









-
Tiegun yam, first documented in the 'Henei County Annals' in 1838[1], has become a widely cultivated medicinal herb, primarily due to its superior quality and high market demand. In the Jiaozuo area of Henan Province, China, the annual planting area of Tiegun yam exceeds 8,000 hectares, yielding approximately 260,000 tons. As a result, Tiegun yam has emerged as the largest and most premium variety of Huai yam. Huai yam refers to the dried rhizome of Dioscorea opposita Thunb. (D. opposita), a species in the Dioscoreaceae family, and is renowned as one of the 'Four Huai Medicines'. Research indicates that Huai yam, particularly Tiegun yam, exhibits a range of physiological and pharmacological properties, including immunomodulation, anti-tumor effects, hypoglycemic, antioxidant activity, and anti-aging benefits[2−4]. Moreover, studies have shown that the total flavone and flavonoid content in the epidermis of Tiegun yam exceeds that in the flesh, with the epidermal part demonstrating greater effectiveness in reactive oxygen species (ROS) scavenging test and anti-tumor properties in mice[5].
The name 'Tiegun' (meaning 'iron rod') derives from the yam's firm, powdery texture and slender, elongated shape. Additionally, the rhizome's epidermis often displays irregular brownish-red spots, referred to as 'Rusty spot' (Rs), which is considered a distinguishing feature of high-quality Tiegun yam. These characteristics set Tiegun yam apart from other yam varieties. However, there is limited research on the correlation between the presence of Rs and the quality or authenticity of Tiegun yam. Furthermore, the chemical composition and formation mechanism of the Rs have not been extensively studied. Based on years of observation, we noted that Rs begins to appear when the plant enters dormancy after the Frost's Descent (FD), a unique physiological trait and external manifestation of Tiegun yam, not a result of pests or disease damage. Recent studies in our laboratory have identified the chemical composition of Rs pigments as anthocyanins[6], which are known to possess antioxidant, anti-cancer, anti-cardiovascular, and other health-promoting properties[7]. This finding supports the notion that the presence of Rs is a significant marker of high-quality Tiegun yam.
Anthocyanins, a class of flavonoid metabolites, play crucial roles in plant physiology, including attracting pollinators, aiding in seed dispersal, and protecting plants from ultraviolet radiation and oxidative stress[8]. Additionally, anthocyanins have beneficial effects on human health. Studies have shown that the synthesis of anthocyanins in plants is influenced by environmental factors, including cold, light, drought, and salt stress[9,10]. The biosynthesis pathway and molecular regulation of anthocyanins have been well-characterized in the model plant Arabidopsis thaliana[10]. Anthocyanin biosynthesis involves both enzyme catalysis by structural genes and regulation by transcription factors. Key enzymes in this pathway include Chalcone Synthase (CHS), Chalcone Isomerase (CHI), Flavanone-3-Hydroxylase (F3H), Dihydroflavonol-4-Reductase (DFR), Anthocyanidin Synthase (ANS), and Glycoside Transferase (UFGT)[9−14]. Furthermore, transcription factors, particularly the MBW complex (MYB, basic helix-loop-helix (bHLH), and WD40 family), play a pivotal role in regulating these enzymes[15−18]. However, the biosynthesis mechanism of anthocyanins in Tiegun yam Rs remains largely unexplored, primarily due to the limited molecular biology research on this species.
To conduct comprehensive molecular biology research on Tiegun yam, a reference genome is essential. Although Tiegun yam is a prominent medicinal material, its genetic background remains unclear, and a reference genome is not yet available. In contrast, the reference genome of Dioscorea alata (Greater yam), a closely related species, has been published[19], which could serve as a preliminary resource for studying anthocyanin biosynthesis in Tiegun yam. However, significant morphological differences between these species suggest that their genomes may also differ substantially. Notably, Dioscorea alata (D. alata) has typically 40 chromosomes (2n = 40)[20], whereas Tiegun yam has 120 chromosomes[21], complicating the use of D. alata's genome for accurate analysis of Tiegun yam's Rs synthesis and regulation. To address these challenges and better understand the genetic foundation of Tiegun yam, we conducted genomic assembly, metabolomic, and transcriptomic sequencing analyses in this study. These efforts aim to uncover the regulatory mechanisms of anthocyanin biosynthesis in Tiegun yam Rs, explore the genetic traits associated with superior quality, and provide foundational knowledge for breeding improvements and the resolution of production challenges.
-
A total of 10 g of Tiegun yam tissues, including bulbil, stem, leaf, rhizome epidermis (with or without Rs), were weighed and placed in a conical flask. Subsequently, 50 mL of a 0.5% hydrochloric acid methanol solution was added. The flask was sealed, and the mixture was extracted in a constant temperature water bath set to 50 °C. The extraction process was repeated three times, each for 3 h. The extracts from each cycle were combined, centrifuged at 4,000 rpm for 5 min, and the supernatant was collected as the anthocyanin extract. The extract was then concentrated to 1/3 of its original volume for future use[22].
The pretreated D101 macroporous resin was first equilibrated with distilled water. The anthocyanin extract was added to the resin column and eluted sequentially with distilled water containing 0.5% HCl, 10% methanol containing 0.5% HCl, and 50% methanol containing 0.5% HCl, using five column volumes for each elution step. Finally, the column was eluted with methanol containing 0.5% HCl, and then the 50% methanol eluate was collected[23]. The collected elution was concentrated under reduced pressure on a rotary evaporator (EYELA, Japan), followed by freeze-drying to obtain purified anthocyanins.
The anthocyanin extraction solution was analyzed for its absorbance value at a wavelength of 526 nm. A similar procedure was used to extract and purify anthocyanins from another rhizome epidermis with Rs. The same was then sent to Sanshu Biotechnology Co., Ltd. (Nantong, Jiangsu Province, China) for anthocyanin component determination.
UPLC-ESI-HRMS analysis of anthocyanins
-
Anthocyanin extracts were analyzed using ultra-high-performance liquid chromatography coupled with high-resolution mass spectrometry (UPLC-HRMS) employing electrospray ionization (ESI). Chromatographic separation was performed on a Waters HSS T3 column (100 mm × 2.1 mm i.d., 1.8 μm particle size) maintained at a flow rate of 0.3 mL/min. The injection volume was 2 µL, and detection was carried out at 254 nm. The mobile phase comprised (A) 0.1% (v/v) aqueous formic acid, and (B) 0.1% (v/v) formic acid in acetonitrile. The gradient elution program was: 5% B (0–2 min), increasing linearly to 30% B (2–25 min).
Mass spectrometric analysis was conducted in positive-ion mode using a Thermo Scientific Q Exactive mass spectrometer. The mass resolution was set to 70,000 (FWHM at m/z 200). The ESI source parameters were optimized as follows: sheath gas flow, 40 arb; auxiliary gas flow, 10 arb; spray voltage, 3.0 kV; capillary temperature, 350 °C.
