









-
Native to the southern regions of China, Yulania liliiflora is often associated with classical Chinese poetry and art, where it is depicted as a metaphor for feminine beauty and grace. It has been increasingly introduced to urban centers in Europe and the United States since 1780. This species is not only of high ornamental value as a landscape plant but also has excellent medicinal properties[1]. The flower buds of Y. liliiflora have been used in Chinese traditional medicine for over 2,000 years. Previous research has confirmed that the leaves of Y. liliiflora could be considered a highly promising by-product and affordable source of lignans for the pharmaceutical, food, and agricultural industries[2]. Additionally, research has found that the essential oil of Y. liliiflora exerts a positive effect in the treatment of pregnant women who have decubitus ulcers[3]. The utilization of essential oil and leaf extracts sourced from Y. liliiflora stands as an exceptional strategy for hindering the proliferation of food spoilage microorganisms and foodborne pathogens[4].
Magnoliids are one of the largest clades of flowering plants, second only to the eudicots and monocots, which have a global distribution and are highly valued for their economic, ornamental, and ecological attributes. Magnoliids, positioned as the third-largest branch with a species count exceeding 10,000, represent a minority, contributing to less than 3% of the overall angiosperm species[5]. As a dicotyledonous plant family featuring primitive flowers, Magnoliaceae is among the largest families within the Magnoliids[6]. Within Magnoliaceae, species like Y. liliiflora represent early diverging lineages, playing a crucial role in advancing our understanding of plant evolution and phylogeny. This information aids in unraveling the evolution route of present-day angiosperms. Previous studies have provided an overview of the phylogenetic research on Magnoliids and explored the possible reasons for the differences in their evolutionary status, such as diversity in phylogenetic data, diversity of sampling, variations in methodology, Long-Branch attraction, and incomplete lineage sorting[5,7]. Nevertheless, achieving a comprehensive understanding of the underlying molecular mechanisms has proven challenging, mainly attributable to the insufficient availability of critical genetic resources. Specifically, there is a dearth of reported mitogenomes for Y. liliiflora.
Higher plant cells inherently possess semi-autonomous genetic systems, specifically, the mitochondrial and chloroplast genomes, which function as vehicles for crucial genetic information[8,9]. Primarily serving as the cellular 'energy factories', mitochondria are responsible for converting biomass energy into chemical energy in living cells[10]. Mitochondrial genomes (mitogenomes), owing to the polymorphism exhibited within their genomes, which encompasses a spectrum of genetic variations such as single nucleotide polymorphisms, insertions, deletions, and gene rearrangements, serve as valuable resources for studying the mechanisms of origin and maintenance of genetic diversity in plants[11]. However, owing to the existence of numerous repetitive sequences and mitochondrial plastid sequences (MTPTs), the assembly of plant mitogenomes is often fragmented, making it difficult to obtain a complete mitogenome. To December, 2023, the National Center for Biotechnology Information (NCBI) database has documented nearly 13,000 complete plastid genomes, and a limited 673 plant mitogenomes[12]. Ongoing research highlights the diverse structural forms of plant mitogenomes, including circular molecules, linear conformations, branched structures, and polycyclic molecules[13−15]. The size of plant mitogenomes varies significantly, spanning from 66 kb (Viscum scurruloideum) to ~19 Mb (Cathaya argyrophylla)[16,17], with the majority falling within the range of 200−800 kb[18]. The gene contents of angiosperm mitogenomes exhibit considerable variation, with notable conservation observed in 24 core protein-coding genes (PCGs), whereas other variable genes display substantial diversity among diverse species[19]. Spermatophyte mitogenomes are rich in repetitive sequences, which include simple sequence repeats (SSRs), tandem repeats, and dispersed repeats. Large repeats greater than 1,000 base pairs (bp) in length and displaying high sequence similarity are typically more prone to frequent recombination, whereas medium-sized repeats spanning 100 to 1,000 bp recombine intermittently, and small repeats under 100 bp are rare[20−22].
In this study, we assembled the complete mitogenome of Y. liliiflora using the HiFi sequencing data. We further annotated and analyzed the Y. liliiflora mitogenome. To gain a deeper insight into the evolution and origin of the Y. liliiflora mitogenome, available data from the other three Magnoliids mitogenomes were used for comparison. The results presented in this study supplement the existing genetic knowledge relating to the genus Yulania. Simultaneously, they offer a promising avenue for conducting more comprehensive genomic investigations of Y. liliiflora.
-
Fresh leaves of Y. liliiflora were collected from the campus of Nanjing Forestry University, Nanjing, Jiangsu Province, China (32°04'41" N, 118°48'23" E). The leaves were promptly frozen and preserved in liquid nitrogen at a temperature of −80 °C. Total genomic DNA was extracted using the Hi-DNAsecure Plant Kit (Tiangen DP350). After the integrity and purity of the isolated DNA were checked by agarose gel electrophoresis and a Nanodrop 2000 ultraviolet spectrophotometer (ThermoFisher), a sequencing library was constructed by the SMRTbell Express Template Prep Kit 2.0 (PacBio Biosciences, CA, USA). The PacBio HiFi reads, achieved through the application of Circular Consensus Sequencing, exhibit an error rate of 1% or below, ensuring a high degree of accuracy[23,24]. HiFi sequencing data were obtained from the PacBio Revio platform[25].
Assembly and annotation of the Y. liliiflora mitogenome
-
The mitogenome of Y. liliiflora was assembled using two distinct approaches. Initially, the 'autoMito' mode of PMAT v1.3.0 was used for mitogenome assembly, with the parameters '-st HiFi -g 2200M'[26]. An alternative approach involved manually selecting three contigs (contig01680, contig00761, and contig114302) from the output file of the PMAT's 'autoMito' mode, based on their congruence with the depth of the mitogenome sequencing. Validation of the PMAT assembly was performed using oatk with the parameters '-k 1000 -c 5'[27]. Consequently, we acquired a preliminary draft mtDNA, a complex multibranched and closed-loop conformation (Supplementary Fig. S1). For the convenience of description, we manually simplified the graphical representation utilizing bandage v0.8.1[28]. During this simplification process, we excluded the chloroplast nodes. Simultaneously, we retained a subset of chloroplast nodes specifically for the analysis of MTPTs (Supplementary Fig. S1). Finally, we processed Y. liliiflora mitogenome into a linear molecule (Supplementary Fig. S1). Of course, we emphasize that the treatment is not the only form because the configuration of mitochondrial DNA is in dynamic transformation mediated by repeats. The mitogenomes of Y. liliiflora were initially annotated using the web-based tool IPMGA (
https://www.lkmpg.cn/mgavas ). Subsequently, the tRNA and rRNA genes were further examined using tRNAscan-SE v2.0[29] and BLASTn[30], respectively. The intron contents of the Y. liliiflora mitogenome were identified by PMGmap (http://www.1kmpg.cn/pmgview ). Manual verification was performed on all PCGs, tRNA, and rRNA genes, and intron contents using MacVector v18.5 (https://macvector.com/ ). Finally, the mitogenome map of Y. liliiflora was constructed using OGDraw vl.3.1, an online tool provided by Greiner et al.[31].Analysis of relative synonymous codon usage (RSCU) and repeat elements
-
The RSCU for PCGs was analyzed and calculated using Mega 11.0 software[32]. To identify the SSRs in the Y. liliiflora mitogenome, the MISA web service (
https://webblast.ipk-gatersleben.de/misa/ ) was utilized[33]. For mono-, di-, tri-, tetra-, penta-, and hexa-nucleotides, minimum repetition thresholds were established at 10, 5, 4, 3, 3, and 3 respectively[34]. Furthermore, a maximum sequence length of sequence between two SSRs was set at 100 bp. The tandem Repeats Finder v4.09 software was utilized to detect any tandem repeats present in the assembled mitogenome[35]. The minimum alignment score was established as 60, while the maximum period size was defined as 500. The identification of dispersed repeats within the Y. liliiflora mitogenome was conducted using the online program REPuter (https://bibiserv.cebitec.uni-bielefeld.de/reputer )[36]. The analysis considered a hamming distance of 3, a minimal repeat size of 50, and a maximum computed repeat count of 5,000. The distribution of dispersed repeats in the Y. liliiflora mitogenome was visualized using the Advanced Circos module in TBtools vl.132[37].Identification of the MTPTs
-
To facilitate the identification of MTPTs, the chloroplast genome of Y. liliiflora was extracted from the whole genome sequencing data[38]. BLASTn analysis was subsequently conducted to detect homologous fragments between the plastid and mitogenomes of Y. liliiflora[30]. The MTPTs with matching rates > 90% and lengths > 50 bp were selected for further investigation. All MTPTs were manually annotated to examine the genes. Subsequently, the MTPTs were visualized using the Advanced Circos module in TBtools v1.132[37].
