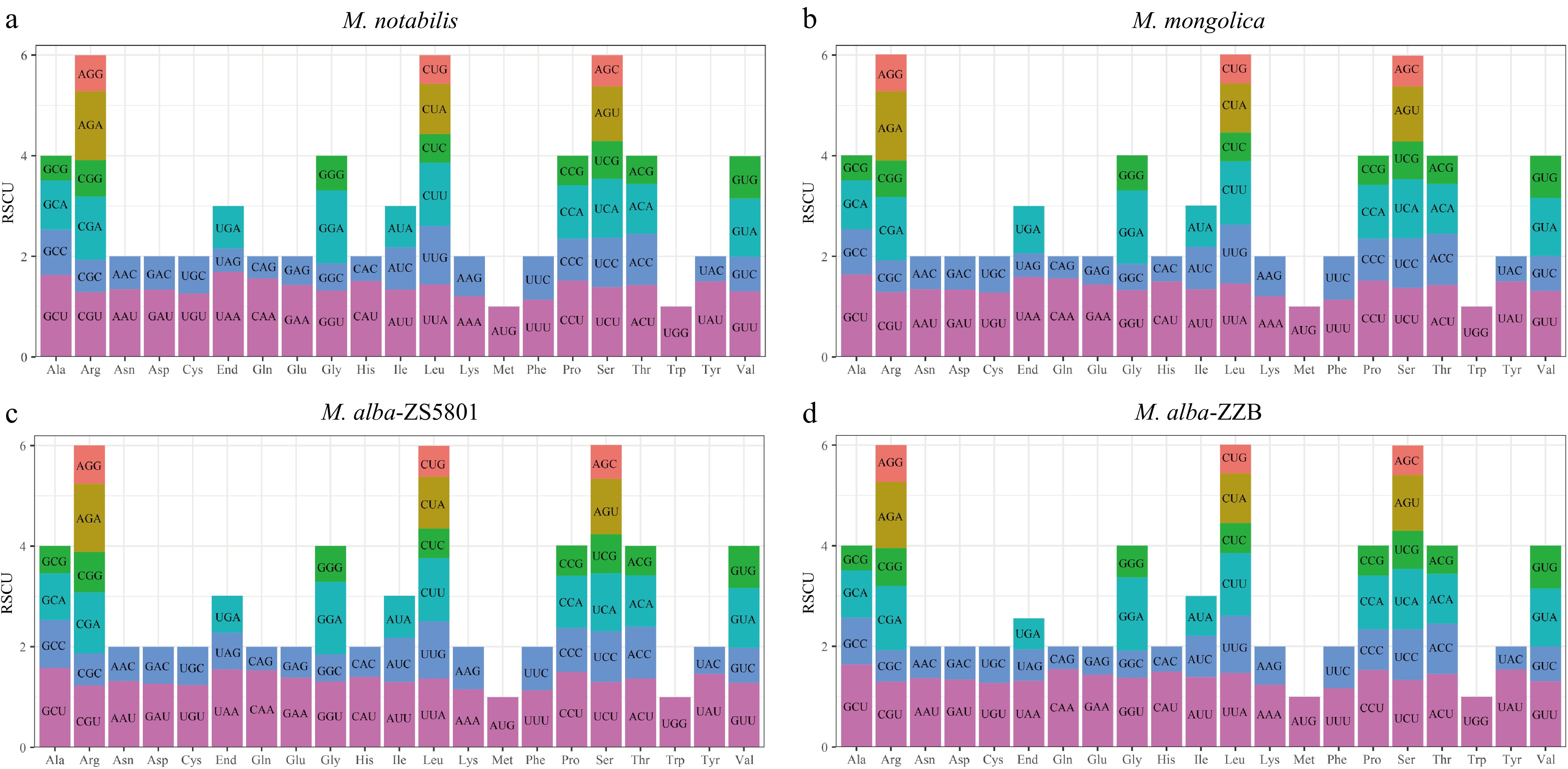
Figure 4.