DNA extraction and sequencing of Tiegun yam (D. opposita) tissue samples
-
High-quality genomic DNA was extracted from leaf using a modified CTAB method. For each sample, 1 μg of DNA served as input material for library preparation. Sequencing libraries were constructed using the VAHTS Universal DNA Library Preparation Kit for MGI (Vazyme, Nanjing, China) following the manufacturer's protocol, with unique index codes assigned to attribute sequences. Library quantification and size distribution were assessed using a Qubit 3.0 Fluorometer (Life Technologies, Carlsbad, CA, USA) and a Bioanalyzer 2100 system (Agilent Technologies, CA, USA). Sequencing was performed on the DNBSEQ-T7 platform (Frasergen Bioinformatics Co., Ltd., Wuhan, China). Raw genomic sequencing data were filtered using Trimmomatic (v0.38) with parameters: 'LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:15'[24]. Data quality was evaluated using FastQC (v0.11.7) under default settings[25].
Genome assembly and annotation
-
The D. opposite genome was assembled using Hifiasm (v0.19.5). To generate monoploid-representative sequences, parameters '-l3 –n-hap 6' were applied, and sequence graphs in GAF format were converted to FASTA format using gfatools (
https://github.com/lh3/gfatools ). PacBio Sequel II/Revio sequencing yielded 28.75 Gb (~ 68 × coverage) of high- accuracy HiFi reads (> Q20, > 99% accuracy). All HiFi reads were utilized for de novo assembly[26]. The final assembly comprised 349 contigs totaling 424.05 Mb, with an N50 of 6.16 Mb.Complementary methods were employed to evaluate the quality of the genome assembly. First, completeness was assessed using conserved embryophyte genes from the BUSCO embryophyta_odb10 database. Second, Illumina short-reads and TGS long-reads were mapped to the assembly using BWA (v0.7.17; mem -t 50)[27] and minimap2 (v2.21), respectively, to determine coverage and average depth. Third, structural integrity was evaluated with LTR_retriever (v2.9.0)[28]. Fourth, consensus quality (QV) and completeness were assessed using Merqury (v1.3)[29].
Chromosome assignment using Hi-C
-
Hi-C reads were trimmed with Trimmomatic (v0.38)[24] to remove adapters and low-quality bases. Filtered reads were aligned to contigs using Juicer (v1.6;
https://github.com/aidenlab/juicer ) to calculate the contact frequency. Scaffolding and misjoin correction were performed with 3D-DNA (v180922)[30] using two iterative rounds (-r1). The resulting scaffolds were oriented, and interaction matrices were generated with Juicer for manual validation and correction in Juicebox (v1.11.08)[31].Genome comparison and evolution analysis
-
Protein sequences were obtained from 15 plant species: Ipomoea batatas (GCA_002525835.2), Cajanus cajan (GCA_000340665.2), Arobidopsis thaliana (TAIR10), Dioscorea zingiberensis (GCA_026586065.1), Dioscorea Cayenensis (GCA_009730915.2), Dioscorea rotundata (GCF_009730915.1), Dioscorea alata (GCA_020875875.1), Dioscorea opposita (this study), Dioscorea dumetorum (GCA_902712375.1), Zingiber officinale (GCA_018446385.1), Elaeis guineensis (GCA_000442705.1), Phoenix dactylifera (GCA_009389715.1), Allium cepa (GCA_030765085.1), and Dendrobium catenatum (GCA_001605985.2). Orthologous groups were identified using BLASTP (E-value < 1e-7) (
https://blast.ncbi.nlm.nih.gov/Blast.cgi ) and clustered with OrthoFinder (v2.5.4; inflation parameter: 1.5)[32]. Single-copy orthologs were aligned with MUSCLE[33], and a phylogenetic tree was reconstructed using RAxML (v8.2.12)[34]. Divergence times were estimated with MCMCTree in PAML (v4.10.0) packages[35], calibrated using four fossil constraints from TimeTree (www.timetree.org ): Monocots-Dicots (173–148 Mya), A. thaliana−Populus. trichocarpa (109–97 Mya), A. thaliana−Vitis. vinifera (115–105 Mya), and Helianthus. annuus−Solanum. lycopersicum (107–93 Mya). Gene family expansion/contraction was analyzed with CAFÉ (v4.2.1)[36].Flow cytometry detection of D. opposita chromosome ploidy
-
Fresh leaf tissue (0.2 g) was dissected in a Petri dish containing 500 μL of nuclear extraction buffer. Samples were homogenized with a razor blade and incubated for 60 s to release intact nuclei. Homogenates were filtered through a 50-μM nylon mesh, and nuclei were stained with 2,000 μL of DAPI solution in the dark for 2 min. Ploidy analysis was performed using a CyFlow Space flow cytometer (Sysmex PartecTM, Görlitz, Germany).
-
The chemical components responsible for the characteristic Rs on Tiegun yam (D. opposita) rhizomes are anthocyanins. Tiegun yam, a distinguished variety within the Huai yam category, is primarily cultivated for its rhizome. The presence of Rs on the rhizome epidermis serves as a specific external marker and an authentic characteristic of Tiegun yam. Our recent study established the anthocyanin nature of these Rs pigments through microscopic examination, ultraviolet spectral absorption scanning, and liquid chromatography analyses[6]. Field observation over multiple years indicates that Rs formation initiates after the FD period (Fig. 1a) and is confined exclusively to the rhizome epidermis, with no occurrence in subepidermal tissues (Supplementary Fig. S1). Quantification revealed significantly elevated anthocyanin levels within Rs compared to adjacent Rs-free epidermal regions (Fig. 1b). Furthermore, metabolomic profiling identified the predominant anthocyanins in Rs as cyanidin, peonidin, and delphinidin, along with several of their glycoside derivatives (Fig. 1c; Table 1). The anthocyanin profile of Tiegun yam Rs differs notably from that of Greater yam (D. alata), suggesting distinct biosynthetic pathways and regulatory mechanisms for anthocyanins between these two yam species. Collectively, these findings demonstrate that anthocyanin biosynthesis occurs in specific areas of the Tiegun yam rhizome epidermis following FD, resulting in the formation of the characteristic Rs.
