









-
Orchidaceae, one of the largest families of angiosperms, represents approximately 10% of all seed plants[1]. Orchidaceae possesses significant medicinal, and ornamental value, and has a key role in advancing botanical and ecological research. Orchids often establish symbiotic relationships with mycorrhizal fungi, which play an essential role in supporting their growth, nutrient uptake, and adaptation to environmental changes[2,3]. Recently, increasing attention has been directed toward the mitochondrial genome (mitogenome) of orchids, a critical component of their genetic makeup. Through genome assembly and annotation, researchers have identified the distinctive structural features and remarkable diversity of orchid mitogenomes[4,5]. Compared to the chloroplast and nuclear genomes, research on the mitogenome of orchids began relatively late. Nevertheless, the mitogenome is crucial for understanding plant biological functions, evolutionary history, and fungal symbiosis mechanisms[6−8].
Recent studies on the mitogenomes of Dendrobium species and related orchids have revealed significant differences in gene content, genome size, and structural organization[9,10]. These mitogenomes are often more complex than those of other plant families, characterized by frequent gene rearrangements and occurrences of horizontal gene transfer (HGT) and endosymbiotic gene transfer (EGT)[1,3]. Such phenomena are common within Orchidaceae, suggesting that the mitochondrial genomes of orchids have undergone extensive gene flow and structural changes throughout their evolution. A deeper understanding of these genomic features is essential for developing effective conservation strategies and expanding the use of orchids as ornamental and medicinal crops. Furthermore, orchid mitogenomes often contain complex repeat sequences and subgenomic structures, as highlighted in recent studies[5,11]. While these features complicate genome assembly, they also provide unique insights into plant genome evolution and functionality.
Mitochondria are essential energy supplier units and 'signaling hubs' in eukaryotic cells, providing 90% of the energy required for cellular activities in the form of ATP. They also play a crucial role as signal integrators during plant cell growth, development, and stress response[12,13]. Mitochondria, functioning as semi-autonomously replicating organelles with independent genomes, possess considerable research significance[14]. Mitogenomes are integral to processes such as protein synthesis, transcription, and replication, playing a crucial role in the study of the origin and evolution of organisms[15]. In the evolution of plant mitogenomes, it has been observed that structural evolution occurs more rapidly than sequence evolution. The rate of synonymous substitutions varies across plant genomes. The nuclear genome evolves the fastest, followed by the plastid genome, which evolves at approximately half the rate of the nuclear genome. In contrast, plant mitochondrial genomes evolve several times more slowly than plastid genomes[16,17]. Analyzing sequence variation in the mitogenome enables the differentiation of species and enhances understanding of their speciation mechanisms, as well as the structure and sequence variation of the genomes during evolution.
Plant mitogenomes are characterized by their large genome size and structural variation, which includes sub-genomic circular structures, polycyclic structures, and complex configurations that can be challenging to resolve. These variations are predominantly composed of non-coding DNA sequences. Due to the unique repair mechanisms in the mitogenome, exogenous DNA sequences can be easily incorporated, leading to the formation of numerous repeated sequences that facilitate homologous recombination[18]. This recombination between homologous sequences generates smaller linear, circular, and multi-branched subgenomes, which coexist alongside the intact primary genome within the cell. The complex structural features of plant mitogenomes have posed significant challenges for their assembly, slowing down sequencing efforts compared to chloroplast DNA (cpDNA).
Despite progress, significant challenges remain in orchid mitochondrial genome research. High structural variability and pronounced differences between species have made the complete assembly of mitogenomes a formidable task. So far, only a few high-quality assemblies have been completed for species within the Dendrobium, focusing primarily on preliminary analyses and annotations of key species. However, the available data remain limited, leaving many Dendrobium species unstudied. This gap underscores the vast potential for further genomic exploration and phylogenetic analysis. In this study, Dendrobium chrysotoxum served as a model for the first assembly and annotation of its mitogenome. D. chrysotoxum is a perennial medicinal plant belonging to the genus of Dendrobium[19]. Recent research has thoroughly investigated the ornamental and medicinal properties of D. chrysotoxum, revealing its considerable potential for development. It is increasingly considered as a promising floral crop with significant value in ornamental, economic, and medicinal sectors within modern agriculture. We conducted a detailed analysis of its structural features, codon bias, RNA editing, HGT, and intracellular gene transfer, comparing the PCGs with those of 21 other angiosperms. Furthermore, we performed collinearity and phylogenetic analyses. The results of this study will establish a solid theoretical foundation for future research, offering new insights into the symbiotic relationships between orchids and fungi. This is crucial for maintaining orchid diversity, horticultural cultivation, and medicinal production.
-
Sample plants of D. chrysotoxum were cultivated at the National Orchid Germplasm Resources Center of Fujian Agriculture and Forestry University. Healthy plants were selected, and young leaves were carefully harvested using sterilized tweezers and scissors. The harvested leaves were placed in 5 mL cryovials, immediately frozen in liquid nitrogen, and stored at −80 °C in an ultra-low temperature freezer. DNA was extracted using the CTAB method. The DNA purity, degradation level, and contamination were assessed using a NanoPhotometer® (IMPLEN, CA, USA) and 1% agarose gel. We measured the concentration with a spectrophotometer, Qubit® 3.0 Fluorometer (Life Technologies, CA, USA). After passing quality checks, we prepared a 350 bp DNA fragmentation library. The insert size was verified using an Agilent 2100 platform. Subsequently, the prepared library was sequenced on the NovaSeq 6000 platform using a paired-end sequencing protocol (PE150), which resulted in reads of 150 bp. For long-read sequencing, we used an SQK-LSK110 sequencing kit (Oxford Nanopore Technologies, Oxford, UK) and the PromethION sequencer (Oxford Nanopore Technologies, Oxford, UK) for library construction and sequencing.
Assembly and annotation of D. chrysotoxum mitogenome
-
In this study, we combine Next-Generation Sequencing (NGS) and Long-Read Sequencing technologies, producing 12.5 GB of raw data from the Illumina platform and 14.6 GB of raw data from the Oxford Nanopore platform. To ensure data quality, we removed adapter sequences and low-quality reads from the raw data. For the third-generation sequencing data, we applied the default parameters of Guppy (v5.0) software for filtering, while the second-generation sequencing data were processed with Trimmomatic (v0.39) software to trim adapters and eliminate low-quality reads[20]. Only the resulting high-quality Clean reads were used for all subsequent analyses. We utilized the Flye (v2.9.2-b1786) software with default parameters to directly assemble long reads, resulting in a graphical representation of the mitogenome[21]. The assembled contigs were then subjected to makeblastdb, using mitochondrial genes from Arabidopsis thaliana as reference sequences. To identify contigs containing mitogenome sequences, we employed the BLASTn program with specific parameters (-evalue 1e-5 -outfmt 6 -max_hsps 10 -word_size 7 -task blastn-short). For visualization and generation of a draft mitogenome, we used the Bandage (v0.8.1) software[22]. Subsequently, both long reads and short reads were aligned to the identified mitochondrial contigs using bwa (v0.7.17) software, retaining only the aligned reads[23]. Finally, we used Unicycler for assembly with default parameters[24], employing a hybrid assembly strategy to construct the mitogenome of D. chrysotoxum. The final mitogenome was then visualized using Bandage (v0.8.1) software[22].
The presence of repetitive sequences often leads to errors in genome assembly, particularly when multiple configurations are possible. In such instances, selecting the correct configuration is crucial. To address repetitive regions within the graphical genome, we prioritize the genome configuration that is supported by the majority of long-reads. When encountering repetitive sequences, especially long reads, we employ the bwa (v0.7.17) software to align them against these sequences[23]. We identify the paths most supported by the long reads to effectively resolve these repetitive areas. This method is also applied to contigs containing multiple junctions to deduce the most probable mitogenome structure. In our case, despite the presence of several repetitive sequences, only one pair of repeats exceeds 1,000 bp - a forward repeat of 2,062 bp located on contig3. Consequently, multiple circular mitochondrial genomes of this species can self-circularize directly without being affected by repetitive sequences. Additionally, the length of our long reads is sufficient to easily span these short repetitive sequences, aiding in obtaining the primary mitogenome configuration. The draft assembled from long-read data was then visualized using the Bandage (v0.8.1) software[22], which resulted in 18 circular genomic DNA sequences.