Identification of RNA editing sites
-
RNA sequencing has provided profound insights into the intricate workings of complex biological mechanisms[39]. The diversification of PCGs sequences in plant mitochondria is intricately linked to the crucial role of RNA editing[40]. In the Y. liliiflora mitogenome, we utilized RNA sequences to assess the prevalence of RNA editing. Specific steps involved the extraction of coding sequences for each PCG, along with 100 bp flanking regions, to systematically construct reference sequences. Subsequently, we used Deepred-mt for the identification of RNA editing sites[41]. Finally, manual confirmation of all identified RNA editing sites was conducted using the IGV software[42].
Whole-genome collinearity analysis
-
To compare the mitogenome structure of Y. liliiflora with other plant species, we downloaded another four closed mitogenomes from NCBI for collinearity analysis including M. officinalis (NC_064401.1), Liriodendron tulipifera (NC_042758), Hernandia nymphaeifolia (NC_063145.1), and Cinnamomum chekiangense (NC_082065.1). The program of BLASTn was used to align the four mitogenomes against the Y. liliiflora mitogenome with a minimum identity > 90% and minimum alignment length > 500 bp[30]. The R package RIdeogram for chromosome data visualization was used to display colinear results generated by BLASTn[43].
Phylogenetic analysis
-
To establish a comparative framework aligned with the kinship of Y. liliiflora, we obtained the mitochondrial PCGs of 16 additional plant mitogenomes from the NCBI Nucleotide database. Cycas taitungensis and Ginkgo biloba were chosen as outgroups. A total of 22 conserved PCGs (atp1, atp4, atp6, atp8, atp9, ccmB, ccmC, cob, coxl, cox2, cox3, mttB, nad3, nad4, nad4L, nad5, nad6, nad7, nad9, rps3, rps4, and rps12) were meticulously selected for mitochondrial multiple sequence alignment, executed by Prank v.170427[44]. For chloroplasts multiple sequence alignment, 45 conserved PCGs (atpA, atpB, atpE, atpF, atpI, ccsA, cloP, matK, ndhA, ndhB, ndhC, ndhD, ndhE, ndhF, ndhG, ndhI, ndhJ, ndhK, petA, petB, psbC, psbD, rbcL, rpl14, rpl20, rpoA, rpoB, rpoC1, rpoC2, rps2, rps4, rps7, rps8, rps11, rps12, rps18, rps19, ycf3, and ycf4). Finally, IQ-TREE v 2.1.4 were used to construct the phylogenetic tree[45]. The maximum likelihood (ML) method was implemented under the GTR + F + I + G4 model chosen for the chloroplast and mitochondrial genome. The online tool iTOL was used to meticulously edit and decorate the phylogenetic tree[46].
-
The Y. liliiflora breach linear mitogenome was assembled, annotated, and submitted to the GenBank nucleotide database with an accession number of NC_085212.1 (Supplementary Fig. S1). The annotated genes' physical arrangements and functional categorizations are presented in Fig. 1a. The Y. liliiflora mitogenome is 865,191 bp in length, resembling the size of most other previously assembled Magnoliids plant mitogenomes. In the Y. liliiflora mitogenome, the nucleotide composition stands at 26.59% A, 26.45% T, 23.40% G, and 23.56% C, with a GC content of 46.95% (Supplementary Table S1). The GC content is higher than the Y. liliiflora chloroplast genome (39.25%)[47]. The total length of PCGs constitutes 4.24% (36,717 bp) of the complete mitogenome, while the lengths of tRNA and rRNA genes account for merely 0.24 and 0.68%, respectively. As illustrated in Fig. 1b, the GC content of PCGs, tRNA, and rRNA genes is 43.14%, 48.63%, and 52.99%, respectively. A total of 65 unique genes were identified in the Y. liliiflora mitogenome, including 41 PCGs (24 core genes and 17 variable genes), 21 tRNA, and three rRNA genes (Table 1). It is found that there are two copies of atp4, nad3, rps7, and rps12, respectively (Table 1). In addition, there are 17 cis-splicing introns in seven genes (nad4, nad7, ccmFc, cox2, rpl2, rps3, and rps10), and five trans-splicing introns in nad1, nad2, and nad5, respectively (Supplementary Fig. S2).
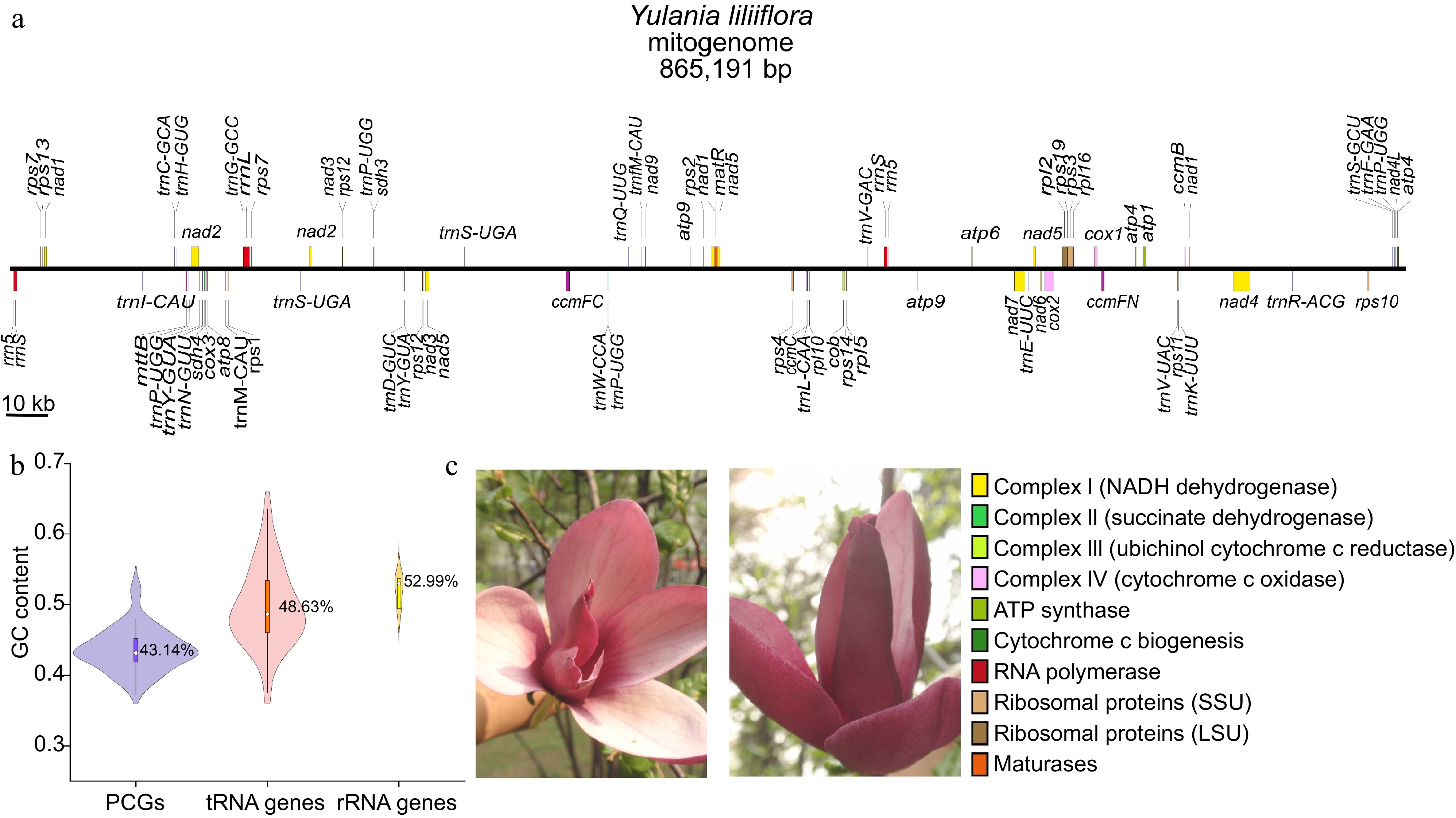
Figure 1.
Summary of the mitogenome of Y. liliiflora. (a) Linear map of the mitogenome of Y. liliiflora. The genes in the upper and lower linear regions are transcribed clockwise and counterclockwise, respectively. (b) The GC content of PCGs, tRNA, and rRNA genes among Y. liliiflora mitogenomes. (c) Y. liliiflora in Nanjing Forestry University (Nanjing, China).
Table 1. The physical placements and functional categorizations of the Y. liliiflora mitochondrial genome (mitogenome).