Relative synonymous codon usage (RSCU) in the four Morus mitogenomes. (a) M. notabilis, (b) M. mongolica, (c) M. alba-ZS5801, and (d) M. alba-ZZB respectively.
Plastid DNA transfer to mitogenome (MTPTs)
-
The plastomes of the four Morus accessions assembled in this study varied slightly in length, ranging from 159,136 bp (M. mongolica) to 159,546 bp (M. notabilis; Fig. 5). To eliminate redundant detections, only a single IR region of each plastome was used for analysis, and 38, 39, 35, and 40 mitochondrial-plastid DNA transfers (MTPTs) were identified in the mitogenomes of M. notabilis, M. mongolica, M. alba-ZS5801, and M. alba-ZZB, respectively (Fig. 5; Supplementary Table S6). These MTPTs had a combined length of 26,024 bp (M. notabilis), 21,398 bp (M. mongolica), 17,698 bp (M. alba-ZS5801), and 10,112 bp (M. alba-ZZB) accounting for 7.19%, 5.68%, 4.86%, and 2.82% of the mitogenomes. Among these MTPTs, the majority of transferred fragments for each accession were shorter than 1000 bp and there was a total of 14 MTPTs longer than 1,000 bp in all the mitogenomes, both the longest (16,238 bp) and shortest (30 bp) MTPTs were found in M. notabilis. These MTPTs were then extracted and annotated. Five intact tRNA genes were discovered (trnP-UGG, trnW-CCA, trnD-GUC, trnM-CAU, and trnN-GUU) and were identified as being transferred from plastomes to the mitogenomes for all accessions. Two intact tRNA genes including trnL-CAA and trnR-ACG transferred to three of the four mitogenomes except M. alba-ZZB, trnI-GAU, and trnV-GAC were only transferred in M. notabilis as well as trnA-UGC was transferred in M. notabilis and M. mongolica. In addition, trnF-GAA and trnQ-UUG might have experienced rapid sequence divergences during the migration process due to partial gene fragments identified in the four mitogenomes. For PCGs, only some fragments of rps12 were transferred to the four mitogenomes.
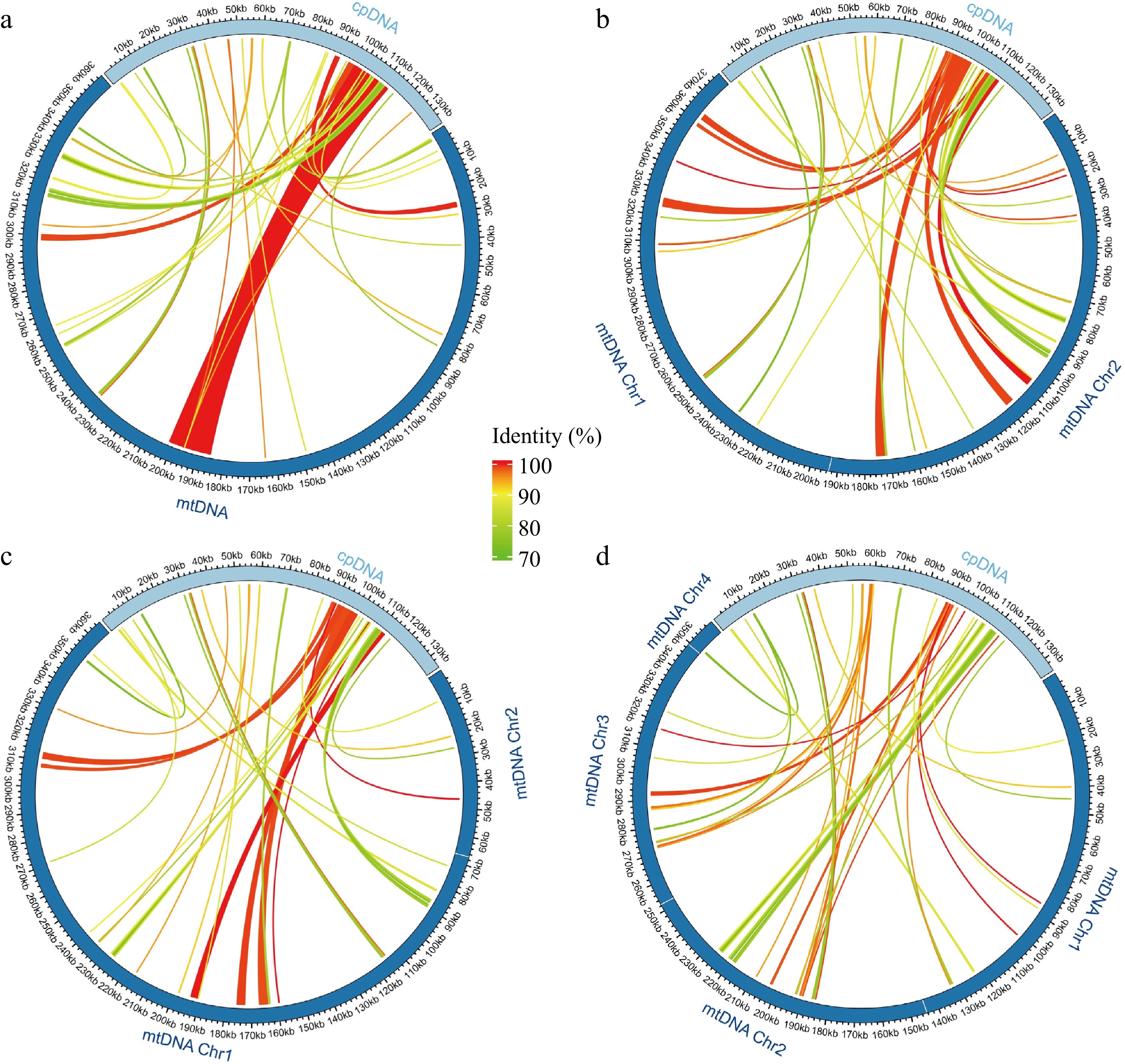
Figure 5.