The mitogenome of D. chrysotoxum was annotated using Geseq (v2.03) software, with reference to genomes from Arabidopsis thaliana (NC_037304) and Liriodendron tulipifera (NC_021152.1)[25]. Additionally, we further annotated the mitogenome of D. chrysotoxum using the online tool IPMGA (
www.1kmpg.cn/ipmga ) and integrated these results with those obtained from the Geseq software. IPMGA was particularly adept at identifying splice sites and trans-splicing genes[26]. tRNA were annotated with the tRNAscan-SE (v.2.0.11) software[27], and rRNAs were annotated using the BLASTN (v2.13.0) software[28]. Manual corrections of annotation errors were made through the Apollo (v1.11.8) software to ensure meticulousness in annotation accuracy and consistency with established standards in mitogenomics research[29].Analysis of relative synonymous codon usage and repeated sequences codon bias analysis
-
We extracted the PCGs from the mitogenome using Phylosuite (v1.1.16)[30]. We performed codon bias analysis and calculation of the relative synonymous codon usage (RSCU) values using MEGA (v7.0)[31]. We employed online tools including MISA (v2.1) (
https://webblast.ipk-gatersleben.de/misa/ ), TRF (v4.09) (https://tandem.bu.edu/trf/trf.unix.help.html ), and the REPuter web server (https://bibiserv.cebitec.uni-bielefeld.de/reputer/ ) to identify repetitive sequences in the mitogenome of D. chrysotoxum[32−34]. These tools facilitated the detection of three types of repetitive sequences in mitogenome, simple sequence repeat (SSR), tandem repeats, and dispersed repeats. We visualized the results using Excel (2021) and RCircos (v0.69.9)[35].Identification of mitochondrial plastid DNA sequences and prediction of RNA editing sites
-
To identify DNA sequences that migrated from the cpDNA to the D. chrysotoxum mitogenome, we assembled the cpDNA using GetOrganelle (v1.6.0)[36]. We used CPGAVAS2 (
http://47.96.249.172:16019/analyzer/annotate ) to annotate the cpDNA[37], and subsequent corrections were made using CPGView (www.1kmpg.cn/cpgview )[38]. We identified and analyzed candidate homologous fragments between the mitogenome and cpDNA of D. chrysotoxum using BLASTN (v2.13.0). The results were visualized with RCircos (v0.69.9)[28,35]. Default parameter settings were used for all software.To predict C to U RNA editing sites in the 37 PCGs of the mitogenome, we used Deepred-mt, a convolutional neural network (CNN)-based tool. The cutoff value was set at 0.9, and we only considered results with probability values exceeding 0.9 this threshold was considered significant[39].
Identification of mitochondrial horizontal gene transfer (HGT) and gene transferred or lose
-
To validate the horizontal gene transfer (HGT) events involving fungal genes and tRNA genes, we implemented a multi-step validation approach. Initially, following the method outlined by Sinn & Barrett[3], we downloaded the Ustilago maydis (DQ157700) mitogenome from the NCBI database as a reference sequence to search for HGT regions. We sourced the small fungal mitochondrial DNA (FMT) region reference sequences for Vanilla odorata and Apostasia shenzhenica from Sinn & Barrett[3], while the HGT regions for U. maydis and Vanilla planifolia were identified by Valencia-D et al.[40] (Supplementary Table S1). To further substantiate our findings, we also referenced large FMT regions from D. loddigesii, Cyrtochilum meirax, and Podochilus_sp, also identified by Valencia-D et al.[40], and the large FMT region sequence of D. officinale was identified[3] (Supplementary Table S2). Sequence alignment was conducted using MAFFT (v7.505) to obtain the D. chrysotoxum HGT region.
To explore whether these lost genes were transferred to the nuclear genome, we employed the identification method described by Wang et al.[9] to search for these genes within the published nuclear genome of D. chrysotoxum[19].
Colinear and phylogenetic analysis
-
To investigate species evolution, we conducted a comprehensive analysis of collinearity and phylogeny of D. chrysotoxum. Using the BLASTn program with the following parameters (e-value 1e-5, word_size 9, gapopen 5, gapextend 2, reward 2, and penalty 3), we identified conserved homologous sequences exceeding 500 bp between the mitogenomes of D. chrysotoxum and five other Dendrobium species. To visualize the relationships among these genomes, we employed MCscanX (
https://github.com/wyp1125/MCScanX ) to generate a Multiple Synteny Plot[41].To analyze phylogeny, we obtained the complete mitogenome sequences from NCBI for a total of 26 species across five angiosperms (Asparagales, Liliales, Arecales, Alismatales, and Ranunculales). Two species from the Ranunculales, Anemone maxima (NC 053368.1), and Aconitum kusnezoffi (NC 053920.1), served as outgroups. We used PhyloSuite (v1.1.16) software to extract the conserved amino acid sequences of common genes from the mitogenomes of 27 species[30], and multiple sequence alignments were performed using MAFFT (v7.505)[42]. The maximum likelihood (ML) phylogenetic tree was constructed with the IQ-TREE (v1.6.12) software using the parameters (alrt = 1,000, -B = 1,000) to construct the maximum likelihood (ML) phylogenetic tree[43], and the results were visualized using ITOL (v6)[44].
-
In this study, we employed a combination of long and short reads to construct the high-quality polycyclic mitogenome of D. chrysotoxum. By excluding repetitive regions from the nanopore data, we successfully assembled the multiple-circular mitogenome of D. chrysotoxum, revealing 18 circular chromosomes totaling 582,418 bp with a GC content of 43.35% (Fig. 1 & Supplementary Table S3). The lengths of these 18 circular chromosomes vary from 21,961 to 57,950 bp, with the GC content ranging from 39.77% (chromosome 7) to 46.23% (chromosome 4).
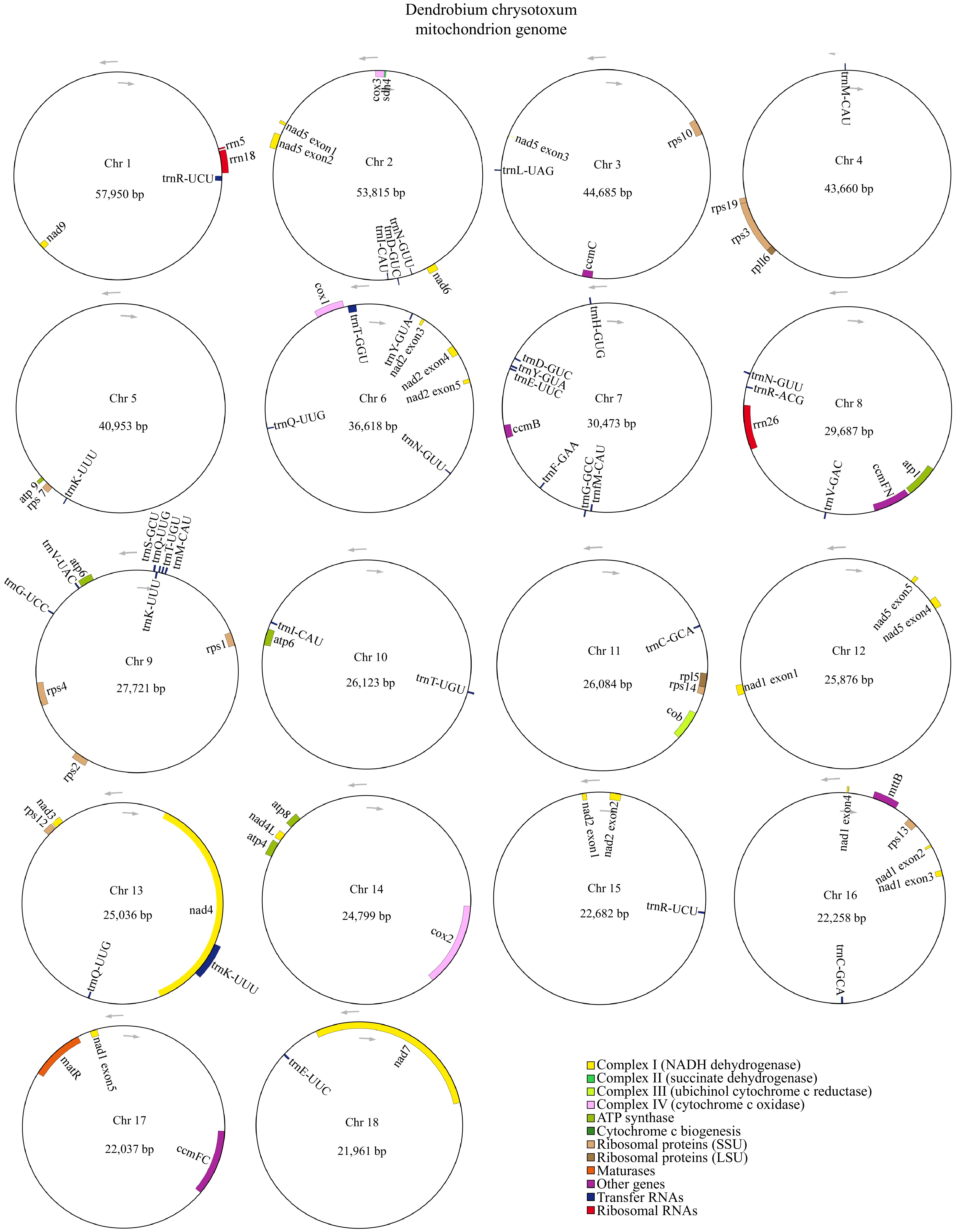
Figure 1.