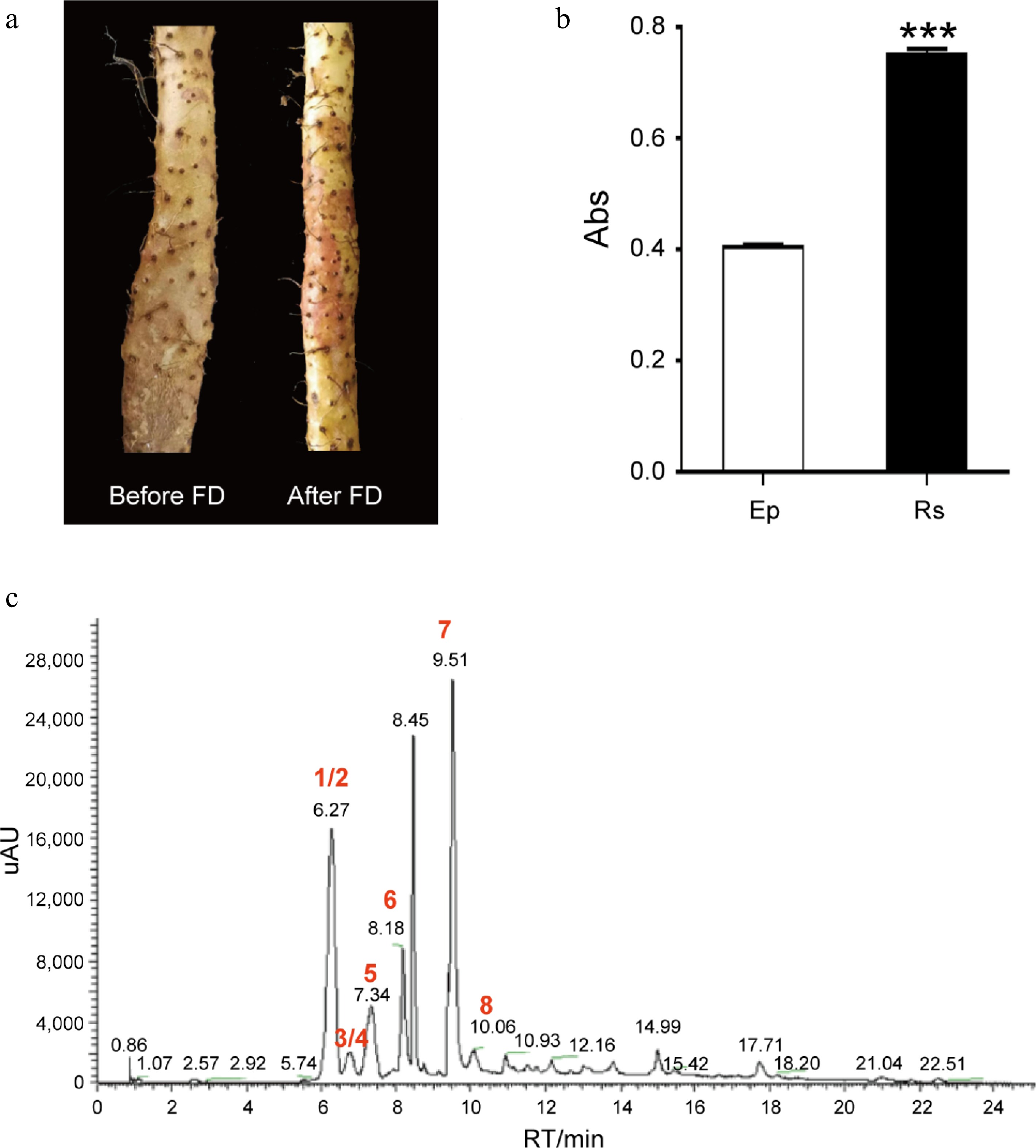
Figure 1.
Rs is the authentic characteristic of D. opposita. (a) Rhizome epidermis of D. opposita before and after Frost's Descent (FD). (b) Determination of anthocyanin content in rhizome epidermis without Rs (Ep) or with Rs by UV spectrum, Values are Mean ± SD, Student test, *** p < 0.001 compared with Rs (n = 3). (c) HPLC analysis of the main types of anthocyanins in Rs. Numbers 1–8 represents compounds cyanidin-3,5-O-diglucoside (1), cyanidin-3-O-sophoroside (2), cyanidin-3-O-galactoside (3), cyanidin-3-O-glucoside (4), peonidin-3,5-O-diglucoside (5), delphinidin (6), cyanidin (7), and peonidin (8), respectively. Among them, 1/2 (and 3/4) means either one of them could be the possible compound. UV: Ultraviolet, HPLC: High Performance Liquid Chromatography.
Table 1. The main types of anthocyanins in D. opposita Rs.
No. Compound Chemical formula Ionization mode m/z (Predicted) m/z (Measured) RT/min Fragment ion-1 Fragment ion-2 1 Cyanidin-3,5-O-diglucoside C27H31O16 M+ 611.16066 611.16034 6.29 449.10837 287.05466 2 Cyanidin-3-O-sophoroside C27H31O16 M+ 611.16066 611.16034 6.29 287.05466 − 3 Cyanidin-3-O-galactoside C21H21O11 M+ 449.10784 449.10745 6.84 287.05453 − 4 Cyanidin-3-O-glucoside C21H21O11 M+ 449.10784 449.10745 6.84 287.05453 − 5 Peonidin-3,5-O-diglucoside C28H33O16 M+ 625.17631 625.17596 7.41 − 301.07007 6 Delphinidin C15H11O7 M+ 303.04993 303.04956 8.25 303.04944 − 7 Cyanidin C15H11O6 M+ 287.05501 287.05466 9.57 287.05466 − 8 Peonidin C16H13O6 M+ 301.07066 301.07028 10.14 301.07025 − 9 Petunidin C16H13O7 M+ 317.06558 317.06509 19.25 317.06522 − RT: Retention Time of HPLC, SLD: Second-Level Debris of TIC, −: Fragment ion was not detected. Sequencing, assembly, and annotation of D. opposita genome
-
Due to the absence of a reference genome for D. opposita, the study of genes regulating anthocyanin synthesis in Tiegun yam Rs has been limited. To better understand the authentic characteristics of Tiegun yam and elucidate the molecular mechanisms of anthocyanin biosynthesis regulation in Rs, we have initiated the sequencing and assembly of the D. opposita genome.
Tiegun yam, the primary cultivated variety of Huai yam was selected for the assembly of the D. opposita genome, generating 276.78 Gb of paired-end reads from BGI (Table 1 and Supplementary Tables S1). K-mer analysis indicated a genome size of 424.05 Mb (Supplementary Table S2). Additionally, 28.75 Gb (68x) of PacBio-HiFi reads (N50 = 19,533 bp) were produced, which were integrated into 349 contigs (424.05 Mb, ContigN50 = 6.16 Mb) and 272 scaffolds (24.05 Mb, scaffold N50 = 19.06 Mb) using the HiFiasm assembly pipeline (Supplementary Tables S2 and S3). The average GC content of D. opposita (38.50%) is higher than that of T. zeylanicus (36.5%)[37] and D. rotunda (35.83%)[38], but lower than that of N. ossifragum (42.5%) (PRJEB71485).