Schematic representation of the distribution of MTPTs between plastome and mitogenomes for (a) M. notabilis, (b) M. mongolica, (c) M. alba-ZS5801, and (d) M. alba-ZZB. The MTPTs on the plastid IR regions were counted only once. Different colors of ribbons represent different identities. The length of each MTPT and the genes it contains can be found in Supplemental Table S6.
Phylogenetic analyses
-
For the phylogenetic inference, 25 individual gene alignments of PCGs were concatenated to generate a combined matrix comprising 42 accessions with a length of 24,627 bp. The best-fit nucleotide substitution model determined for the concatenated matrix was GTR + I + G. The tree topologies reconstructed by ML and BI analyses were completely consistent (Fig. 6). In the phylogenetic trees, most nodes were strongly supported with maximal ML bootstrap (BS) or Bayesian posterior probability (PP), and all the species were grouped into six families with different species of the same genus forming monophyletic clades. In the Rosales, the six families were divided into two clades with maximal support (BS/PP = 100/1). Two subfamilies of Rosaceae, Amygdaloideae, and Rosoideae, comprised one clade. In the other clade (BS/PP = 100/1), the family Rhamnaceae was the first diverging lineage, followed by a clade comprising two Hippophae species from the family Elaeagnaceae. For the remaining three families, Moraceae and Cannabaceae first formed a monophyletic group (BS/PP = 100/1) which was subsequently sister to the family Ulmaceae. Within the family Moraceae, ten species from five genera grouped into two clades. Four individuals of three Morus species formed one of the clades (BS/PP = 100/1), M. notabilis was firstly diverged and two individuals of M. alba were not resolved as monophyletic. Ficus carica occupied the basal position of the other clade with moderate support (BS/PP = −/0.76), followed by a maximal supported clade (BS/PP = 100/1) comprising of Malaisia scandens and Allaeanthus kurzii sister then to the genus Broussonetia. For the three Broussonetia species, B. papyrifera was well-supported to be sister to the B. kaempferi and B. monoica clade.