The mitogenome of D. chrysotoxum was represented as a multiple-branching map, consisting of 18 circular chromosomes. Genes transcribed clockwise are depicted inside each circle, while those transcribed counterclockwise are shown outside the circles. Genes are color-coded according to their functional classification.
A total of 63 genes were annotated, comprising 37 unique PCGs, 23 tRNA genes (14 of which are multicopy), and three rRNA genes. The 37 unique PCGs were made of 24 core genes and 13 variable genes. We categorized the core genes into seven functional types, including five ATP synthase genes (atp1, atp4, atp6, atp8, and atp9), nine NADH dehydrogenase genes (nad1, nad2, nad3, nad4, nad4L, nad5, nad6, nad7, and nad9), four cytochrome C biogenesis genes (ccmB, ccmC, ccmFC, and ccmFN), three cytochrome C oxidase genes (cox1, cox2, and cox3), one protein transport subunit gene (mttB), one maturases (matR), and one cytochrome b (cob). The variable genes encompass two ribosomal protein large subunit genes (rpl5, and rpl16), 10 ribosomal protein small subunit genes (rps1, rps2, rps3, rps4, rps7, rps10, rps12, rps13, rps14, and rps19), one succinate dehydrogenase gene (sdh4) (Table 1).
Table 1. Gene contents of the mitogenome of D. chrysotoxum.
Group of genes Name of genes ATP synthase atp1, atp4, atp6 (×2), atp8, atp9 NADHdehydrogenase nad1, nad2, nad3, nad4, nad4L, nad5, nad6, nad7, nad9 Cytochrome b cob Cytochrome c biogenesis ccmB, ccmC, ccmFC, ccmFN Cytochrome c oxidase cox1, cox2, cox3 Maturases matR Protein transport subunit mttB Ribosomal protein large subunit rpl5, rpl16 Ribosomal protein small subunit rps1, rps2, rps3, rps4, rps7, rps10, rps12, rps13, rps14, rps19 Succinate dehydrogenase sdh4 Ribosome RNA rrn5, rrn18, rrn26 Transfer RNA trnC-GCA×2, trnD-GUC×2, trnE-UUC×2,
trnF-GAA, trnfM-CAU, trnG-GCC, trnG-UCC, trnH-GUG, trnI-CAU×2, trnK-UUU×3, trnL-UAG, trnM-CAU×2, trnN-GUU×3, trnQ-UUG×3,
trnR-ACG, trnR-UCU×2, trnS-GCU, trnS-GGA, trnT-GGU, trnT-UGU×2, trnV-GAC, trnV-UAC, trnY-GUA×2We also examined the conservation of PCGs across 22 angiosperms. To this end, we integrated a variety of factors to select Amborella trichopoda as the most basal extant flowering plant. Additionally, Liriodendron tulipifera, which has 17 variable genes, and monocotyledon species of Phoenix dactylifera, Allium cepa, and Asparagus officinalis were selected. Furthermore, representative species from five subfamilies of Orchidaceae and eight published species of Dendrobium (as of July 1, 2024) were included for comparison. This analysis focused on mitochondrial gene content differences between D. chrysotoxum and the other 21 species (Fig. 2). The number and types of PCGs varied across species. For example, L. tulipifera had the highest number of PCGs (41), while A. cepa had the lowest (24). We found that the rps8 gene was found to be either completely lost or pseudogenized in all 22 examined species. The sdh3, sdh4, and rpl10 genes were universally absent in orchid mitogenomies, except for sdh4, which was fully identified in Paphiopedilum micranthum and D. chrysotoxum. Among the nine Dendrobium species analyzed here, we found all 24 core genes were present, with the only exception being a single pseudogene mttB in D. amplum. Additionally, both rps11 and rpl12 genes were absent from the mitogenome of D. chrysotoxum.
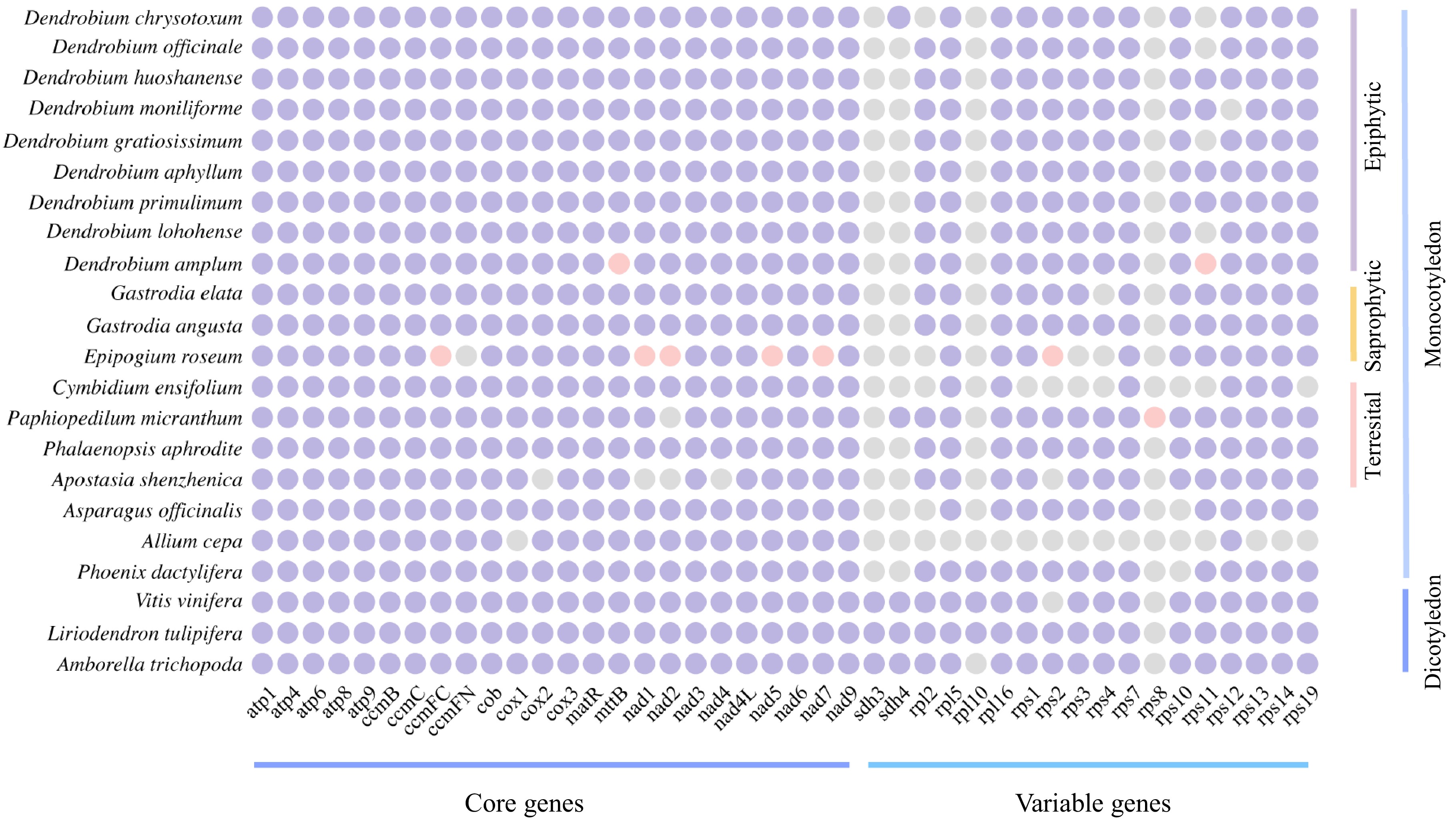
Figure 2.