To assess the quality and completeness of the genome, we compared the BGI data, achieving an alignment rate of 97.02%. The BUSCO (v5.2.2) gene set used was embryophyta_odb10, and the completeness was assessed to be 93.5%. The results of Hi-C chromosome-assisted assembly showed that the D. opposita genome was anchored on 20 pseudochromosomes (n = 20). Long terminal repeat (LTR) annotation showed that the LTR assembly index (LAI) score was 13.46 (Supplementary Fig. S2), meeting the reference standard[39].
We utilized 3D-DNA software for clustering, constructing interaction matrices, and creating interaction maps. Subsequently, we employed JuiceBox to correct errors in contig ordering and direction during the assembly process. Ultimately, 97 contigs were anchored to 20 pseudochromosomes, with a total length of 395,509 Mb, representing 95.52% of the original genome length. The longest and shortest pseudochromosomes were No. 5 (28.73 Mb) and No. 9 (15.29 Mb), respectively (Supplementary Table S4).
Using de novo prediction, homology annotation, and transcriptome prediction, 24,405 genes were anchored to the 20 chromosomes, with 20,168 genes showing differential expression across tissues (Fig. 2; Table 2). The average lengths of genes, coding sequences (CDS), and exons in D. opposita were 4,501, 1,206, and 5.16 bp, respectively, and the gene density across the genome was approximately 17.38 Kb. Functional annotation revealed that 96.20% of the genes could be annotated based on the databases: InterPro (72.71%), GO (70.23%), KEGG (91.46%), Swissprot (69.00%), TrEMBL (91.78%), and NR (96.16%) (Supplementary Table S5). We also identified 2,577 noncoding RNAs (ncRNA), including 65 microRNAs (miRNAs), 359 transfer RNAs (tRNAs), 2,058 ribosomal RNAs (rRNAs), and 95 small nuclear RNAs (snRNAs) (Supplementary Table S6). The repetitive sequences of D. opposita were annotated, with a total length of 254,089 Mb, of which 8.18% were DNA transposons, 51.58% were retrotransposons, and 0.03% were simple repeats and satellite DNA.
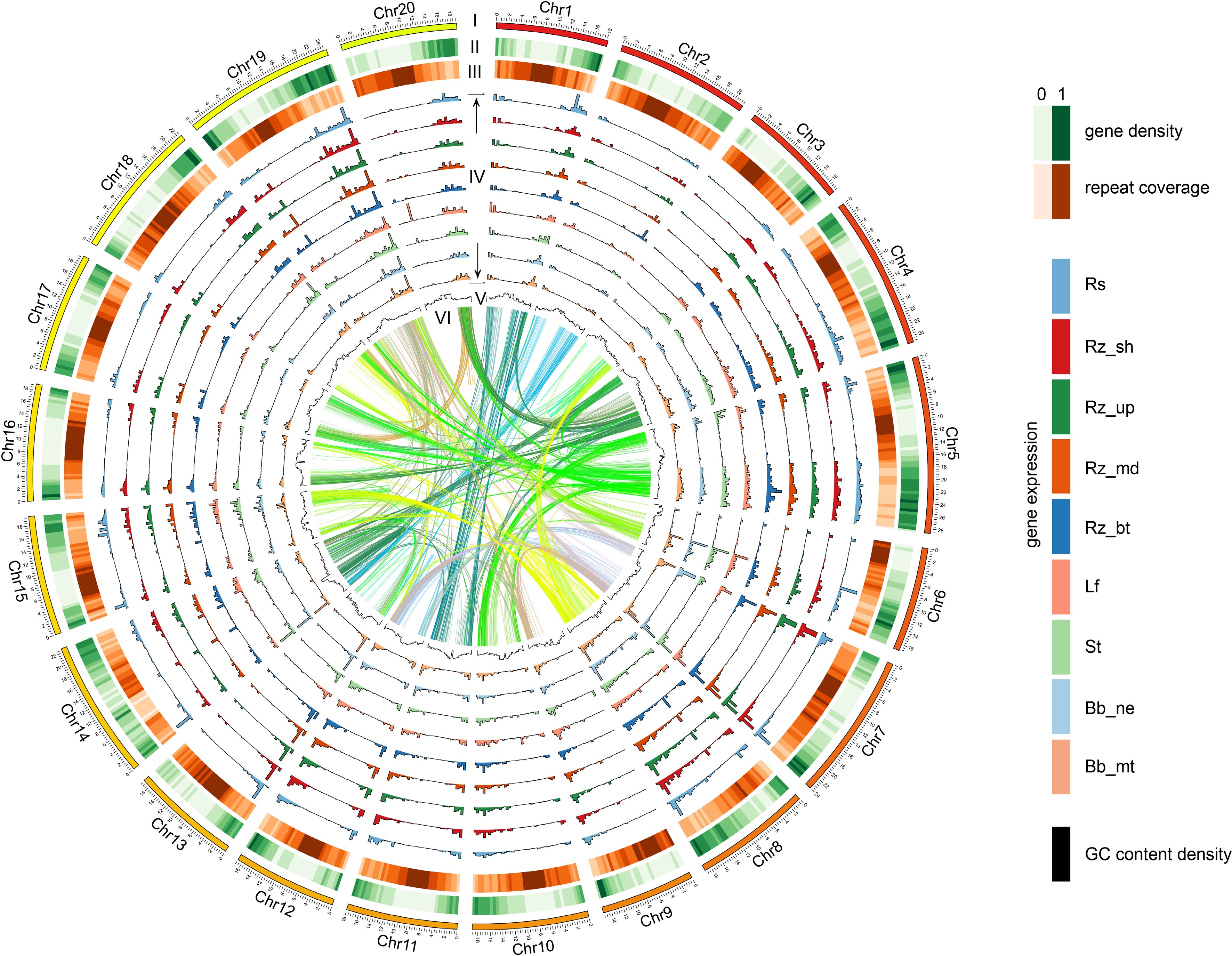
Figure 2.
The chromosomes of D. opposita. Twenty chromosomes were ordered by size (megabases) and featured in 500 kb intervals across the chromosomes. Tracks displayed are: I. chromosome; II. gene density; III. repeat coverage; IV. tissue specific expression level (from outer to inner: Rs, Rz_sh, Rz_up, Rz_md, Rz_bu, Lf, St, Bb_ne, Bb_mt); V. GC content density; VI. relationship between syntenic blocks, as indicated by lines. Each line represents a syntenic block; block size = 3 kb. Rs: rhizome peel with Rs, Rz_sh: rhizome shoot, Rz_up: rhizome upper part, Rz_md: rhizome middle part, Rz_bt: rhizome bottom part, Lf: leaf, St: stem, Bb_ne: new bulbil, Bb_mt: mature bulbil.
Table 2. Statistics for the D. opposita genome and gene prediction.