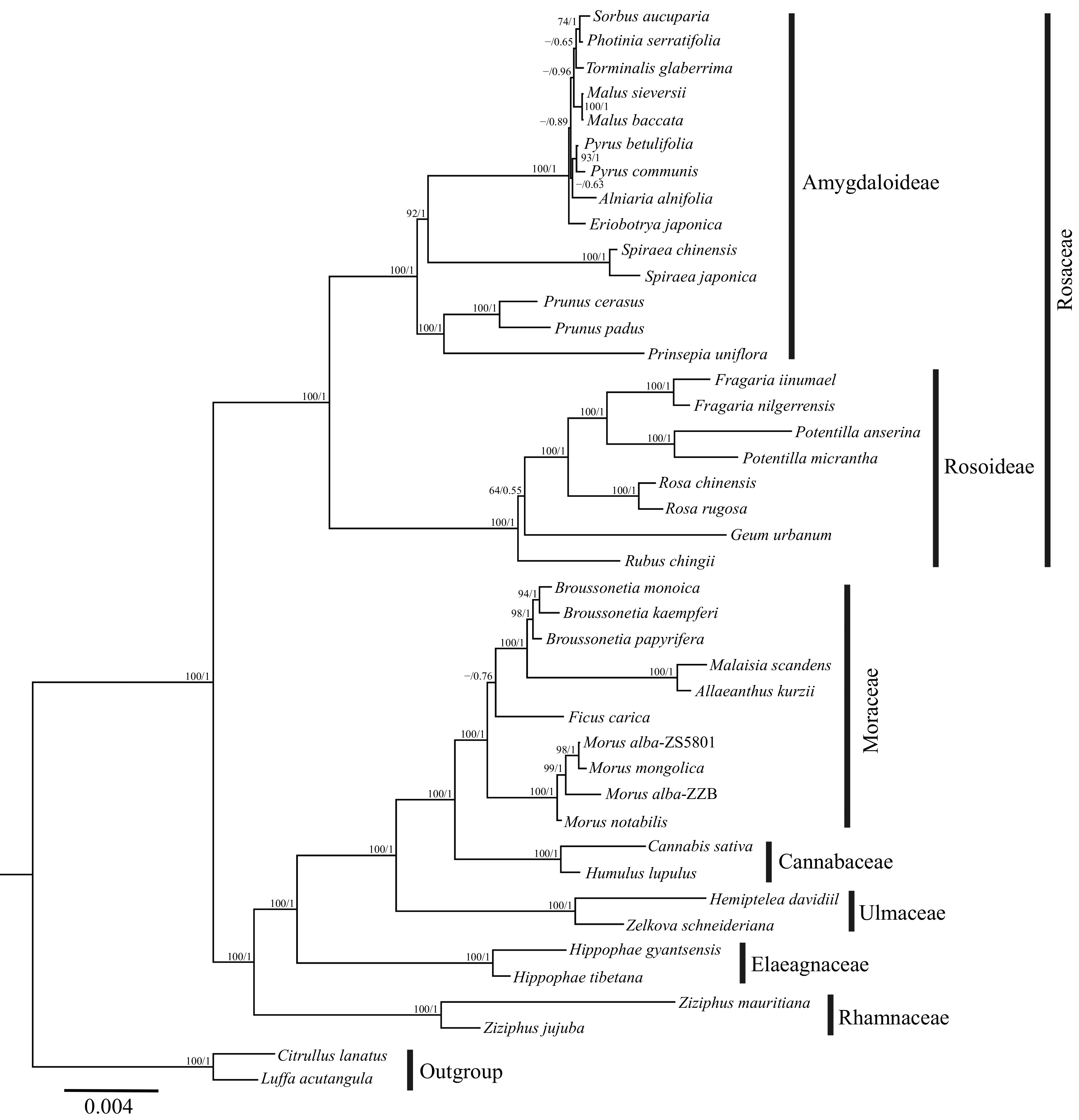
Figure 6.