Group of genes Gene name Length (bp) Start codon Stop codon Amino acids ATP synthase atp1 1,530 ATG TGA 509 atp4 × 2 582 ATG TAA 193 atp6 720 ATG CAA 239 atp8 480 ATG TAA 159 atp9 225 ATG TAA 74 NADH dehydrogenase nad1**++ 978 ACG TAA 325 nad2***+ 1,467 ATG TAA 488 nad3 × 2 357 ATG TAA 118 nad4*** 1,488 ATG TGA 495 nad4L 303 ACG TAA 100 nad5**++ 2,013 ATG TAA 670 nad6 735 ATG TGA 244 nad7**** 1,185 ATG TAG 394 nad9 573 ATG TAA 190 Cytohrome c biogenesis ccmB 621 ATG TGA 206 ccmC 960 ATG TAA 319 ccmFc* 1,329 ATG CGA 442 ccmFn 1,806 ATG TAG 601 Maturases matR 1,959 ATG TAG 652 Ubichinol cytochrome c reductase cob 1,182 ATG TGA 393 Cytochrome c oxidase cox1 1,584 ACG TAA 527 cox2** 765 ATG TAA 254 cox3 798 ATG TGA 265 Transport membrance protein mttb 768 ACG TGA 255 Ribosomal proteins (LSU) rpl2* 1,665 ATG TAG 554 rpl5 561 ATG TAA 186 rpl10 471 ATG TAA 156 rpl16 435 GTG TAA 144 Ribosomal proteins (SSU) rps1 693 ATG TAG 230 rps2 657 ATG TAA 218 rps3* 1,572 ATG TAA 523 rps4 1,071 ACG TAA 356 rps7 × 2 450 ATG TAA 149 rps10* 360 ACG TGA 119 rps11 516 ATG CAA 171 rps12 × 2 378 ATG TGA 125 rps13 351 ATG TGA 116 rps14 303 ATG TAG 100 rps19 282 ATG TAA 93 Succinate dehydrogenase sdh3 330 ATG TAA 109 sdh4 447 ATG TGA 148 Transfer RNAs trnC-GCA 71 − − − trnD-GUC 74 − − − trnE-UUC 72 − − − trnF-GAA 74 − − − trnfM-CAU 74 − − − trnG-GCC 72 − − − trnH-GUG 74 − − − trnI-CAU 74 − − − trnK-UUU 73 − − − trnL-CAA 74 − − − trnM-CAU 73 − − − trnN-GUU 72 − − − trnP-UGG 75 − − − trnQ-UUG 72 − − − trnR-ACG 79 − − − trnS-GCU 88 − − − trnS-UGA 87 − − − trnV-GAC 72 − − − trnV-UAC 73 − − − trnW-CCA 74 − − − trnY-GUA 83 − − − Ribosomal RNAs rrn5 118 − − − rrnL 3,710 − − − rrnS 2,087 − − − The numbers following the gene names indicate the number of copies. * Indicates the number of cis-splicing genes, while + indicates the number of trans-splicing genes. Codon usage analysis of PCGs
-
The total length of PCGs in Y. liliiflora is 36,717 bp encoding 12,239 codons excluding termination codons. The analysis of codon usage was performed on 45 PCGs in the mitogenome of Y. liliiflora, with the codon usage amino acid displayed in Fig. 2. This analysis reveals preferential codon usage patterns that could aid in understanding the underlying evolutionary mechanism in plants. It sheds light on potential selective pressures and evolutionary influences on mitochondrial protein synthesis in Y. liliiflora[48,49]. Previous research has strongly demonstrated that mutational bias, translational selection, and gene functional bias should work on affecting the RSCU of single-exon genes in Oryza sativa[49,50]. RSCU values exceeding 1 are indicative of amino acid preferences, while values below 1 suggest underutilization of the codon[51]. Notably the initial codons AUG (Methionine) and UGG (Tryptophan), both with RSCU values of 1. The PCGs of Y. liliiflora mitogenome display a common codon preference. GUU (Valine) possesses the highest RSCU value at 1.46, followed by GGA (Glycine) at 1.44 (Supplementary Table S2). However, cysteine (Cysteine), lysine (Lysine), and phenylalanine (Phenylalanine) exhibit RSCU values below 1.2, indicating minimal codon preferences (Supplementary Table S2). The results of this study indicate that A/U ending codons are commonly overabundant, while G/C ending codons are frequently under represented. Codon usage analysis of the Y. liliiflora mitogenome reveals leucine (Leucine), serine (Serine), and arginine (Arginine) as the most prevalent amino acids, while methionine (Methionine) and tryptophan (Tryptophan) are less common (Fig. 3).
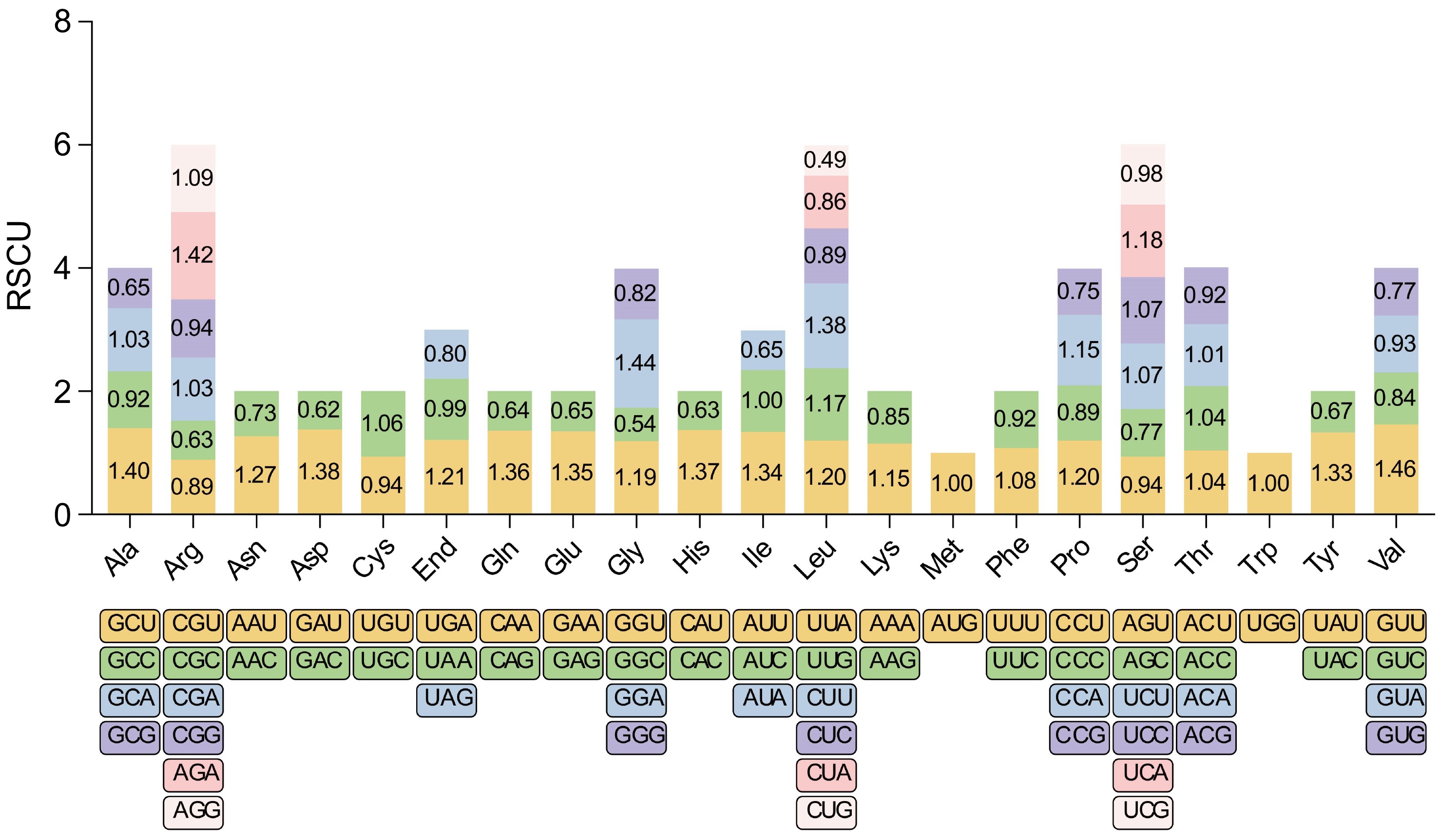
Figure 2.
RSCU in the Y. liliiflora mitogenome. The x-axis of the graphical representation depicts codon families, while RSCU values on the y-axis signify the frequency of codons relative to the expected frequency for uniform synonymous codon usage. This analysis reveals preferential codon usage patterns, shedding light on potential selective pressures and evolutionary influences on mitochondrial protein synthesis in Y. liliiflora.