Feature Number Length (Mb) Percentage Assembly feature Estimated genome size 2,405.58 Assembled scaffold sequences
(> 1 kb)272 424.05 100% N50 scaffold 19.06 N90 scaffold 16.83 Max. scaffold 28.73 Assembled contig sequences
(> 1 kb)349 424.01 100% N50 contig 6.16 N90 contig 1.42 Max. contig 18.98 GC content 38.45% Insert gap 77 3.85 0.01% Chromosome 20 405.00 95.52% Anchored and oriented scaffolds 20 405.00 95.52% Genome annotation Total repetitive sequence 1,463,958 276.39 65.18% Genes 24,405 109.47 25.82% Genes in a chromosome 22,905 106.74 25.17% Noncoding RNAs 2,577 5.62 1.33% Additionally, flow cytometry analysis revealed that the DNA content of D. opposita was 1.5 times that of the closely related tetraploid D. zingiberensis (Supplementary Fig. S3), indicating that D. opposita is hexaploidy. It has been reported that the chromosome count of in D. opposita is 120[21], thus confirming that its chromosome ploidy is 6n = 120.
Comparative analysis of the D. opposita genome
-
The Dioscoreaceae family is an important family within the angiosperms, containing many economically significant plants such as yam, sweet potato, and Dioscorea species. According to Fig. 3a, the origin of Dioscoreaceae plants can be traced back to approximately 140 million years ago. Earliest Dioscoreaceae species, such as D. zingiberensis (Ginger yam), and D. cayenensis (Sweet yam) began to evolve in different ecological niches early on, with divergence times around 158.3 million years ago and 88.9 million years ago, respectively. This indicates that the ancestors of these species underwent a long evolutionary history and began to spread globally early on. Earlier-diverging species like D. alata (Greater yam) and D. rotundata (White yam) have divergence times of approximately 25.3 million years ago and 9.7 million years ago, respectively. D. alata is mainly distributed in tropical and subtropical regions, while D. rotundata is more commonly found in Africa. These species adapted to different environmental conditions, reflecting the independent evolution of Dioscoreaceae plants in different geographical regions.
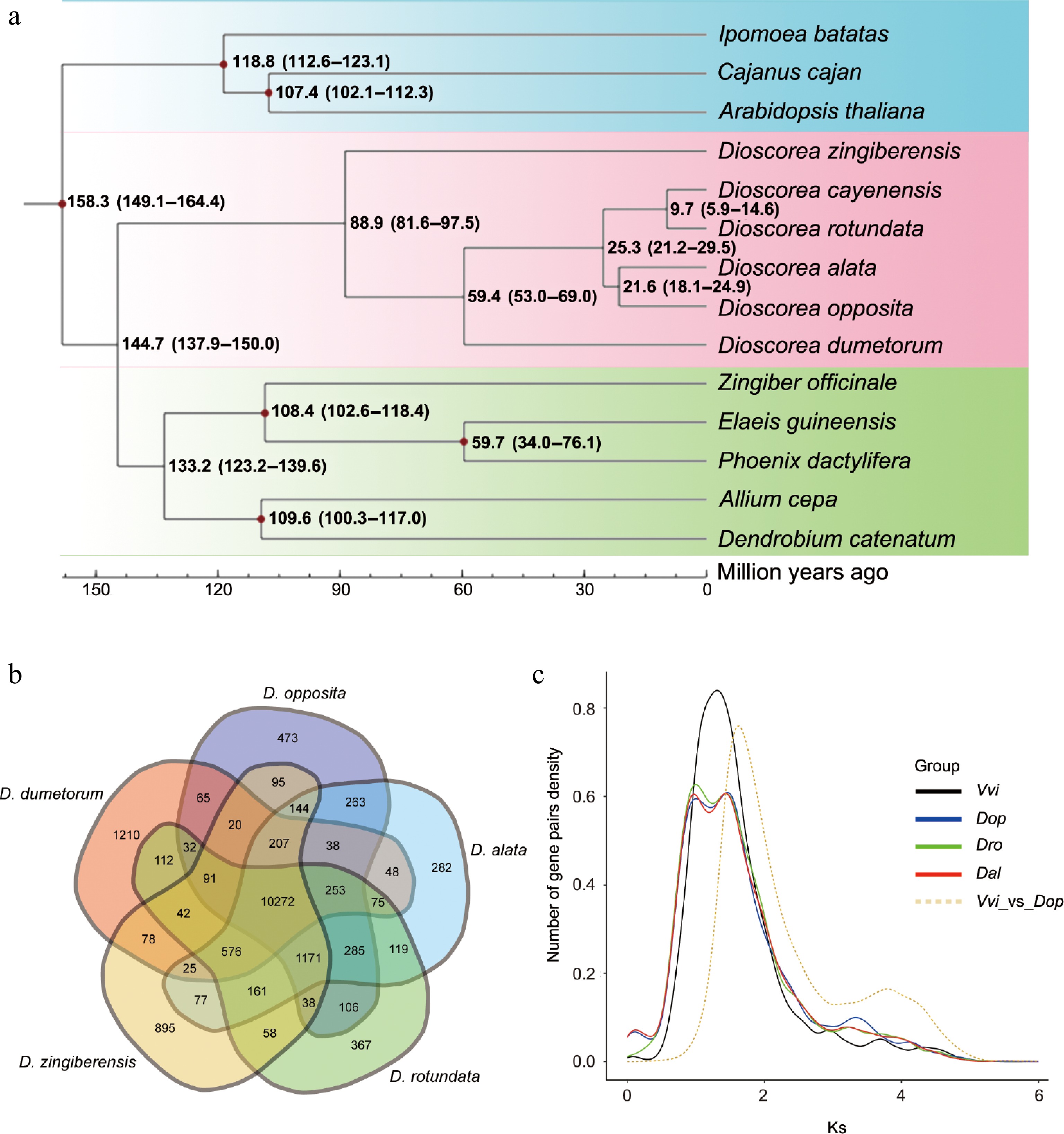
Figure 3.
Evolutionary and gene family analysis of D. opposita. (a) Evolutionary analysis shows the divergence time. (b) Venn diagram displays shared and different gene families among D. opposita, D. alata, D. rotundata, D. zingiberensis, and D. dumetorum. (c) Ks distribution of paralogs identified from V. vinifera (Vvi), D. opposita (Dop), D. alata (Dal), and D. rotundata (Dro).
The divergence time of D. opposita is about 21.6 million years ago, which is relatively young. This suggests that different Dioscoreaceae species exhibit high spatiotemporal diversity during their evolutionary process. D. opposita is a very important edible plant today, especially in China. Its divergence is closely related to environmental changes, such as climate shifts and geographic isolation, which may have contributed to the evolutionary independence of this species. Modern Dioscorea opposita is commonly found in temperate regions, and is becoming a major edible yam in those areas (Fig. 3a).