Phylogenetic trees of Rosales estimated using RA × ML from 25 PCGs of the mitogenomes. Numbers above the branches represent ML bootstrap/Bayesian posterior probability (BS/PP), ML bootstrap values with lower than 50 are indicated by the dash.
-
Compared to the mitogenome of animals and the plastome of plants, the plant mitogenome underwent dramatic structural changes during evolution, which involve huge genome size variations, genomic rearrangements, repeat-mediated recombination, low gene density, and gene transfer[58,59]. Due to the presence of repeat-mediated recombination, plant mitogenomes usually exhibit multiple conformations[59,60], including circular (e.g. Crataegus spp.)[24], linear and branched (e.g. Lactuca spp.)[29], and numerous smaller circular molecules (e.g. Actinidia chinensis)[61]. Previous studies have reported that the variations in mitogenomes are not only between plant species but also can be within the same species[62,63], and even plant mtDNA may simultaneously exist in different genome conformations within the same individual[64]. Given the importance of the plant mitogenome in understanding the phylogeny, germplasm resource identification, genetic diversity conservation and adaptive evolution, four representative accessions of three Morus species were sequenced and comparatively analyzed in this study.
The complete mitogenome sequences of the four Morus accessions ranged from 359,062 to 376,846 bp in length, and encoded a different set of genes with the number ranging from 49 to 55 (Fig. 2). The size and gene content of mitogenomes within Morus varied more than that of plastomes sequenced in this study and compared with those published by a previous study[5]. Upon further confirmation, it was found that the variation of mitogenome size of Morus species was mainly caused by the impact of repetitive sequences within it (Fig. 3), rather than foreign sequences transferred from the plastomes (Fig. 5). Compared with the reported mitogenomes of other Moraceae species, the relatively medium size of Morus was larger than that of Broussonetia (276,967−325,822 bp)[65] but smaller than Ficus (480,902 bp)[66].
Previous studies have shown that the plant mitogenome has a dynamic structure due to recombination events that are commonly facilitated by repetitive sequences dispersed across the mitochondrial DNA[67−69]. In the assembly of the Morus mitogenomes, repeat-mediated recombination activity was also found. One or three repeat sequences were confirmed that mediated recombination and generated multiple genome conformations in Morus (Fig. 1). Back mapping of long reads to these repeats verified the different conformations for each accession and that the varied conformations did indeed exist simultaneously. The existence of these various conformations and structural variations in Morus mitogenomes may potentially influence their evolutionary dynamics, and their ability to adapt to environmental changes, and can also offer increased flexibility for interactions with the nuclear genome.[70] Although the repeat-mediated recombination events combined with several gene transfer events from plastomes resulted in a dramatic genome structure rearrangement (Fig. 1; Supplemental Fig. S3), the remaining 30 PCGs were shared across the four Morus mitogenomes with the exception of rps3 being lost in M. alba-ZZB and rps13 in M. alba-5801 (Supplemental Fig. S2). It is worth noting that the five exons of two PCGs including nad1 and nad5, which encode subunits 1 and 5 of the NADH-ubiquinone oxidoreductase complex were annotated into different genomic molecules, suggesting a layered recombination and gene loss mechanism for the creation of such unique mixed genome-derived chimeric genes. The mechanism by which these genes function normally would require deeper study.
Phylogenetic relationships of Rosales
-
For the past three decades, plant systematists have used DNA sequences to characterize and classify plant diversity, which have had a profound influence on our understanding of plant phylogenetic relationships and patterns of diversification[71]. With the increasing availability of genomic resources held in public databases and many newly developed pipelines for identifying orthologous genes (OrthoFinder, HybPiper)[72,73] or assembling plastomes (NOVOPlasty, GetOrganelle)[33,74], a large number of plastomes[75,76] and nuclear genes[5,21] have been used in phylogenomic inference for different plant lineages. Unlike plastomes or nuclear genes, the mitogenome is rarely used in phylogenetic inference mainly due to the slower substitutional rates of genes and difficult mitogenome assembly caused by frequent genomic rearrangements and foreign gene transfer[77,78]. However, the advent of third-generation sequencing and the development of fast and accurate software for assembling mitogenomes have promoted mitogenomes for phylogenetic analyses at higher taxonomic levels[24,64,65].