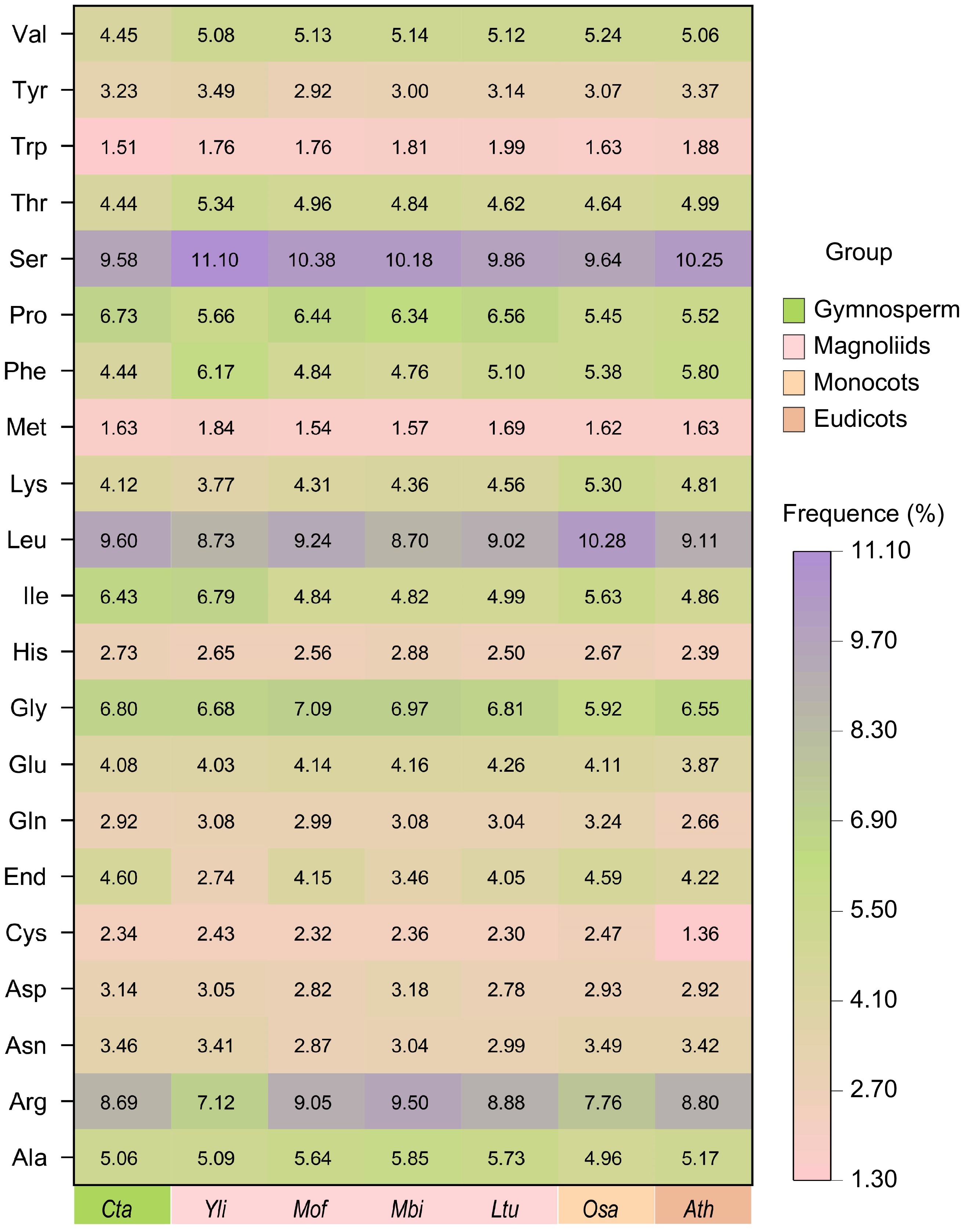
Figure 3.
Codon usage frequence of the Y. liliiflora mitogenome compared with C. taitungensis, M. officinalis, M. biondii, L. tulipifera, O. sativa, and A. thaliana. The x-axis represents different species, while the y-axis depicts amino acid families. Color intensity reflects the percentage of each codon relative to the total codons coding for a specific amino acid, ranging from pink (1.30%) to deep purple (11.10%). The gradual color transition signifies increasing percentages, providing a visual representation of the relative abundance of each codon in the context of all mitochondrial proteins.
Repetitive sequences of the Y. liliiflora mitogenome
-
Based on sequence characteristics, repetitive sequences are categorized into three types: simple repetitive sequences, tandem, and dispersed repeats. In the Y. liliiflora mitogenome, a total of 303 SSRs are found, with monomeric and dimeric SSRs occupying 43.23% of the total SSRs (Fig. 4a, Supplementary Table S3). Among the 68 monomer SSRs, it should be emphasized that adenine (A) and thymine (T) monomer repeats constitute 30.88% and 44.12% respectively, while TA repeats are the most frequent dimeric types, comprising 20.63%. Tetrameric SSRs are the most common, representing 34.65% of the total SSRs. However, only seven hexamer SSRs were observed in the mitogenome. In the mitogenomes of M. biondii, M. officinalis, and L. tulipifera, we identified 273, 327, and 234 SSRs respectively. The prevalent SSR type is tetranucleotide, while the infrequent SSR type is hexanucleotide.
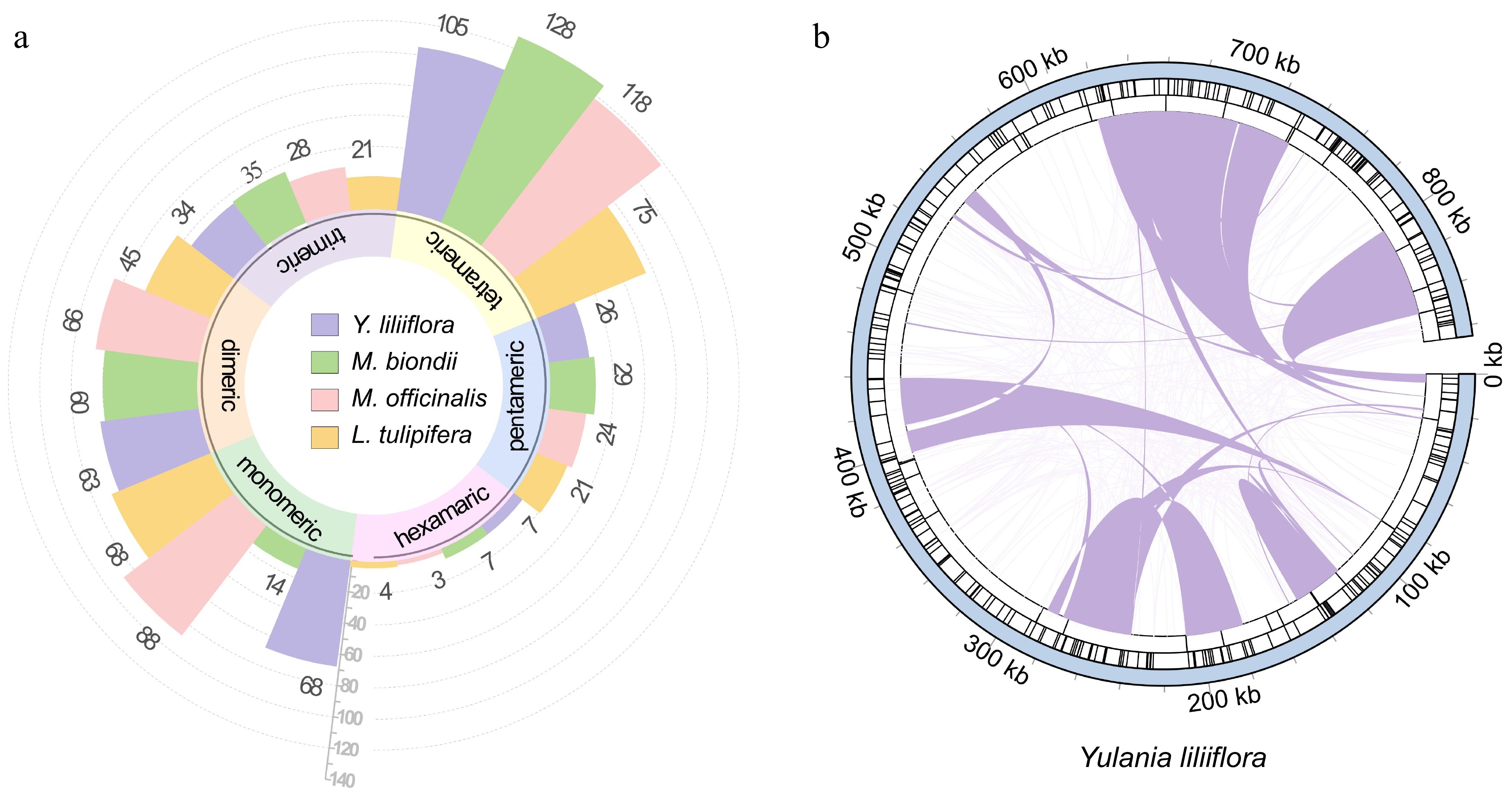
Figure 4.