Comparison of the mitogenome PCGs content of D. chrysotoxum and selected angiosperms. Purple indicates present, grey indicates lost, and pink indicates pseudogenes.
Additionally, we used the GetOrganelle (v1.6.0) software to assemble the complete cpDNA of D. chrysotoxum. The cpDNA displayed a monocyclic structure, totaling 150,607 bp in length. Unlike the mitogenome, the cpDNA features a simpler cyclic structure, consisting of a large single copy (LSC) region, a small copy (SSC) region, and two inverted repeat (IR) sequences (IRA, and IRB). We annotated 110 genes in the cpDNA, including 73 PCGs categorized into 12 functional types, nine rRNA with four rRNA genes (with four duplicated), and 28 tRNA (with 10 tRNA duplicated) (Supplementary Fig. S1 & Supplementary Table S4).
Codon usage of PCGs in D. chrysotoxum
-
Codon usage bias stems from a relative equilibrium within the cell that develops over the long-term evolution of a species. We calculated the relative synonymous codon usage (RSCU) for 37 PCGs in the D. chrysotoxum mitogenome. An RSCU value above 1 indicates a preference for a specific amino acid codon. Our analysis revealed that, except the start codon AUG, the tryptophan (Trp) codon UGG, and the serine (Ser) codon AGU (all of which have RSCU values of 1), the mitogenome PCGs of D. chrysotoxum generally exhibited a preference for codon usage (Fig. 3 & Supplementary Table S5). Among the mitochondrial PCGs, alanine (Ala) displayed the highest preference with the GCU codon achieving an RSCU value of 1.61. This was followed by the glutamine (Gln) codon CAA and the histidine (His) codon CAU, each with RSCU values of 1.5. A total of 29 A/U-ending codons presented RSCU values above 1, excluding UUG. Additionally, there were 32 codons with RSCU less than 1, with the exceptions of AUA and CUA which ended with C/G bases.
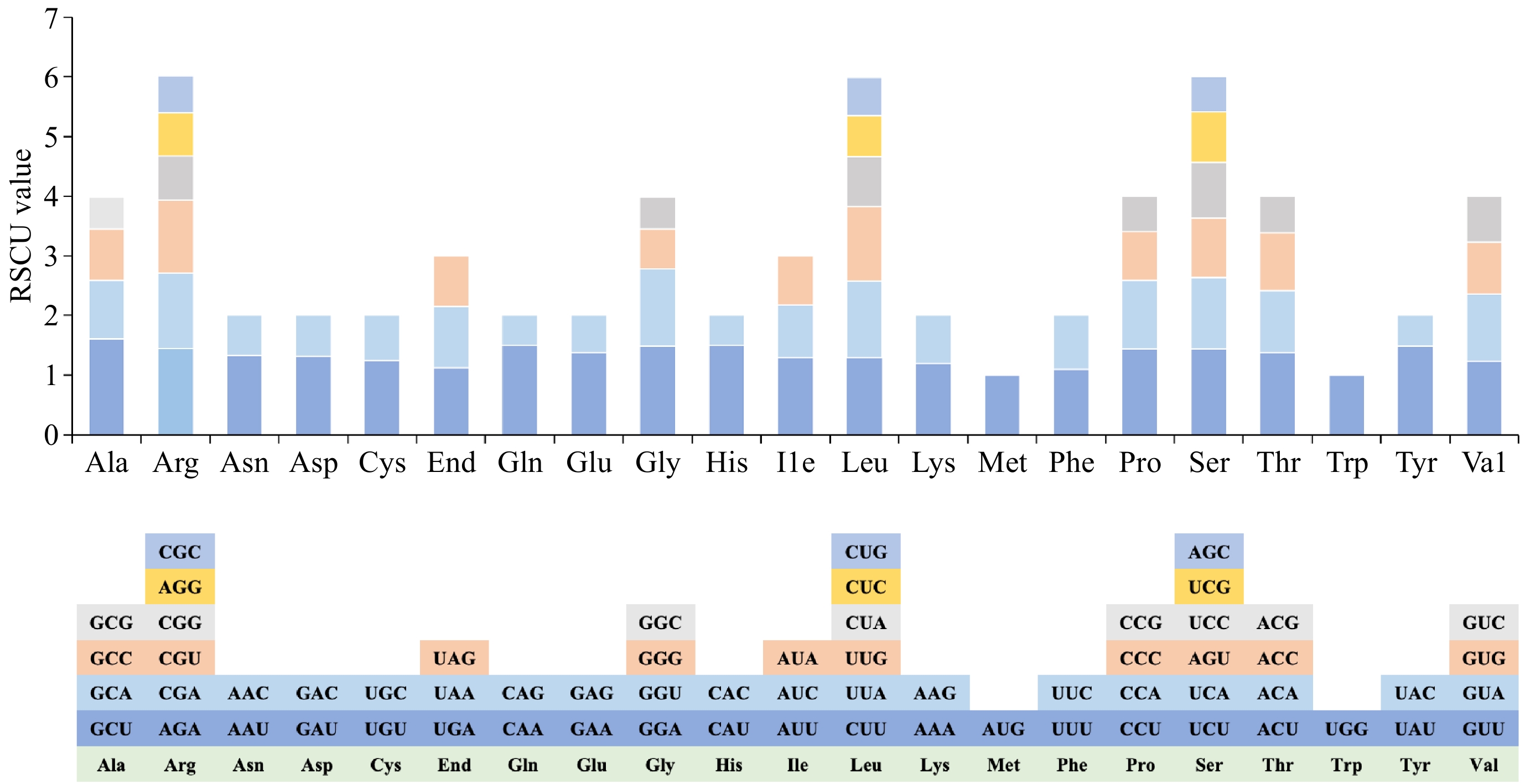
Figure 3.
Analysis of relative synonymous codon usage (RSCU) from D. chrysotoxum mitogenome.
Repeated sequences in the mitochondrial genome of D. chrysotoxum
-
SSRs, a type of tandem repeat sequence with commonly used inheritance characteristics, are frequently utilized in the development of molecular markers. In this study, we identified a total of 148 SSRs from 18 chromosomes. We found that the distribution of these SSRs varied, with chromosome 3 exhibiting the highest number (17) of SSRs (Fig. 4a). The identified SSR repeat fragments were classified based on the number of bases into monomers (A/T), dimers, trimers, tetramers, pentamers, and hexamers. Tetrameric SSRs were the most abundant, comprising 26 types and 51 instances, which accounted for 34.46% of the total SSRs while monomers, dimers, and trimers accounted for 27.03%, 20.95%, and 15.54% of the total SSRs, respectively. Additionally, two pentamers (GATGG/ATCAA) and one hexamer (CATTTC) were identified (Supplementary Table S6).