Here, the 40 species of Rosales including six of the nine families were selected for phylogenetic inference, representing the first use of mitogenomes to investigate the phylogenetic relationships of Rosales. In Rosales, the six families were divided into two clades based on ML and BI methods (Fig. 6). Two subfamilies of Rosaceae including Amygdaloideae and Rosoideae comprised one clade, which was consistent with the previous classification results of Rosaceae using plastid genes and nuclear genes[79,80]. However, the phylogenetic position of Dryadeae cannot be confirmed at the mitochondrial level due to the lack of samples from this subfamily. The remaining five families formed the other clade and the family Rhamnaceae was recovered as the first diverging lineage within it (Fig. 6), which was the most striking difference with previous studies. Whether phylogenetic analyses relied on a small number of plastid genes[81,82], whole plastomes[75,83], or even numerous nuclear genes[22,84], the phylogenetic analyses all showed that the family Rhamnaceae was resolved as a sister to the family Elaeagnaceae and the clade was subsequently sister to a clade including Moraceae, Cannabaceae and Ulmaceae. As is known, the plastid and mitogenomes typically are both maternally inherited in angiosperms[85,86], but the different placement of Rhamnaceae from mitochondrial and plastid data suggest that the evolutionary histories of these two subcellular compartments are unlinked. However, the biparental inheritance of organellar genomes has been documented[87,88], and paternal inheritance of plastomes has been reported in Turnera ulmifolia[89] and Medicago sativa[90], which were closely related to Rosales in the phylogenetic tree[21]. Therefore, it was deduced that the identical placement of Rhamnaceae in nuclear and plastid phylogenies due to the paternal inheritance of plastomes of this family, and the conflicting topologies of Rhamnaceae in plastid and mitochondrial phylogenies were mainly caused by ancient hybridization. The above speculation however needs to be further confirmed via expanded sampling of Rhamnaceae in future work. It is worth noting that the two cultivars of M. alba, 'Zhongsang5801' and 'Zhenzhubai', did not cluster together due to the embedding of M. mongolica, suggesting frequent hybridization in Morus species. This result was also recovered in the previous study based on plastome data[5].
-
In the present study, four high-quality mitogenomes of M. notabilis, M. mongolica, M. alba 'Zhongsang5801' and M. alba 'Zhenzhubai' were assembled based on a hybrid strategy, and various aspects explored including genome structure, gene transfer, and phylogenetic implications. The mitogenomes were structurally heterogeneous across four Morus accessions and multiple genome conformations existed simultaneously for each species. The gene content was relatively conserved especially for PCGs, but the repeat sequences and foreign gene transfer sites varied widely in the four mitogenomes. The phylogeny of Rosales was investigated based on mitogenome sequences, the phylogenetic position of Rhamnaceae showed a strong difference compared to earlier plastid and nuclear phylogenies, which provided us with new insights into the evolution of Rosales.
This work was financially supported by the Xinyang Academy of Ecological Research Open Foundation (Grant No. 2023DBS09), the Henan Province Science and Technology Research Project (NO. 232102320272), the Henan Province Major Research Fund of Public Welfare (Grant No. 201300110900), and the Key Technology Research and Development Program of Zhejiang Province (Grant No. 2023C03138).
-
The authors confirm contribution to the paper as follows: conceptualization, project administration, resources, supervision: Liu L, Li P; data curation: Long Q, Qian J, Shi Y; formal analysis, investigation: Long Q, Lv W; methodology: Qian J, Shi Y; writing – original draft: Long Q, Lv W; writing - review and editing: Liu L, Egan AN, Li P; funding acquisition: Liu L. All authors reviewed the results and approved the final version of the manuscript.