Repetitive sequences of the Y. liliiflora mitogenome. (a) Ribbon bar graphs presenting SSRs. The legend is in purple, green, pink, and yellow for Y. liliiflora, M. biondii, M. officinalis, and L. tulipifera respectively, with every four bars clustered into one category starting from the 0 scale, representing monomers, dimers, trimers, tetramers, pentamers, and hexamers. (b) The repeats of Y. liliiflora mitogenome. The color line on the inner circle connects two repeated dispersed repeat. The deep purple lines represent ≥ 500 bp repetitive fragments. The tandem repeat is represented by short bars in the intermediate circle, while the SSRs are shown in the outer circle.
Tandem repeats, are a common phenomenon in eukaryotic genomes, with sporadic instances, also observed in certain prokaryotic genomes. In the mitogenome of Y. liliiflora, we identified a total of 31 tandem repeats, spanning a length range of 31 to 127 bp, and exhibiting a high degree of sequence similarity, exceeding 90%. As depicted in Fig. 4b, the Y. liliiflora mitogenome exhibits 328 pairs of dispersed repeats, each having a length equivalent to, or exceeding 50 bp. The analysis revealed 164 palindromic repeat pairs and 161 forward repeat pairs, whereas no complement or reverse repeats were detected among them (Supplementary Table S4). Among them, there are 25 dispersed repeats of more than 500 bp (Supplementary Table S5), and the longest forward repeat is 48,270 bp, whereas the longest palindromic repeat is 105,147 bp.
Migration sequences of Y. liliiflora
-
The transfer of genes from chloroplasts to mitochondrial genomes is a prevalent phenomenon observed during the extended evolution of angiosperm plants[52,53]. The mitogenome of Y. liliiflora contains 35 DNA fragments homologous to cp genomes (Fig. 5, Supplementary Table S6). The length of these fragments is 37,517 bp and constitutes 4.34% of the total length of the mitogenome. These findings substantiate the inference that mitochondrial DNA can be attributed to the chloroplast genome, constituting approximately 1%−10.3%[54]. Among them, there are a total of 19 MTPTs that are larger than 100 bp. The longest fragment is 5,866 bp. There are 10 complete chloroplast genes be encompassed in these homologous fragments, including nine PCGs (ndhJ, petG, petL, psbE, psbF, psbL, rpl14, rps7, and rps8), and one tRNA gene (tRNA-His).
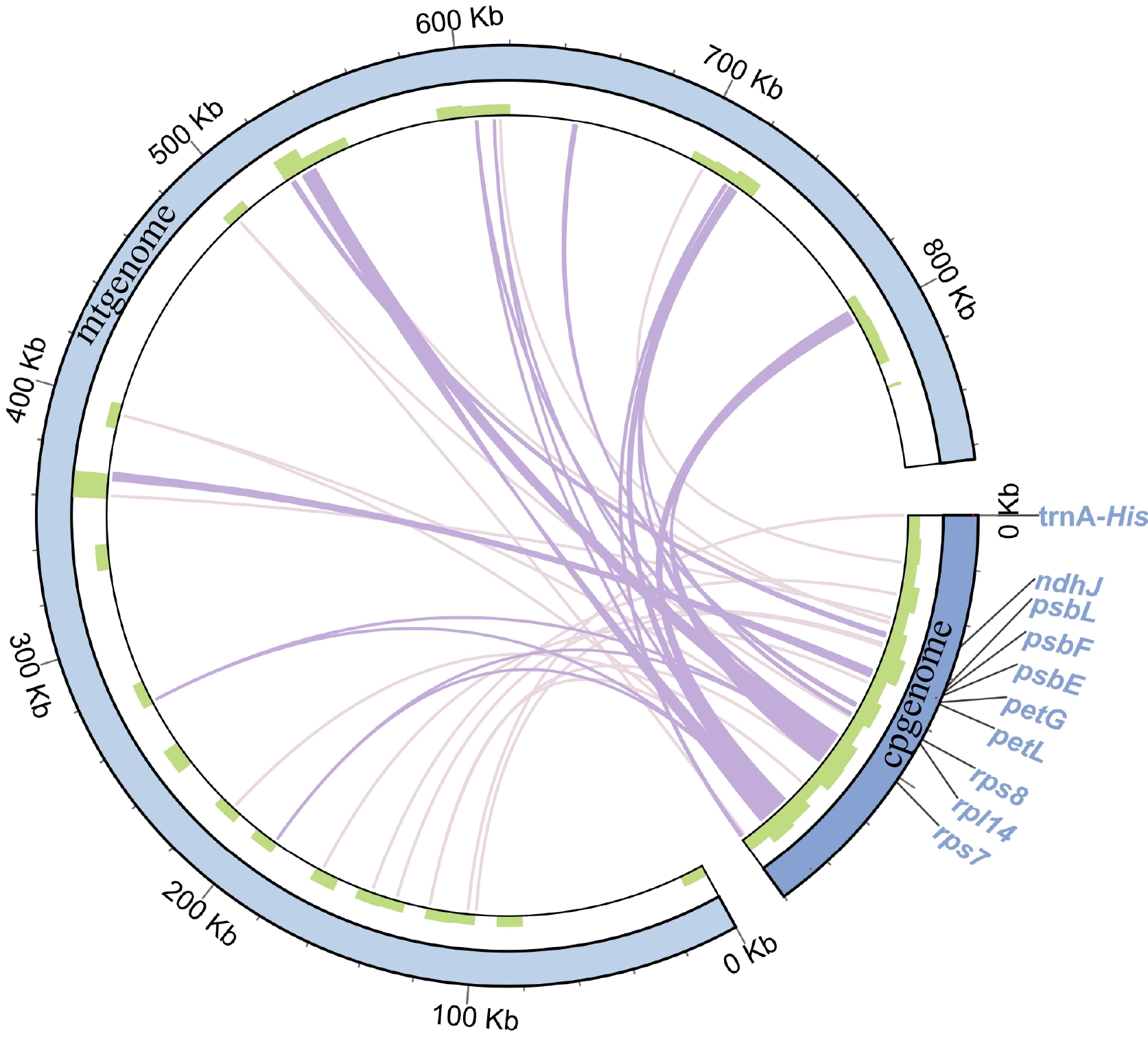
Figure 5.
Migration sequences of the Y. liliiflora mitogenome. The pale purple lines on the inner circle represent fragments that transfer from chloroplasts to mitochondria. The diagram displays transfer fragments of ≥ 100 bp as deep purple lines, while the sequence length is shown on the outside circle.
Prediction of RNA editing sites in PCGs
-
Within plant systems, RNA editing is crucial for modulating gene expression, specifically through the conversion of cytidine (C) to uridine (U) in mitogenomes[55]. By utilizing the Deepred-mt software to analyze the variations observed between the DNA and RNA sequences of the Y. liliiflora mitogenome, we predicted 894 potential C-U RNA editing sites distributed across 45 PCGs. Figure 6 shows that the nad4 gene has 68 identified editing sites, establishing it as the most extensively edited gene. In close succession, the nad5 gene displays 48 instances of RNA editing, while the rpl2 and rps14 genes exhibit only two editing events each. Most PCGs start with the typical ATG codon, although specific genes such as nad1, nad4L, cox1, mttB, rps4, and rps10 initiate with ACG. This phenomenon is plausibly a result of C to U RNA editing taking place at the second site. It should be noted that rpl16 uses GTG as its start codon, given that the actual start codon has not yet been definitively determined. As for PCGs, four distinct termination codons were identified: TGA, TAA, CAA, and CGA. This can be attributed to the observed phenomenon of C-to-U RNA editing, which is prevalent among the genes atp6, ccmFc, and rps11.