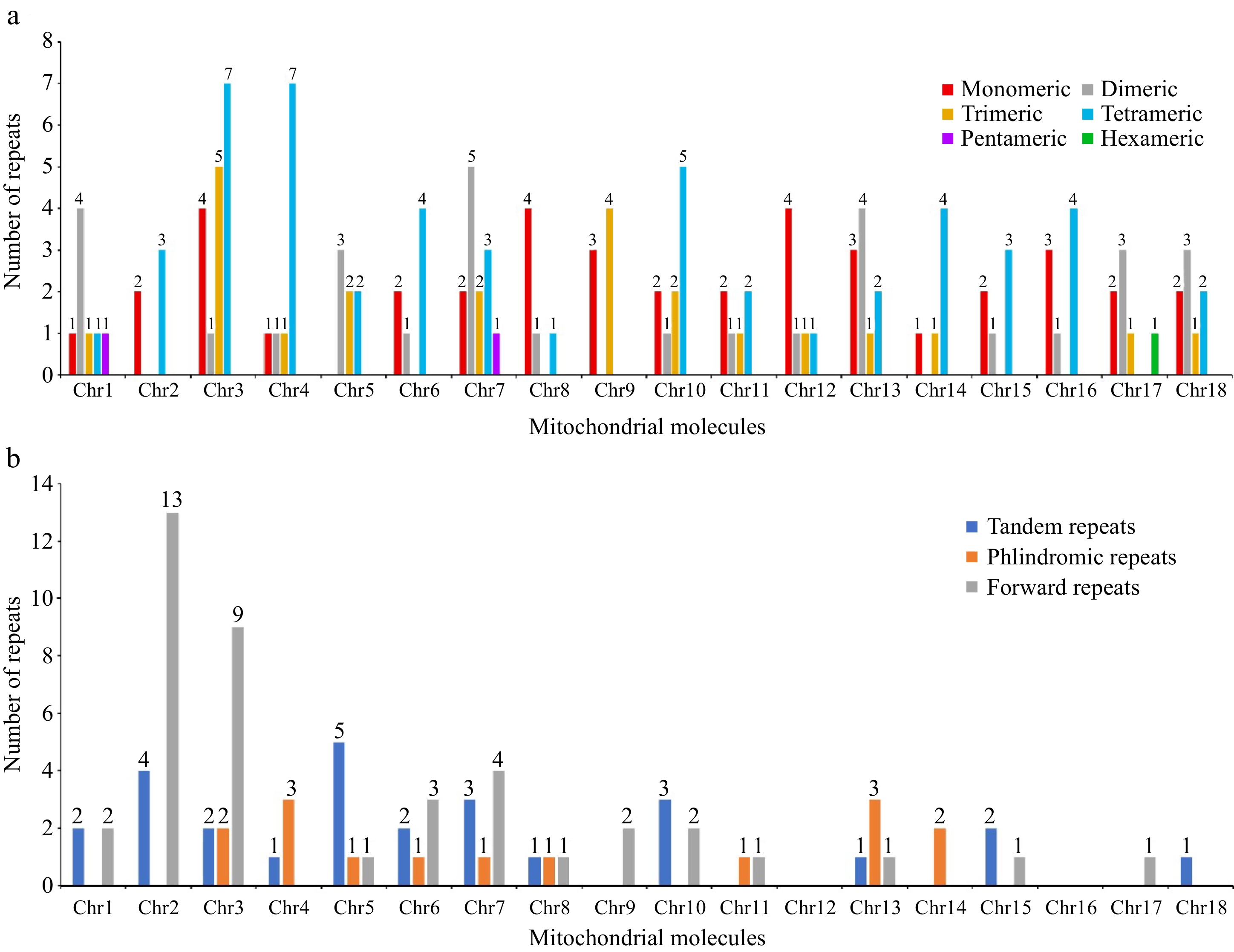
Figure 4.
The repeated sequences in the mitogenome of D. chrysotoxum. (a) The SSRs identified in the D. chrysotoxum mitogenome. The x-axis indicates mitochondrial molecules. The y-axis indicates the number of repeat fragments. The red legend indicates monomeric SSRs, the gray legend indicates dimeric SSRs, the orange yellow legend indicates trimeric SSRs, the blue legend indicates tetrameric SSRs, the purple legend indicates pentameric SSRs, and the green legend indicates hexametric SSRs. (b) The tandem and dispersed repeats identified in the D. chrysotoxum mitogenome. The blue legend said tandem repeats, the yellow legend says palindromic repeats, and the gray forward repeats.
The three primary types of repetitive sequences in plant cells are SSRs, tandem repeats, and dispersed repeats. In this study, we identified a total of 27 tandem repeat sequences, ranging in length from 24 to 179 bp and in copy number from 1.9 to 5.6. We found that chromosome 5 exhibited the highest number of tandem repeat sequences, totaling 18.52% of all identified tandem repeats. Further, we observed 54 pairs of dispersed repeats (≥ 30 bp) unevenly distributed across 15 chromosomes (Supplementary Table S7). Of these, 41 pairs (75.92%) were direct repeat sequences and 15 pairs (27.78%) were palindromic repeat sequences, with lengths varying from 30 to 2,062 bp. Most dispersed repeats (43 out of 54, 79.63%) had lengths of 30 to 49 bp, with 13 pairs classified as intermediate-sized repeats (50–500 bp) and one pair as a large repeat (2,062 bp) (Fig. 4b & Supplementary Table S8). Additionally, some SSRs, interspersed repeats, and tandem repeats were found to overlap (Supplementary Fig. S2).
RNA editing events of D. chrysotoxum in 37 PCGs
-
A total of 605 potential RNA editing sites, converting C to U, were identified across 37 PCGs. The editing sites were unevenly distributed across the genes. The nad4 gene had the highest number of editing sites (57), whereas the rps7, rps10, and sdh4 genes each contained only two. Seven genes (ccmB, ccmC, ccmFN, nad1, nad2, nad5, and nad7) each contained 30 or more editing sites (Fig. 5). To assess whether RNA editing exhibited codon position preference, we analyzed the percentage of RNA editing sites as a function of their codon positions. Among all edits observed, first codon positions were edited 182 times (30.08%), second codon positions were edited 387 times (63.97%), and third codon positions were edited 36 times (5.95%). A total of 44 codons underwent RNA editing events, and the number of edits per codon varied from 1 to 84. The most common transition was from UCA to UUA, which occurred 84 times. Conversely, transitions such as GUC to GUU, AAC to AAU, UAC to UAU, and GGC to GGU occurred only once each in the D. chrysotoxum mitogenome. Following RNA editing, the encoded proteins changed as follows, Ser to Leu and Phe, Pro to Ser, Leu, and Phe, Arg to Cys, End, and Trp, His to Tyr, Thr to Ile, and Met, Leu to Phe, Ala to Val. Before the codon shift, the usage frequency of Ser was the highest at 234 (38.6%), while after the codon shift, the usage frequency of Leu was the highest at 268 (44.2%). Additionally, a specific amino acid Arg (CGA) was modified into a stop codon, End (UAA) (Supplementary Table S9).
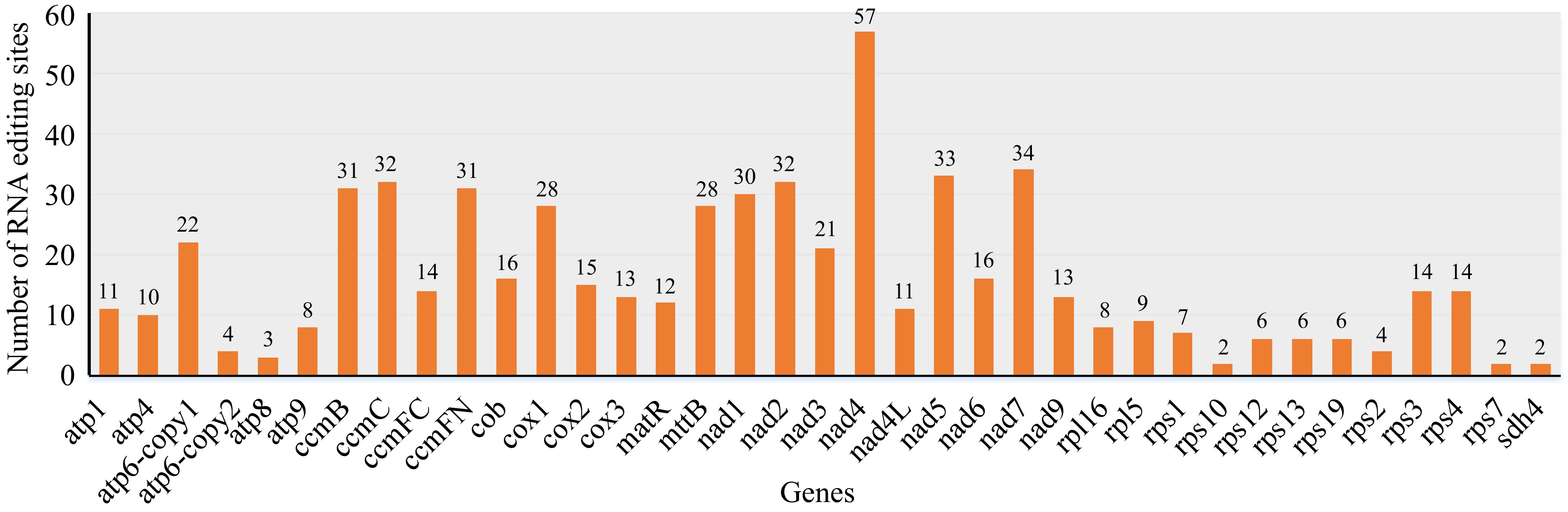
Figure 5.
Prediction of RNA editing sites in mitogenome of D. chrysotoxum. The horizontal and vertical coordinates indicate the gene name and the number of RNA edits, respectively.