Whole-Genome Duplication (WGD) analysis
-
To estimate WGD timing in D. opposita, we used grape (Vitis vinifera, V. vinifera; one documented polyploidization event, γ-WGD) as a reference for Ks calculation[40,41]. Synonymous substitution rate distributions revealed concave peaks at proximate Ks positions for D. opposita, D. alata, and D. rotundata (Fig. 3c), suggesting concurrent WGD events. An additional flat peak at Ks ≈ 3.34 implies an earlier WGD in D. opposita. Using the formula T = Ks/2r (r = 6.98 × 10−9 substitutions/site/year)[42,43], WGD events were dated to approximately 71, 104, and 239 Mya. These results indicate more complex in D. opposita polyploidization than in related species, potentially explaining its elevated ploidy.
Synteny and gene family evolution
-
Synteny analysis demonstrated higher collinearity between D. opposita and D. alata (92.5% and 83.45%) than between D. opposita and D. rotundata (84.79% and 77.39%) (Fig. 4c), confirming closer homology with D. alata.
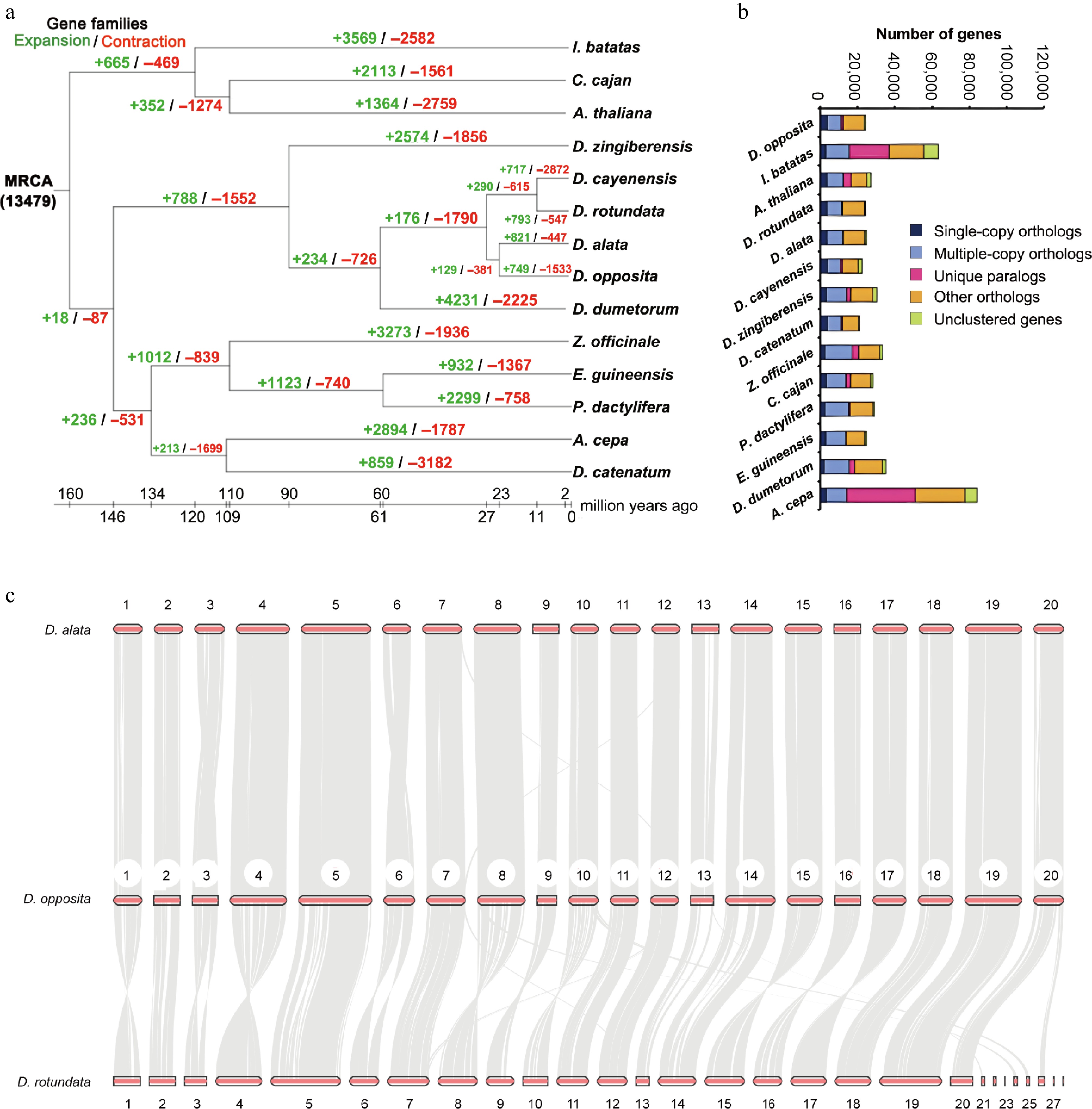
Figure 4.
Genomic comparative analysis. (a) Gene family expansion and contraction of multiple species. (b) Gene numbers of multiple species. (c) Synteny analysis between D. opposita and D. alata, or D. opposita and D. rotundata.
Among 13,553 gene families identified in D. opposita, 10,272 were shared with four related Dioscorea species, including D. alata, D. rotundata, D. zingiberensis, and D. dumetorum (Fig. 3b). Post-diversification, 749 families expanded while 1,533 contracted (Fig. 4a), indicating predominant gene loss. Single-copy and multi-copy genes each constituted ~50% of the genome (Fig. 4b), likely resulting from historical hybridization events during evolution and domestication.
Anthocyanin-related gene expression in D. opposita
-
The epidermis of the Tiegun yam rhizome forms Rs after FD, whereas other tissues, such as the bulbil (Bb), stem (St), and leaf (Lf) do not exhibit this phenomenon (Fig. 5a). Anthocyanin content determination revealed that the concentration of anthocyanins in Rs was significantly higher than in Bb, St, and Lf (Fig. 5b). KEGG pathway analysis indicated that genes with high expression in Rs, relative to bulbil, stem, and leaf, were enriched in the anthocyanin biosynthesis pathway (Fig. 5d, e). Furthermore, Gene Ontology (GO) enrichment analysis revealed that genes highly expressed in Rs, as compared to other tissues, were associated with pathways involved in light capture, light response, photosynthesis, and other signaling pathways (Supplementary Fig. S4a−c).