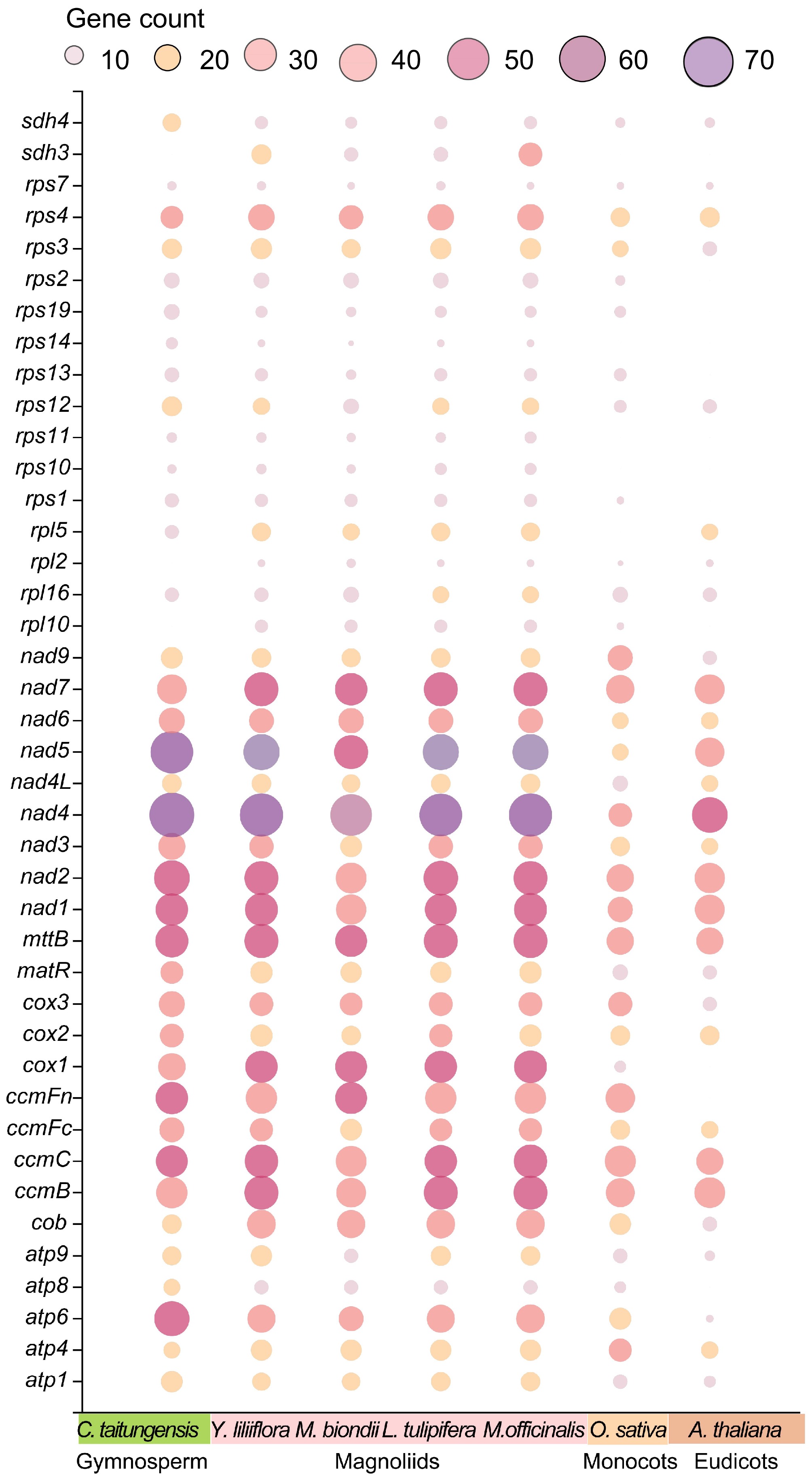
Figure 6.
Bubble plot of the number of RNA editing sites in mitogenome PCGs of C. taitungensis, Y. liliiflora, M. officinalis, M. biondii, L. tulipifera, O. sativa, and A. thaliana.
As shown in Fig. 6, it exhibits similar editing site numbers of PCGs in Magnoliids. For example, Y. liliiflora, M. biondii, and L. tulipifera contained 894, 734, and 834 C-U editing sites, respectively. The PCG copies of M. officinalis are notably abundant, resulting in the inclusion of 1184 C-U editing sites. The number of RNA editing sites exhibits a gradual decline, progressing from gymnosperms to angiosperms, from Magnoliids to monocots, and further to eudicots. Additionally, specific variable genes exhibit an absence of editing sites in A. thaliana, O. sativa, and C. taitungensis. The RNA editing events in plant mitogenomes result in alterations to start and stop codons, potentially influencing mitochondrial function and cellular metabolism. However, due to the lack of transcriptome data, the specific impact on Y. liliiflora remains to be further elucidated.
Phylogenetic and synteny analysis
-
To elucidate the phylogenetic classification of Y. liliiflora, we retrieved 16 plant mitogenomes and chloroplast genomes from GenBank. Phylogenetic trees were constructed using 22 mitochondrial PCGs derived from 17 species, spanning 12 orders of angiosperms (Amborellales, Nymphaeales, Austrobaileyales, Laurales, Magnoliales, Alismatales, Arecales, Poales, Malpighiales, Brassicales, Solanales, and Apiales) and two orders of Gymnospermae (Cycadales, Ginkgoales). G. biloba and C. taitungensis are designated as outgroups (Supplementary Tables S7 & S8). As depicted in Fig. 7, every node within the constructed tree boasts bootstrap support values surpassing 80%, and it is noteworthy that 10 nodes, specifically, enjoy a unanimous support of 100%. The phylogenetic trees originated from the chloroplast and mitogenomes align with the latest Angiosperm Phylogenetic Group (APG IV) classification[6]. The phylogenetic results provide robust support for the grouping of Y. liliiflora. The phylogenetic tree provides support for situating Y. liliiflora within the Magnoliids and delineates its associations with eudicots and monocots.
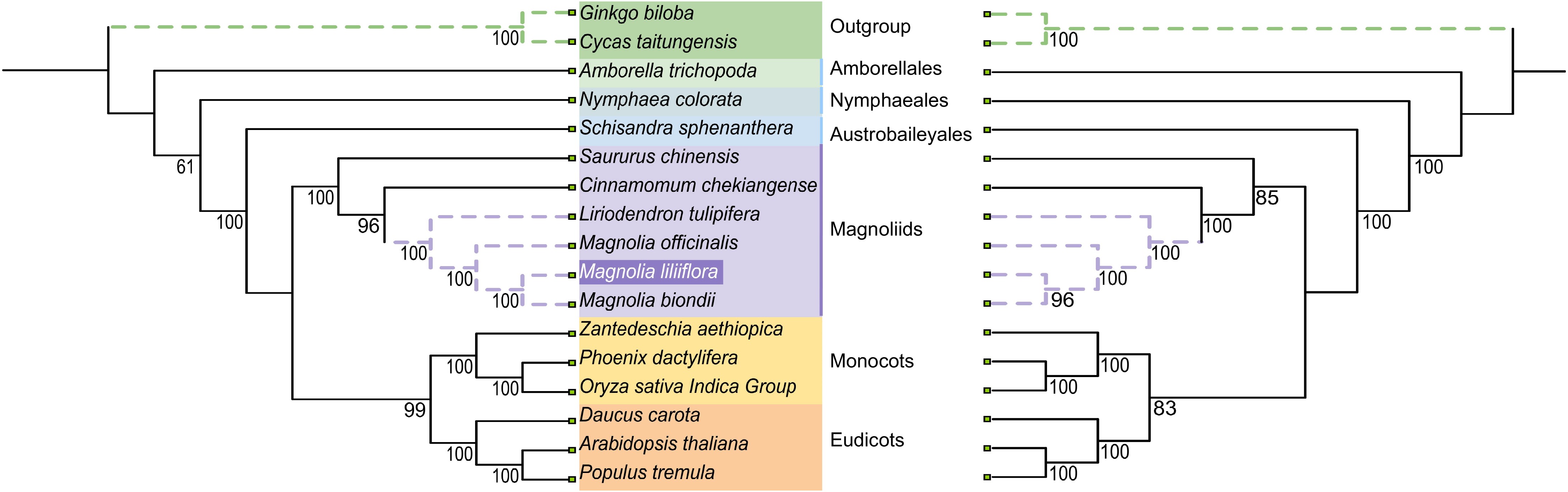
Figure 7.
The relationships of Y. liliiflora with the 16 other represented land plants are shown based on the chloroplast (left) and mitochondrial (right) genomes. Bootstrap support values are presented at each node.
The study identified and analyzed mitogenome collinearity among five Magnoliid species with a matching degree greater than 90 and a length of more than 500 bp. To analyze the structural collinearity of Y. liliiflora, it was compared with M. officinalis, L. tulipifera, C. chekiangense, and H. nymphaeifolia. The principal factor driving the evolution of plant mitogenomes is frequent rearrangement. As depicted in Fig. 8, between the mitogenomes of Y. liliiflora and M. officinalis, a total of 96 local colinear blocks are present, accounting for 95.78% (828,710 bp) of the whole Y. liliiflora mitogenome. Between the mitogenomes of Y. liliiflora and L. tulipifera, we detected 91 local colinear blocks (total length: 443,814 bp; 51.30% of the mitogenome). Comparing the mitogenome of Y. liliiflora with those of C. chekiangense and H. nymphaeifolia, the colinear sequences only accounts for 27.55% and 23.26% of Y. liliiflora whole mitogenome, respectively. The colinear length and ratio of C. chekiangense, and H. nymphaeifolia are lower than those of the other two species (M. officinalis and L. tulipifera) of Magnoliids. While the degree of collinearity remains significant, it is noteworthy that the complex distribution of collinear segments across the contigs exhibits considerable diversity in sequence orientation among the various species. However, the study detected numerous similar segments between Y. liliiflora and the four species, indicating a distinct relationship (Fig. 8).