Identification of mitochondrial plastid sequence and horizontal gene transfer in the mitogenome of D. chrysotoxum
-
During the evolution of mitochondria, sequences from cpDNA have migrated into the mitogenome. In this study, we compare sequences between the mitogenome and cpDNA of D. chrysotoxum using BLASTn (v2.13.0) and identified potential mitochondrial plastid DNA sequence (MTPTs) fragments. A total of 87 MTPT fragments were identified, varying in length from 29 bp (MTPT4) to 9,321 bp (MTPT19) with a cumulative length of 74,266 bp, accounting for 12.75% of the total mitogenome length. We found that these homologous sequences were unevenly distributed across 16 chromosomes, excluding Chr11 and Chr16 (Fig. 6a & Supplementary Table S10). Additionally, we validated the HGT event between the U. maydis mitogenome and the D. chrysotoxum mitogenome (Fig. 6b). We also conducted a comparative analysis of the HGT regions of D. chrysotoxum with that of A. shenzhenica and D. officinale. The identified HGT regions of D. chrysotoxum not only included fungal genes consisting of 270 bp from U. maydis but also four tRNAs (trnM-CAU, trnS-GCU, trnV-UAC, and trnG-UCC) and one ATP synthase gene (atp6).
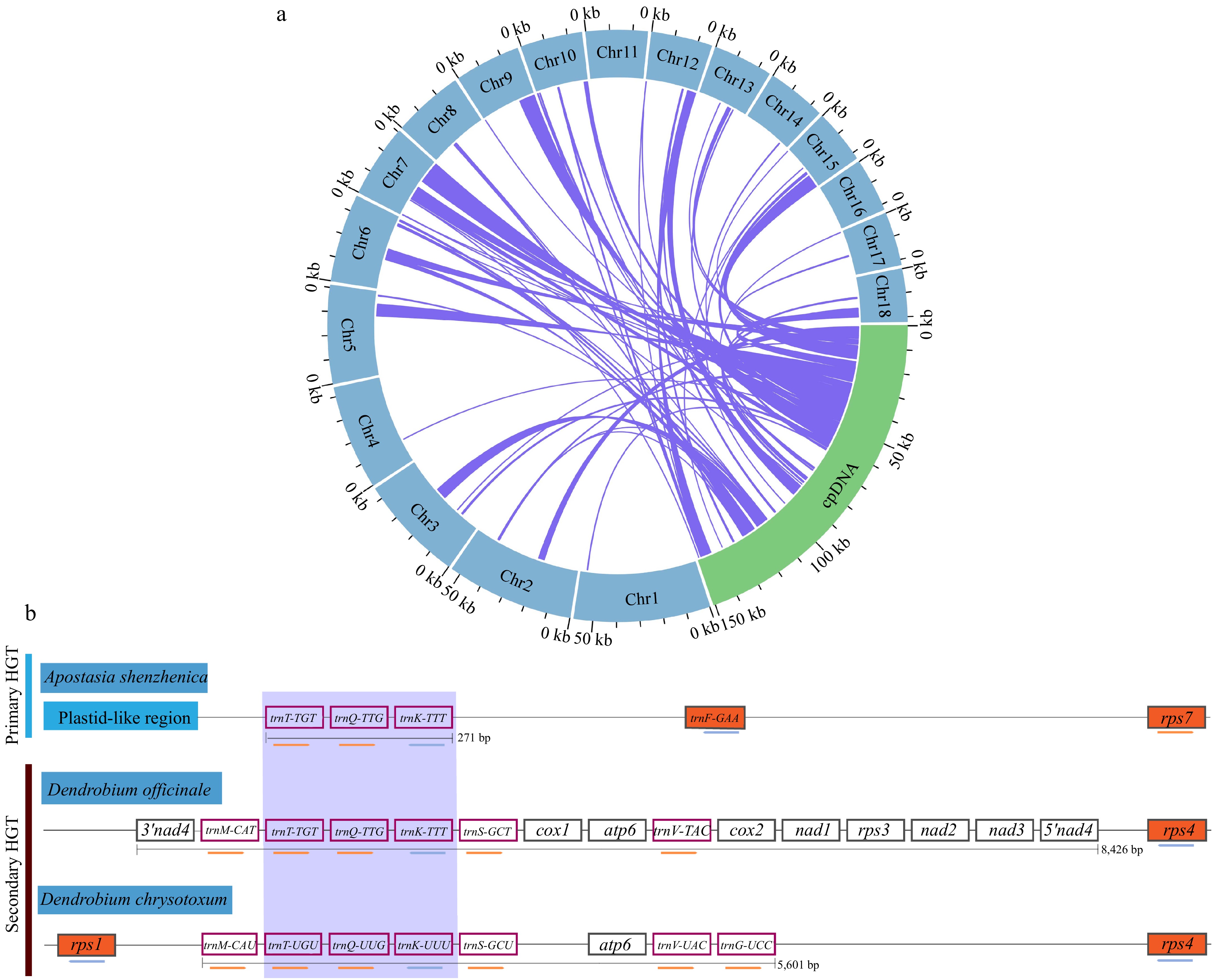
Figure 6.
The intracellular gene transfer and horizontal gene transfer from fungi in the mitogenome of D. chrysotoxum. (a) Gene transfer between the cpDNA and mitogenome of D. chrysotoxum. Blue represents the mitogenome, the green arc represents cpDNA, and the homologous fragments are indicated using the purple lines between the blue and green arcs. (b) Horizontal gene transfer from fungi in the mitogenome of D. chrysotoxum. The purple box highlights the transfer of RNA genes common to all HGTs in three orchids. Orchids plant mitochondrial genes, or nearby plastid-like mitogenomic sequence, is depicted in vermilion, while genes with white backgrounds represent genes of fungal origin, and the coral box represent transfer RNA. The maps are not drawn to scale.
By annotating these MTPT fragments, it was revealed that their origins were both PCGs and tRNA genes within the plastid genome. We identified a total of 31 complete genes, including 20 PCGs (accD, atpB, atpE, ccsA, ndhD, ndhE, petN, psaA, psaB, psaC, psaI, psbI, psbM, psbZ, rbcL, rpl23, rpoB, rps14, rps16, and ycf3) and 11 tRNA genes (trnD-GUC, trnE-UUC, trnfM-CAU, trnG-UCC, trnH-GUG, trnI-CAU, trnL-UAG, trnM-CAU, trnN-GUU, trnQ-UUG, and trnY-GUA) (Supplementary Table S10). In addition, our analysis of the diversity of migration sequences revealed 24 intergenic spacers (IGS) within the MTPT fragments, such as trnF-GAA-ndhJ, ndhJ-ndhJ, trnT-GGU-psbD, trnH-GUG-trnH-GUG, rps4-rps4, trnN-GUU-trnN-GUU, trnN-GUU-rpl32, psbJ-psbJ, trnR-UCU-trnR-UCU, trnG-UCC-trnfM-CAU, psbK-psbI, rpoB-trnC-GCA, ndhJ-trnV-UAC, rpl32-trnL-UAG, rbcL-accD, ndhB-rps7, rps7-ndhB, ndhJ-trnV-UAC, trnS-GGA-trnG-UCC, trnS-GGA-trnG-UCC, trnT-GGU-psbD, trnT-UGU-trnL-UAA, matK-rps16, and trnN-GUU-trnN-GUU. In summary, DNA exchange between the mitogenome and cpDNA of D. chrysotoxum is significant.
The phylogenomic and colinear of D. chrysotoxum
-
To more accurately describe the evolution of the D. chrysotoxum mitogenome, we constructed an ML phylogenetic tree using DNA sequences derived from the 24 core genes. These genes are shared by 26 different angiosperms across five orders, with two mitogenomes from the Ranunculales serving as outgroups (Fig. 7a). The topology of the resulting phylogenetic tree, based on the mitogenome DNA sequence of D. chrysotoxum, aligns with the latest Angiosperm Phylogeny Group (APG) classification. Our results demonstrate that D. chrysotoxum and D. officinale cluster in the same branch, suggesting a more recent common ancestor and indicating a closer relationship between them. In addition, we conducted a collinearity analysis of the mitogenomes of D. chrysotoxum and five closely related species using the MCScanX software, focusing on sequence similarity. To enhance the clarity of the results, we excluded collinear blocks smaller than 0.5 kb. Red arcs represent regions of inversion, while gray regions denote highly conserved homologous areas (Fig. 7b). A substantial number of homologous collinear blocks were observed among D. chrysotoxum and the five related species. The findings here reveal that the arrangement of collinear blocks across the six mitogenomes is inconsistent, suggesting that D. chrysotoxum has undergone genomic rearrangements, resulting in a highly non-conserved order of mitogenome sequences among these species.