-
The raw sequencing reads of the HiFi and Illumina for M. mongolica mitogenome and plastome assembly have been deposited in the NCBI database with BioProject IDs PRJNA1141701 and PRJNA1140905, and the data are available under the accession numbers SRR30031178 and SRR30012719. The mitogenome and plastome sequences of the four Morus accessions have been submitted to the NCBI database, and the GenBank accession numbers for mitogenomes are shown in Supplemental Table S2, while the GenBank accession numbers for M. notabilis, M. mongolica, M. alba-ZS5801 and M. alba-ZZB are PQ083379, PQ083380, PQ083381, and PQ083382, respectively.
-
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
-
# Authors contributed equally: Luxian Liu, Qian Long
- Supplementary Table S1 The detail information of public data downloaded from the National Genomics Data Center for mitogenome assembly in this study.
- Supplementary Table S2 The GenBank numbers of mitogenome for phylogenetic inference used in this study.
- Supplementary Table S3 Statistics of dispersed repeats in four Morus mitogenomes.
- Supplementary Table S4 The detailed information of tandem repeats distributed in the four Morus mitogenomes.
- Supplementary Table S5 Frequency of amino acids and relative synonymous codon usage (RSCU) in the four Morus mitogenomes.
- Supplementary Table S6 The BLASTn results searched between the plastome and mitogenome for the four Morus species.
- Supplementary Fig. S1 Sequencing depth (count of mapping reads) of Morus mitogenomes in this study. A, B, C, and D represents M. notabilis, M. mongolica, M. alba-ZS5801 and M. alba-ZZB, respectively. Counts of Illumina and HiFi reads are indicated by black and blue lines, respectively.
- Supplementary Fig. S2 Gene content of the mitochondrial genomes for Morus species. Different colors represent the number of genes present in each mitogenome. a, b, c represents PCGs, rRNAs, and tRNAs respectively.
- Supplementary Fig. S3 Mauve alignment of four Morus mitogenomes. Within each of the alignment, local collinear blocks are represented by blocks of the same color connected by lines.
- Copyright: © 2024 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Liu L, Long Q, Lv W, Qian J, Egan AN, et al. 2024. Long repeat sequences mediated multiple mitogenome conformations of mulberries (Morus spp.), an important economic plant in China. Genomics Communications 1: e005 doi: 10.48130/gcomm-0024-0005 

Long repeat sequences mediated multiple mitogenome conformations of mulberries (Morus spp.), an important economic plant in China
- Received: 24 October 2024
- Revised: 15 November 2024
- Accepted: 20 November 2024
- Published online: 28 November 2024
Abstract: Mulberries (genus Morus; Moraceae) hold significant economic value in sericulture and have great potential in the horticultural industry, food industry, and human health arenas worldwide. Since the advent of the genomic era, biological macromolecules of Morus species such as whole genome and plastome sequences have been reported, but mitochondrial genome sequences are relatively scarce which greatly hinders the comprehensive understanding of the evolutionary history and processes at work with Morus. Here, four Morus mitogenomes were assembled using Illumina and PacBio HiFi data. The results elucidated that the structure of the four mitogenomes was greatly heterogeneous due to the presence of different numbers of repeat-mediated recombination events with multiple conformations existing simultaneously in the mitogenome for each species. The genome size ranged from 359,062 to 376,846 bp. The repeat sequences and gene transfers from plastome to mitogenome varied widely among the four mitogenomes, which was the main cause of variation in mitogenome size. Finally, the evolutionary history of Rosales was inferred based on the mitogenome sequences. The analyses revealed a strong difference in the phylogenetic placement of Rhamnaceae compared to earlier plastid or nuclear phylogenies, likely due to the effects of ancient hybridizations. Overall, the results presented here will provide important genetic resources for the utilization of this important economic plant.
-
Key words:
- Mulberry /
- Mitogenome /
- Repeat-mediated recombination /
- Phylogeny /
- Rosales