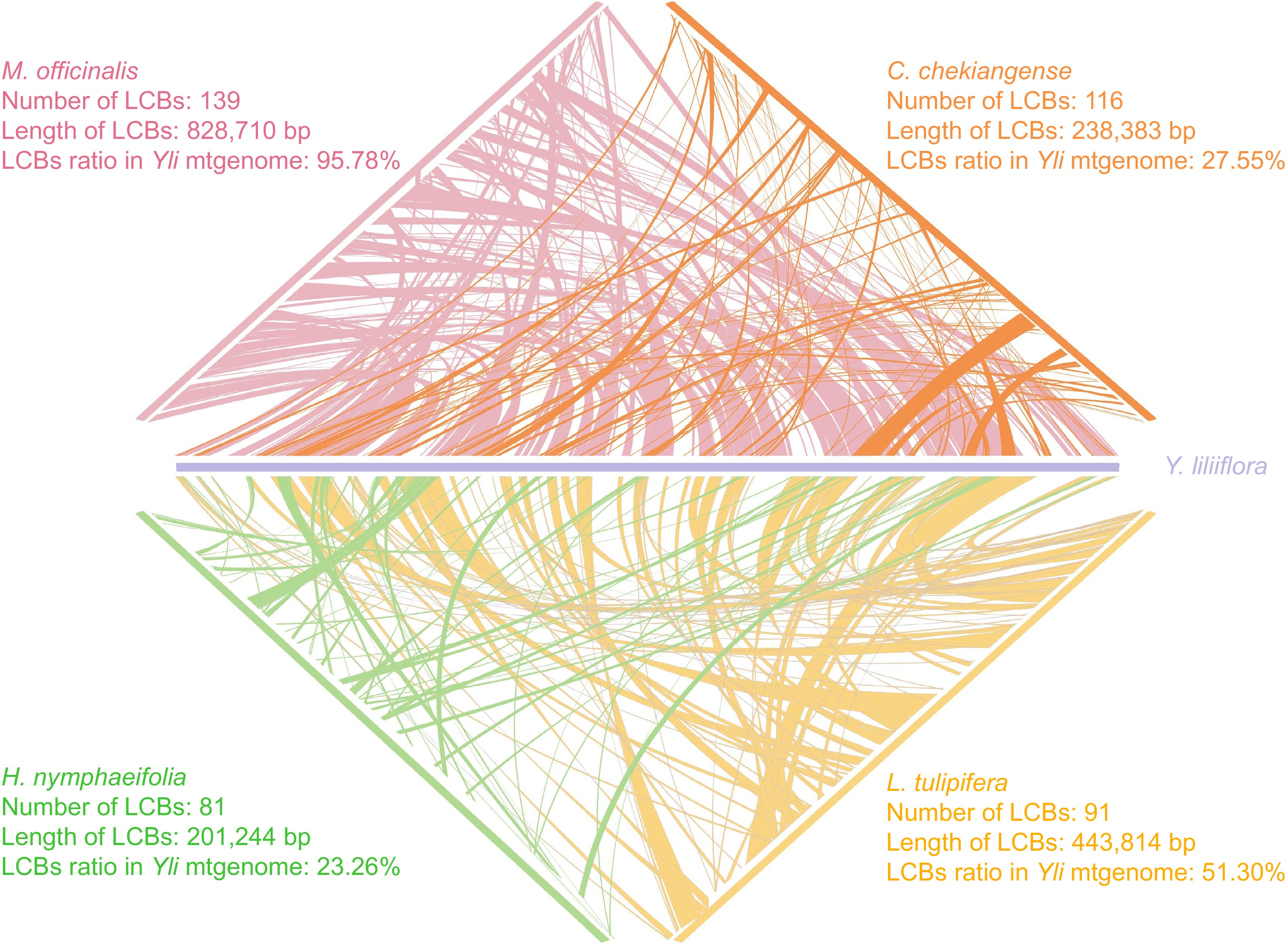
Figure 8.
Mitogenome synteny of five species. The purple, pink, orange, yellow, and green legends represent Y. liliiflora, M. officinalis, C. chekiangense, L. tulipifera, and H. nymphaeifolia, respectively.
-
The complete mitogenome of Y. liliiflora was assembled into a breach linear structure, with 865,191 bp in length (GenBank accession number: NC_085212.1), reflecting the average size of Magnoliids mitogenomes. Notably, within the Magnoliid clade, mitogenome sizes exhibit substantial variability, extending from a minimum of 535,805 bp in H. ymphaeifolia to a maximum of 967,100 bp in M. biondii[56]. However, the size of most mitogenomes of angiosperms ranges from 200 to 800 kb, with Magnoliid being on the large side[18]. Research into structure and size in plant mitogenomes is important because the evolution of plant mitogenomes involves notable episodes of breakage and rejoining, accompanied by DNA sequence gain and loss, more likely driven by non-adaptive selection[57−59]. During this process, repeats, and MTPTs may occur. Previous studies observed repeats within vascular plants tend to be larger and occur with greater frequency[59,60], such as four larger repeats (63.9, 10.6, 9.1, and 2.5 kb) in Gossypium raimondii mitogenome, and intramolecular recombination in the evolution of higher plants has been identified as an active process[61]. Meanwhile, some large repeats (> 1 kb) were identified in Y. liliiflora mitogenome, and this phenomenon was also reported in L. tulipifera (Magnoliids)[59,62]. Research has indicated that the presence of repeats in vascular plants is related to the size and complexitity of mitogenomes[59]. In addition, MTPT also has a significant impact on the size of the mitogenome. For example, Amborella trichopoda has acquired foreign mitochondrial DNA from sources such as green algae, mosses, and other angiosperms[63]. Nevertheless, further examination is necessary to explore their potential influence on the phenotypic traits of Y. liliiflora.
Structure and intron contents of the Y. liliiflora mitogenome
-
Ever since the initial discovery of the mitogenome in the land-dwelling Marchantia polymorpha[13], we have been intrigued by the classical circular DNA structure. Emerging evidence indicates that, in terms of stoichiometry, multi-chromosomal arrangements comprising circular, overlapping, and linear molecules could outpace the prevalence of the circular mapping architecture exhibited plant mitogenomes[15]. The mitogenome of Ilex macrocarpa and Salix cardiophylla have typical circular structures, while the mitogenomes of Lactuca linnaeus and Nicotiana tabacum have complex structures[64−67]. Indeed, the mitogenome sizes exhibit significant variations across the Magnoliids clade, such as M. biondii[56], C. chekiangense[68] were assembled into a single circular molecule. It is significant to point out that the mitogenome structure of Y. liliiflora is breach linear in shape.
Mitogenomes have been found to occur in complex structures depending on the stage of plastid maturation[69,70]. The abundance of repetitive sequences can enhance recombination events, facilitate exogenous DNA integration, and contribute to the complexity of mitogenome structure, thereby enabling, for instance, distinctive self-replication of three mitochondrial chromosomes is noted in cucumber plant cells[69,71]. The linear structure also may be attributed to the telomeric recombination's significant role in the formation of breached linear organelle chromosomes[14]. In addition, this study identified colinear regions and rearrangements among five Magnoliids mitogenomes, revealing a high degree of homology as depicted in Fig. 8. The mitogenomes of Y. liliiflora and two Magnoliaceae species (M. officinalis and L. lulipifera) showed the highest collinearity (95.78%, 51.30%), while the collinearity between Y. liliiflora and Lauraceae (C. chekiangense, H. nymphaeifolia) are over 20%. The collinearity analysis results indicate that the mitogenome of Magnoliaceae may be more evolutionarily conserved.
Previous studies have found that the phenomenon of trans-splicing has manifested recurrently in numerous lineages of vascular plants, with a significant portion originating from ancestors that employed cis-splicing[18,72−74]. According to research by Guo et al. and Mower, across angiosperms and gymnosperms, there is a shared presence of five mitochondrial trans-spliced group II introns, suggesting their probable evolution in the common ancestor through the fragmentation of a cis-spliced arrangement[72,73], which confirms the fact that nad1, nad2, and nad5 contains trans-spliced in our study. This result also strongly suggests that the mitogenome of Y. liliiflora is highly conserved.