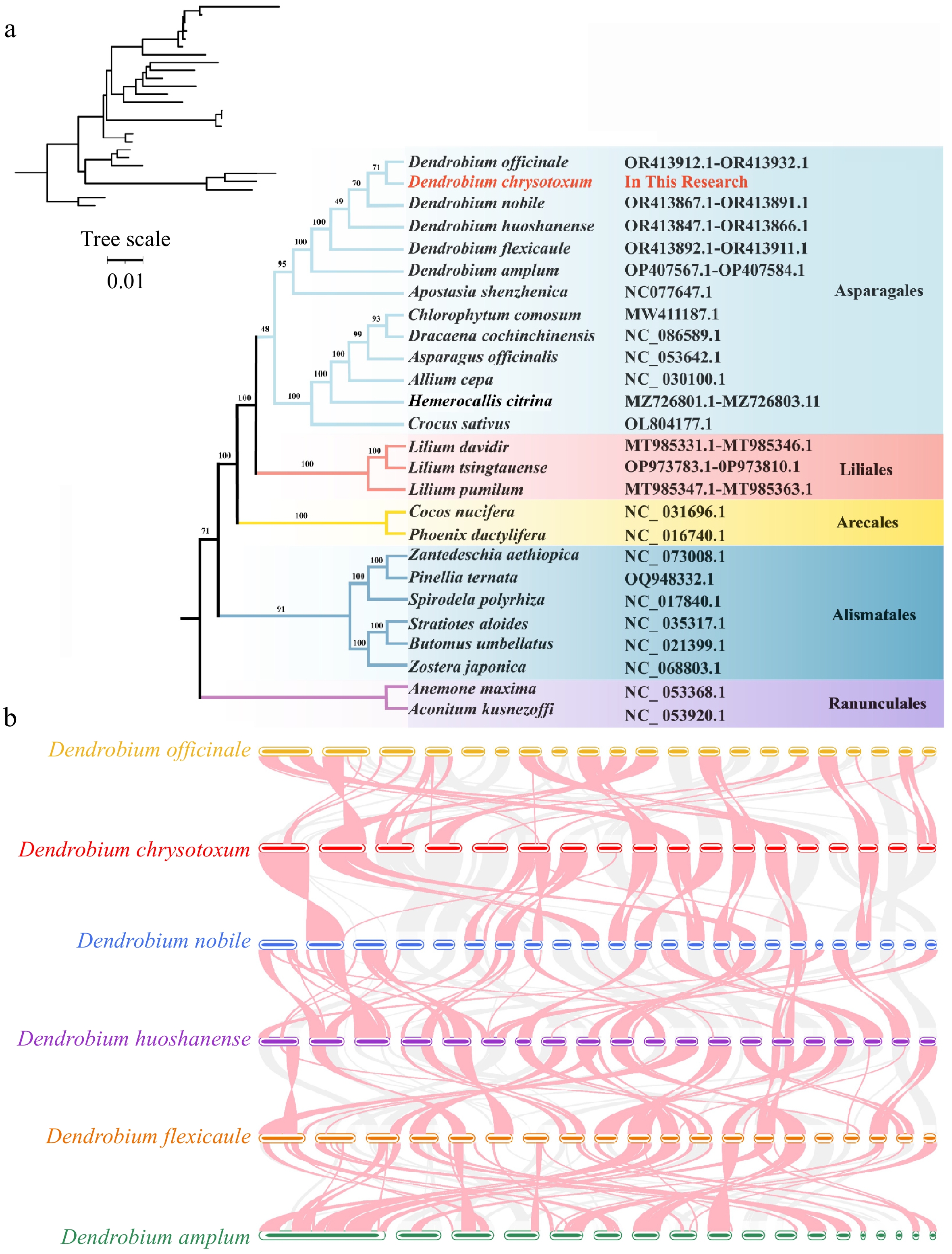
Figure 7.
Phylogenomic and collinear analysis of D. chrysotoxum. (a) Construction of the maximum likelihood (ML) tree based on the 26 species. (b) Collinear analysis of the mitogenome of six Dendrobium species. The red arcs indicate inverted regions, and the gray arcs indicate better homologous regions.
-
The mitogenomes of angiosperms are characterized by abundant repetitive sequences and complex circular structures. Cymbidium ensifolium mitogenome consists of 19 circular chromosomes and 13.78% (77,244 bp) dispersed repetitive sequences[5]. These repetitive sequences impact the assembly quality of plant mitogenomes, making the assembly of angiosperm mitogenomes particularly challenging[45]. In our study, we utilized a hybrid approach combining Illumina and Oxford Nanopore technologies to assemble the first high-quality complete D. chrysotoxum mitogenome of 582,418 bp. Compared to animals (typically a single circular chromosome with < 20 kb) and fungi (17–100 kb)[12,46,47], the size of angiosperm mitogenomes exhibits significant variation, ranging from 66 kb (Viscum scurruloideum) to 11.3 Mb (Silene conica)[45,48]. The mitogenome of Dendrobium showed significant differences from those of other orchids. For example, the mitogenome sizes of various orchids are as follows, Dendrobium (420,538–807,551 bp), Gastrodia elata (1,340,105 bp), P. micranthum (447,368 bp), and E. roseum (414,552 bp)[1,9,49,50]. Notably, the mitogenome of D. chrysotoxum was different from the other orchid species, despite this, its GC content (43.35%) remains relatively consistent compared to other orchids (43.09%–46.2%), suggesting conserved GC content across orchid mitogenomes.
The number of PCGs in these mitogenomes varies from 19 (Viscum scurruloideum) to 41 (Liriodendron tulipifera), and the mitogenome of L. tulipifera, often referred to as a 'fossil' genome contains 17 variable genes and 24 core genes[25,48]. In this study, the mitogenome of D. chrysotoxum lacks at least three PCG genes sdh3, rpl10, and rps11. While sdh3, sdh4, and rpl10 were absent from all assembled orchid genomes, the sdh4 gene is present in the mitogenome of D. chrysotoxum and Paphiopedilum micranthum. Interestingly, the sdh4 gene in the D. chrysotoxum mitochondria was retained with a length of 153 bp. Similar reductions in the sdh4 gene sequence have been observed in the mitogenomes of P. micranthum (204 bp), Cocos nucifera (183 bp), and Asparagus officinalis (222 bp)[1].
We investigated whether the missing genes in the D. chrysotoxum mitogenome had been transferred to the nuclear or cpDNA by examining published core and assembled cpDNAs[19]. We found the rps11 genes in the cpDNA and nuclear genome (ID: Maker45777 and Maker42721), but the rpl10 genes were not in either the nuclear genome or cpDNA. It has been observed that the rpl10 gene is frequently lost or pseudogenized in angiosperms[51] and in sequenced monocot plants[52], a finding that is consistent with this study. These results indicate that both gene loss and transfer was present in the D. chrysotoxum mitogenome. Additionally, the number and sequence conservation of rps genes vary significantly between species[53]. We annotated 10 unique ribosomal protein genes in the mitogenome of D. chrysotoxum. The number of rps genes contained in the mitogenomes of Cymbidium ensifolium and Allium cepa was 5 and 1, respectively, which is considerably fewer than in other species. These differences in PCG numbers across these species may be related to the diversity of ribosomal protein small subunit genes.
Repetitive sequences are prevalent in the nuclear, chloroplast, and mitogenomes of angiosperms, influencing their size and structural variation and even mediating mitogenome recombination[54,55]. SSR, tandem repeat, and dispersed repeat are the three main types of repeat sequences in the mitogenome of angiosperms[56,57]. In our study, we identified a multitude of repetitive sequences in D. chrysotoxum mitogenome, including a pair of large dispersed repeats (2,062 bp), which potentially contribute to genome expansion. These repetitive sequences may expedite the rearrangement process within the mitogenome. In summary, the abundance of repetitive sequences may accelerate mitogenomic rearrangements in D. chrysotoxum.
Angiosperms often include DNA fragments from the plastid genome in their mitogenome[58]. These mitochondrial plastid DNA transfers (MTPTs) play a crucial role in mitogenome assembly[59]. There is a high diversity of MTPTs identified across different species[59,60]. For example, the fragment length and type of MTPTs in the mitogenomes of D. wilsonii (79,909 bp, 216–4,227 bp) and D. henanense (96,511 bp, 263–9,901 bp) differ significantly[61]. We identified 87 MTPTs in the D. chrysotoxum mitogenome, which constitute 12.75% of its total length a proportion that is relatively high compared to other Dendrobium species (D. wilsonii, 10.5%; D. henanense, 12%), and the average level in angiosperms[61]. MTPTs in D. chrysotoxum contain complete PCG sequences, which typically lose functionality[62]. We identified 20 PCGs and 11 tRNA genes in these MTPTs fragments, these findings are consistent with annotations in other angiosperms[59].