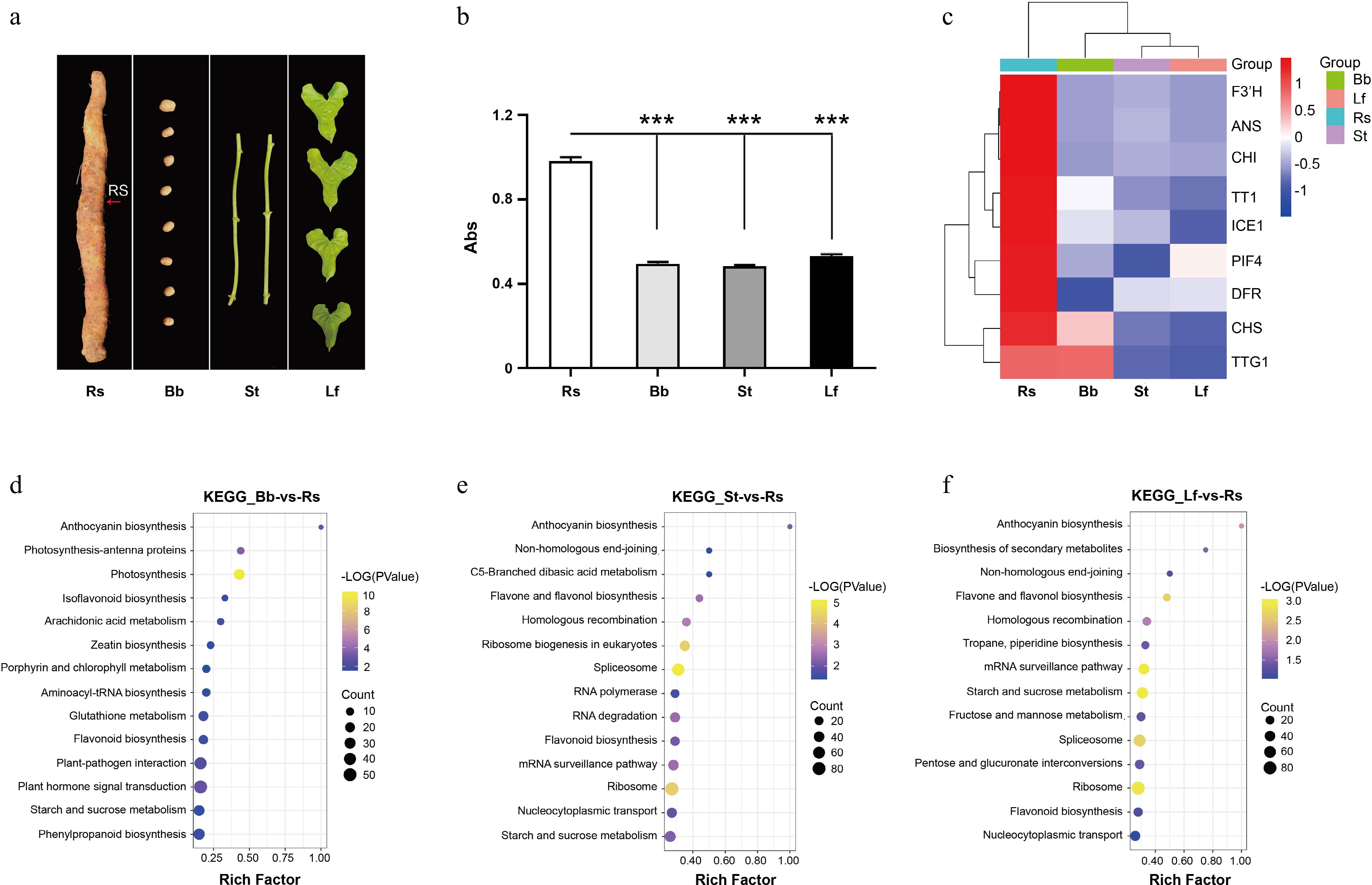
Figure 5.
Transcriptome analysis of D. opposita tissues. (a) Tissues of D. opposita, including rhizome with Rs as the red arrow points, bulbil (Bb), stem (St), and leaf (Lf). (b) UV spectroscopy detection of anthocyanin content in the tissues above, values are Mean ± SD, Student test, *** p < 0.001 compared with Rs (n = 3). (c) Expressions of anthocyanin biosynthesis and regulation related genes in Rs, Bb, St, and Lf of D. opposita. (d) KEGG enrichment analysis of up-regulated DEGs in Rs compared with those in Bb. (e) KEGG enrichment analysis of up-regulated DEGs in Rs compared with those in St. (f) KEGG enrichment analysis of up-regulated DEGs in Rs compared with those in Lf.
Gene expression analysis showed that, in contrast Bb, St, and Lf, the genes highly expressed in Rs included the transcription factors Transparent Testa 1 (TT1)[44] and Transparent Testa Glabra 1 (TTG1)[45], which regulate anthocyanin synthesis. Additionally, key enzymes involved in anthocyanin biosynthesis, including CHS, CHI, F3'H, ANS, DFR, and others, were also highly expressed. In addition, Phytochrome Interacting Factor 4 (PIF4)[46], a gene related to light signaling, and Inducer of CBF Expression 1 (ICE1)[47,48], associated with cold stress, were also highly expressed in Rs (Fig. 5c). These results suggest that the synthesis of anthocyanins in Rs is closely linked to the elevated expression of these related genes.
-
Tiegun yam, one of the most cultivated varieties of Huai yam (D. opposita), possesses significant medicinal and nutritional value, making it a crucial food and medicine variety. The Jiaozuo area of Henan Province, including Wen County, Boai County, Wuzhi County, Qinyang City, etc., is the authentic production area of Tiegun yam, and the geographical coordinates are between 112°51′39″ and 113°13′20″ east longitude and 34°52′ and 35°2′48″ north latitude. Historically, Huai yam has been esteemed in traditional medicine for its 'warming and tonic' properties and 'neutral in nature', solidifying its role as a staple tonic in Huai medicine formulations. As a prime example of 'medicine and food of the same origin', Tiegun yam stands out as the premium variety within Huai yam, commanding high demand in both medicinal and food markets.
Understanding the authentic characteristics of Tiegun yam is paramount for distinguishing it from other yam species and preventing adulteration. We observed that Rs appear as the plant enters dormancy following the FD period (Fig. 1a). As the temperatures drop post-FD in winter, the number and area of Rs progressively increase until harvest. Conventionally, Rs were perceived as lesions indicative of poorer quality. However, our research represents a significant paradigm shift. Through sustained observation and systematic investigation, we have established that Rs are not lesions but constitute a crucial authentic morphological feature of Tiegun yam. A key breakthrough finding underpinning this reclassification is the discovery that Rs are enriched with anthocyanins (Fig. 1b, c and Table 1). This specific association of anthocyanin accumulation with a distinct morphological marker (Rs) is a novel contribution rarely reported before.