PCG content of the Y. liliiflora mitogenome
-
In endosymbiotic evolution, organellar gene content declined, yet photosynthetic and respiratory chains, along with protein synthesis components, remained conserved across lineages[73,75]. The core PCGs consist of five ATP synthase genes (ap1, 4, 6, 8, and 9), four cytochrome C biogenesis genes (ccmB, C, Fc, and Fn), ubiquinol cytochrome c reductase (cob), three cytochrome C oxidase genes (cox1−3), 9 NADH dehydrogenase genes (nadl-7, 9, and 4L), a transport membrane protein (mttB/tatC), and a maturase (matR). The variable PCGs consist of five large subunits of ribosome proteins (rpl2, 5, 6, 10, and 16), 12 small subunits of ribosome proteins (rps1−4, 7, 8, 10−14, and 19), and two respiratory genes (sdh3−4). The mitogenome of Y. liliiflora mainly consists of non-coding sequences, with PCGs only accounting for 4.24% in the mitogenome. As depicted in Supplementary Fig. S3, all 24 core PCGs and 17 variable PCGs were identified in the Y. liliiflora. Almost all of the ancestral 24 core PCGs were identified, such G. biloba (gymnosperm)[76], L. tulipifera (Magnoliids)[62], O. sativa (monocots)[77], and A. thaliana (eudicots). During the evolutionary process from bryophytes to the ancestral line of angiosperms, there was a loss of the rpl6 and rps8 genes. However, the rps11 gene, while retained in gymnosperms, seems to have vanished in both monocots and eudicots, presumably during the split of these groups from the gymnosperms. Following the divergence of monocots and eudicots, a distinct pattern of gene loss emerged, with four genes (sdh3, sdh4, rps10, and rps14) being absent in monocots and rps2 being lost in eudicots. However, variable genes are more severely missing in monocots and eudicots plants. In sharp contrast to gene loss, gene duplication emerges as one of the principal genetic mechanisms supporting the expansion of gene families in mitogenomes[78,79]. In the mitogenome of Y. liliiflora, atp4, nad3, rps7, and rps12 have been duplicated, which is possibly related to the significant physiological functions, but further research is needed to confirm.
Plastid and mitochondrial phylogeny
-
The phylogenetic relationships of the five major lineages of the Mesangiospermae (Ceratophyllales, Chloranthales, eudicots, Magnoliids, and monocots) remain unclear, despite extensive studies of their diversification. As compared to Magnoliids, the phylogenetic trees constructed from organellar genomes currently suggest a closer evolutionary affiliation between monocots and eudicots[80,81]. Intergeneric relationships based on the mitochondrion and chloroplast dataset are generally well supported. The phylogenetic results obtained from the mitochondrial and chloroplast datasets are consistent, indicating that Y. liliiflora is the sister to M. biondii (posterior probability [PP] = 0.96). Y. liliiflora, M. biondii, and M. officinalis constitute the Magnolia genus, and they are sister groups with L. tulipifera. The plastid sequence data, inclusive of our dataset, unambiguously situates Y. liliiflora belongs to Magnoliales order and is closely related to the Laurales. The remaining species form two major clades. Within the updated APG IV classification framework, angiosperms are segregated into two major groups: basal angiosperms and Mesangiospermae[6]. Amborellales, Nymphaeales, and Austrobaileyales constitute the basal angiosperms, known as ANA clade, which is sister to all remaining selected species (PP > 0.99). Another clade formed by eudicots and monocots as successive sister groups to Magnoliids (PP > 0.83). The results show that Magnoliids is sister to the clade consisting of monocots and eudicots, which agrees with previous studies[80,81]. Nevertheless, the phylogenetic associations inferred from nuclear genes demonstrate a notable instability among the five Mesangiospermae clades[81]. This finding points to a potential occurrence of hybridization and introgression during the early evolutionary history of angiosperms.
-
In this study, we assembled and annotated the mitogenome of Y. liliiflora, undertaking comprehensive analyses utilizing both DNA and amino acid sequences derived from the annotated genes. The mitogenome of Y. liliiflora exhibits a breach linear structure, encompassing a length of 865,191 bp. We annotated 69 genes, including 45 protein-coding, 21 tRNA, and three rRNA genes. We detected 17 cis- and five trans-splicing introns in the mitogenome of Y. liliiflora. Furthermore, we conducted analyses on codon usage, sequence repeats, RNA editing, and sequence migration within the Y. liliiflora mitogenome. Phylogenetic analyses demonstrate that Y. liliiflora has the closest genetic relationship with M. biondii of Magnoliaceae, and Magnoliids emerge as a closely related group to the clade encompassing monocots and eudicots. The results presented in this paper supplement the existing genetic knowledge pertaining to the genus Yulania. Simultaneously, they offer a promising avenue for conducting more comprehensive genomic investigations of Y. liliiflora.
The work is supported by the Natural Science Foundation of Jiangsu Province (BK20220414). We thank Assoc. Prof. Kewang Xu from Nanjing Forestry University for collecting the samples of Y. liliiflora. We would also like to express our gratitude to Assoc. Prof. Meng Li from Nanjing Forestry University for providing photos of Y. liliiflora.
-
The authors confirm contribution to the paper as follows: study design and leading: Bi C; research conducting: Chen Y, Qiu J, Liu W; materials collecting: Bi C; DNA for sequencing preparation and analysis: Bi C; data analysis and manuscript writing: Chen Y; manuscript revision: Bi C, Qiu J. All authors reviewed the results and approved the final version of the manuscript.
-
The sequencing data have been deposited in the NCBI under accession number NC_085212.1.
-
The authors declare that they have no conflict of interest.
- Supplementary Table S1 Genome contents of the Y. liliiflora mitogenome.
- Supplementary Table S2 Relative synonymous codon use of codons by individual amino acids in the Y. liliiflora mitochondrial genome.
- Supplementary Table S3 SSRs in the mitochondrial genome of Y. liliiflora.
- Supplementary Table S4 Tandem repeat sequences in the mitochondrial genome of Y. liliiflora.
- Supplementary Table S5 Dispersed repeat sequences in the mitochondrial genome of Y. liliiflora.
- Supplementary Table S6 The homologous DNA fragment in the Y. liliiflora mitogenome.
- Supplementary Table S7 The taxonomy of Y. liliiflora and 16 other selected species with their accession numbers were obtained from the NCBI Nucleotide Database to construct a phylogenetic tree of the mitogenome.
- Supplementary Table S8 The taxonomy of Y. liliiflora and 16 other selected species with their accession numbers were obtained from the NCBI Nucleotide Database to construct a phylogenetic tree of the chloroplast genome.
- Supplementary Fig. S1 (a, b)Mitogenome structure from Y. liliiflora accessions generated using bandage. (c, d) chloroplast genome structure from Y. liliiflora accessions generated using bandage.
- Supplementary Fig. S2 Trans-splicing and cis-splicing genes of Y. liliiflora mitogenome.
- Supplementary Fig. S3 Bubble plot of mitochondrial PCGs content in Y. liliiflora and 16 other terrestrial species, with pink representing 1, yellow representing 2, and green representing 3.
- Copyright: © 2025 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Chen Y, Liu W, Qiu J, Bi C. 2025. Assembly and comparative analysis of the complete mitochondrial genome of Yulania liliiflora: an ornamental plant with high medicinal value. Ornamental Plant Research 5: e019 doi: 10.48130/opr-0025-0017 

Assembly and comparative analysis of the complete mitochondrial genome of Yulania liliiflora: an ornamental plant with high medicinal value
- Received: 25 November 2024
- Revised: 10 February 2025
- Accepted: 03 March 2025
- Published online: 06 May 2025
Abstract: Yulania liliiflora is a highly exploitable plant with medicinal value from its flower buds, leaves, and bark. The species, as an ornamental plant, has significant implications for the phylogeny of land plants and offers promising prospects for scientific research. In this study, the Y. liliiflora mitochondrial genome (mitogenome) was assembled into a breach linear chromosome using PacBio HiFi sequencing, with a length of 865,191 bp and a GC content of 46.95%. A total of 69 genes were identified in the Y. liliiflora mitogenome. The Y. liliiflora mitogenome contains 65 unique genes, including 41 protein-coding genes, 21 tRNA genes, and three rRNA genes. Numerous repetitive sequences have been discovered, revealing the recombination events and the reason for the bigger mitogenome of Y. liliiflora. Many mitochondrial plastid sequences were found, with 12 complete chloroplast genes encompassed in these homologous fragments. Mitogenome from other Magnoliids were also used for collinearity comparison, which suggests a high recombination rate between Y. liliiflora and Magnoliids. Analysis of phylogeny has demonstrated that Y. liliiflora had the strongest genetic relation to Magnolia biondii of the Magnoliaceae, and Magnoliids emerged as a sister group to the clade encompassing monocots and eudicots. The results presented in this paper improve the understanding of the existing genetic knowledge of the genus Yulania. Simultaneously, they offer a promising avenue for conducting more comprehensive genomic investigations of Y. liliiflora.