In addition, the trnT-TGT, trnQ-TTG, and trnK-TTT genes were identified as part of the complete HGT region (270 bp) in both Apostasia shenzhenica and Vanilla planifolia, referred to as the primary HGT. In contrast, the 8 kb fungal mitogenome sequence identified in D. officinale represents a secondary HGT region[3]. In this study, we identified a 5,601 bp secondary HGT fragment in the Chr9 section of the D. chrysotoxum mitogenome, aligning with previous findings[3]. These results suggest that this HGT sequence is conserved. This fragment represents a horizontal transfer from fungal mitogenomes, potentially linked to the symbiotic relationship between orchids and fungi during their early developmental stages. We also examined HGT fragments in other Dendrobium species, identifying them in Chr9 of D. officinale and in Chr10 of D. huoshanense[61]. Therefore, it is hypothesized that D. chrysotoxum and D. officinale may share a common ancestor. In addition, we retrieved whole-genome sequencing (WGS) data of other D. chrysotoxum individuals from the SRA database and subsequently downloaded the PacBio dataset with accession number SRR12823530. This dataset was then aligned to our assembled reference genome, and the results are presented in Supplementary Table S11. Our analysis revealed a substantial number of SNPs and INDELs distributed across both coding and non-coding regions of the genome. Thus, understanding the migration patterns is crucial for elucidating the structural variations of plant mitochondria.
RNA editing is crucial for plant growth and development[39,63]. Additionally, RNA editing facilitates plants' adaptation to various environmental stresses, including drought and high temperatures[64−66]. RNA editing often restores evolutionarily conserved amino acid residues in mRNA or creates translation start and stop codons. This can lead to amino acid substitutions, potentially altering the structure and function of the proteins encoded by the nad4 gene. For instance, the ahg11 mutant in A. thaliana, which lacks the nad4 gene, exhibits reduced sensitivity to ABA due to impaired RNA editing. This may affect the nad4 protein's function by altering its electrostatic potential or hydrophobicity. Additionally, in the mef35-2 Arabidopsis thaliana transgenic plants, the RNA editing ability of the nad4 gene was restored upon introducing the wild-type MEF35 (Mitochondrial Editing Factor 35) gene. In this study, we identified a total of 605 RNA editing sites in the D. chrysotoxum mitogenome, with the highest frequency of edits occurring in the second codon position (63.96%), consistent with previous findings in other medicinal plants[59,67]. RNA editing also modifies start and stop codons, underscoring its significance in the regulation of RNA in mitochondrial organelles. During RNA transcriptional regulation, the transcripts of cox2, nad1, nad7, and rps1 are transferred from ACG to AUG to create the start codon. Similarly, atp8 and ccmFC modify their stop codon through the editing of the CGA codon to UGA. In the mitogenome of Avena longiglumis, the ccmFC gene undergoes RNA editing to produce stop codons[68]. This process may contribute to maintaining the length stability of protein products encoded by atp8 and ccmFC genes. Thus, predicting RNA editing events provides a potential avenue for unraveling gene functions.
-
In this study, we have successfully assembled and annotated the first complete mitogenome of D. chrysotoxum. We found that the mitogenome is comprised of 18 circular structures totaling 582,418 bp with a GC content of 43.35%. It encodes 63 unique genes, including 37 PCGs. Our analysis highlighted significant aspects such as genome reorganization events, gene content, repetitive sequences, phylogenetic relationships, and RNA-editing sites. The presence of repetitive sequences and identified chloroplast migration sequences in the mitogenome of D. chrysotoxum indicate that significant genome reorganization events have occurred during its evolution. The alignment of homologous fragments in the cpDNA, along with the sequence migration fragments identified in this study, suggest extensive DNA exchange. Notably, we identified a 5,601 bp secondary HGT fragment on Chr9, which originated from the U. maydis fungal mitogenome. We predicted a total of 605 RNA editing sites in the 37 PCGs, including two sites producing stop codons, which could be a focus for future studies. The construction of the ML phylogenetic tree indicated that D. chrysotoxum is closely related to D. officinale. These findings enrich the database of Dendrobium mitogenomes, providing valuable insights into interspecific relationships, genetic evolution, and the molecular breeding of orchid plants.
The research was funded by the National Key Research and Development Program of China (2023YFD1600504), and the Teacher Education Research Project of Fujian Provincial Department of Education (JAT210069).
-
The authors confirm contribution to the paper as follows: study conception and design: Zhang CL, Liu X, Lin WJ, Lan SR; data collection: Zhang CL, Tu XD, Wang LY; analysis and interpretation of results: Zhang CL, Chen DQ, Zeng MY; draft manuscript preparation: Zhang CL, Lin WJ; providing suggestions and editing for the manuscript: Yin WL, Liu ZJ. All authors reviewed the results and approved the final version of the manuscript.
-
The assembled mitogenome of Dendrobium chrysotoxum has been deposited to GenBank under the accession numbers PP963656–PP963673. The raw data has been released through NCBI with the following accession numbers: BioProject PRJNA1194330, BioSample SAMN45177729, SRA SRR31614518 (Illumina), SRA SRR31614517 (Nanopore). All data generated or analyzed during this study have been included in this published article and its supplementary files and also available from the corresponding author on reasonable request.
-
The authors declare that they have no conflict of interest.
- Supplementary Table S1 Small FMT region reference sequences of four species and D. chrysotoxum.
- Supplementary Table S2 Large FMT regions reference sequences of five species and D. chrysotoxum.
- Supplementary Table S3 Accession number for the mitogenome of D. chrysotoxum.
- Supplementary Table S4 Gene contents of the chloroplast genome of D. chrysotoxum.
- Supplementary Table S5 Relative synonymous codon usage (RSCU) of individual amino acid pairs of codons in the D. chrysotoxum mitogenome.
- Supplementary Table S6 Types of SSRs detected in the D. chrysotoxum.
- Supplementary Table S7 Tandem repeat sequences in the mitogenome of D. chrysotoxum.
- Supplementary Table S8 Dispersed repeat sequences in the mitogenome of D. chrysotoxum.
- Supplementary Table S9 RNA editing events of 37 PCGs in D. chrysotoxum.
- Supplementary Table S10 The homologous DNA fragment in D. chrysotoxum mitogenome.
- Supplementary Table S11 Variant results of D. chrysotoxum.
- Supplementary Fig. S1 D. chrysotoxum map of the chloroplast genome.
- Supplementary Fig. S2 Chordal diagram of repeated sequences of D. chrysotoxum.
- Copyright: © 2025 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Zhang CL, Tu XD, Wang LY, Chen DQ, Zeng MY, et al. 2025. A comprehensive analysis of the complete Dendrobium chrysotoxum mitogenome reveals a multiple-circular structure and horizontal gene transfer from fungi. Ornamental Plant Research 5: e007 doi: 10.48130/opr-0025-0009 

A comprehensive analysis of the complete Dendrobium chrysotoxum mitogenome reveals a multiple-circular structure and horizontal gene transfer from fungi
- Received: 28 July 2024
- Revised: 16 December 2024
- Accepted: 20 December 2024
- Published online: 25 February 2025
Abstract: Dendrobium chrysotoxum, a perennial medicinal and horticulture plant in the genus Dendrobium within the orchid family (Orchidaceae), holds significant medicinal, ornamental, and scientific value, and thus it is recognized as an innovative horticultural crop. Despite its importance, the mitochondrial genome (mitogenome) of D. chrysotoxum, remains unexamined in the current scientific literature. In this study, we assembled an annotation of the complete mitogenome of D. chrysotoxum, which comprises 18 circular chromosomes with a total length of 582,418 bp. The mitogenome encodes a total of 63 genes including 37 protein-coding genes (PCGs), three rRNA genes, and 18 tRNA genes. Comparative analyses with other angiosperms revealed variations in gene content. Notably, our analysis revealed the retention of sdh4 in the D. chrysotoxum mitogenome, and it has contracted to 153 bp. Using RNA editing sites, we predicted a total of 605 editing sites across all PCGs, with the nad4 gene exhibiting the highest number of sites at 57. Our research further identified the extensive presence of mitochondrial plastid DNA sequences, with 87 fragments accounting for 12.75% of the mitogenome. Phylogenetic and synteny analyses further elucidated the evolutionary dynamics and rearrangement events in D. chrysotoxum as compared to other Dendrobium species. Additionally, we identified a 5,601 bp secondary horizontal gene transfer event involving fungal genes and tRNA genes. Our results provide valuable insights into the symbiotic relationships between orchids and fungi and aids in further research on its potential applications as a medicinal herb with ornamental value.