Progress in elucidating the formation and regulatory mechanism of Rs was historically impeded by the absence of a reference genome for D. opposita. To address this critical gap and enable species-specific research inaccessible with the D. alata genome alone, we sequenced and assembled the first D. opposita genome. Our assembly yielded 424.05 Mb scaffolds and a 424.01 Mb high-quality reference genome sequence. While the haplotype chromosome number (n = 20) is conserved between D. alata and D. opposita, and evolutionary and synteny analyses confirm high homology (> 90% of D. opposita genes map to D. alata; Supplementary Table S7), the D. opposita assembly harbors 24,405 protein-coding genes compared to 25,189 in D. alata (Supplementary Table S7). The established high quality of the D. alata genome[19], and the demonstrated high similarity support the robustness of our D. opposita assembly. The release of this D. opposita genome is a foundational breakthrough: it uniquely enables detailed molecular investigation of species-specific traits like Rs formation and anthocyanins regulation in this authentic medicinal yam, significantly enriching Dioscoreaceae genomics beyond the existing D. alata resource. It provides the essential reference for gene function studies, germplasm resource development, and molecular breeding specifically for D. opposita traits.
Leveraging this novel D. opposita genome, we conducted the first molecular analysis of anthocyanin accumulation within the Rs feature. We confirmed Rs are unique to the rhizome epidermis, and absent in bulbils, stems, or leaves (Fig. 5a, b), with anthocyanin content in Rs vastly exceeding that in other tissues. Transcriptome analysis revealed that genes highly expressed in Rs, compared to other tissues, were significantly enriched in the anthocyanin biosynthesis pathway (Fig. 5d, e). Crucially, we identified the coordinated upregulation of key structural genes, including F3'H, ANS, CHI, DFR, and CHS[49]; and regulatory genes, mainly contain the MBW complex TT1 and TTG1, master regulators of anthocyanin biosynthesis[44,45,50]; genes responsive to environmental cues, notably ICE1 and PIF4 (Fig. 5c). ICE1, a key inducer of anthocyanin biosynthesis under cold stress[47], is up-regulated in the cold season, which is consistent with the formation of Rs under cold weather conditions. As reported, PIF4 down-regulates the expression of CBF genes, thereby reducing cold tolerance in Arabidopsis[46,51]. In contrast, in D. opposita, PIF4 is up-regulated during winter, potentially due to species-specific differences in the function of PIF4. This result indicates the importance of genome assembly of D. opposita for studying the specific molecular regulation of Tiegun yam. The identification of this specific cold-responsive transcriptional network driving localized anthocyanin accumulation to form the Rs is a major mechanistic discovery. Additionally, the finding also highlights the significance of the D. opposita genome assembly for understanding a broader molecular regulatory mechanism in Tiegun yam.
Genome sequencing and assembly are foundational in genomics. The D. opposita genome assembled in this study provides an indispensable species-specific tool, enabling breakthroughs unavailable with the D. alata reference alone. It is critical for deciphering the biosynthesis and regulation of anthocyanins in the Rs, a hallmark of authenticity. Furthermore, this genome will accelerate the construction of genetic maps, facilitate molecular-assisted breeding strategies, and empower the creation of improved Tiegun yam varieties with enhanced anthocyanin content or other desirable traits. Ultimately, this resource is vital for advancing research on the authentic characteristics of Tiegun yam, improving its quality and yield, and unraveling the molecular basis of its unique medicinal and agricultural value.
We thank Ms. Cuihong Lu, Wen County Academy of Agriculture Sciences, for providing Tiegun yam germplasm resources. This work was supported by the National Natural Science Foundation of China (Grant No. 82473890), the Natural Science Foundation of Henan Province (Grant No. 232300421127), and the Open Project Fund of Collaborative Innovation Center of Research and Development on the Whole Industry Chain of Yu-Yao, Henan Province (Grant No. 2024YYXT-KFKT-01).
-
The authors confirm their contributions to the paper as follows: study conception and experiment design: Ma B, Zheng X; funding acquisition and manuscript revising: Ma B; manuscript writing and refining and data analysis: Gao J; preparation of purified anthocyanin samples for the metabolome and determination of anthocyanin content: Liu M; preparation of Tiegun yam samples for genome sequencing, transcriptome sequencing, and determination of anthocyanin content: Ma H, Miao S; preparation of Tiegun yam tissues for phenotypic observation: Wang L, Wen C. All authors reviewed the results and approved the final version of the manuscript.
-
The data that support this study are available in National Center for Biotechnology Information (NCBI), and will be available once the article is accepted. The D. opposita genome assembly has been deposited in the NCBI under BioProject accession code PRJNA1207442. Other sequencing data is deposited under Biosample accession code SAMN46126475.
-
The authors declare that they have no conflict of interest.
- Supplementary Table S1 Quality values of the assembled contigs and scaffolds.
- Supplementary Table S2 Summary of D. opposita genome assembly and annotation.
- Supplementary Table S3 Summary of sequencing data used for genome assembly.
- Supplementary Table S4 Hi-C assembly statistics.
- Supplementary Table S5 Functional annotation statistical results.
- Supplementary Table S6 Non-coding RNA annotation result statistics.
- Supplementary Table S7 Published genome information statistics.
- Supplementary Fig. S1 Rs only exists in Tiegun yam rhizome epidermis.
- Supplementary Fig. S2 Hi-C heat map and distribution map of LAI for each chromosome of D. opposita.
- Supplementary Fig. S3 D. opposita chromosome ploidy detection.
- Supplementary Fig. S4 GO enrichment analysis of Tiegun yam tissues.
- Copyright: © 2025 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Gao J, Liu M, Miao S, Wang L, Ma H, et al. 2025. The Dioscorea opposita genome provides insights into anthocyanin biosynthesis in Tiegun yam. Medicinal Plant Biology 4: e028 doi: 10.48130/mpb-0025-0027 

The Dioscorea opposita genome provides insights into anthocyanin biosynthesis in Tiegun yam
- Received: 15 April 2025
- Revised: 18 July 2025
- Accepted: 24 July 2025
- Published online: 28 August 2025
Abstract: Tiegun yam (Dioscorea opposita Thunb., D. opposita) is the most widely cultivated variety and a premium product among Huai yams, which are considered one of the 'Four Huai Medicines' in China. The presence of 'Rusty spot' (Rs) on the rhizome epidermis of Tiegun yam is a characteristic feature indicative of its superior quality and authenticity. However, the mechanisms underlying the formation and regulation of Rs remain largely unexplored. This knowledge gap is primarily due to the absence of a reference genome for D. opposita, which hinders in-depth molecular research. In this study, we sequenced and assembled the genome of D. opposita, producing a 424.01 Mb high-quality reference genome anchored to 20 pseudochromosomes (n = 20) in haplotype. We identified 24,405 protein-coding genes. Metabolomic and transcriptomic analyses revealed that Rs is rich in anthocyanins, with up-regulated expression of genes involved in the biosynthesis, environment regulation, and signaling pathways of anthocyanins in Rs compared to other tissues. The genomic assembly of D. opposita provides valuable insights into its genetic background, facilitates the identification of genes controlling important agronomic traits, and supports the development of high-quality germplasm resources for Tiegun yam and potentially other yam species.
-
Key words:
- Tiegun yam /
- Dioscorea opposita /
- Genome /
- Anthocyanin biosynthesis